

Abstract
Nicotine and alcohol are often co-abused suggesting a common mechanism of action may underlie their reinforcing properties. Both drugs acutely increase activity of ventral tegmental area (VTA) dopaminergic (DAergic) neurons, a phenomenon associated with reward behavior. Recent evidence indicates that nicotinic acetylcholine receptors (nAChRs), ligand-gated cation channels activated by ACh and nicotine, may contribute to ethanol-mediated activation of VTA DAergic neurons although the nAChR subtype(s) involved has not been fully elucidated. Here we show that expression and activation of nAChRs containing the α6 subunit contribute to ethanol-induced activation of VTA DAergic neurons. In wild-type (WT) mouse midbrain sections that contain the VTA, ethanol (50 or 100 mM) significantly increased firing frequency of DAergic neurons. In contrast, ethanol did not significantly increase activity of VTA DAergic neurons in mice that do not express CHRNA6, the gene encoding the α6 nAChR subunit (α6 knock-out (KO) mice). Ethanol-induced activity in WT slices was also reduced by pre-application of the α6 subtype-selective nAChR antagonist, α-conotoxin MII[E11A]. When co-applied, ethanol potentiated the response to ACh in WT DAergic neurons; whereas co-application of ACh and ethanol failed to significantly increase activity of DAergic neurons in α6 KO slices. Finally, pre-application of α-conotoxin MII[E11A] in WT slices reduced ethanol potentiation of ACh responses. Together our data indicate that α6-subunit containing nAChRs may contribute to ethanol activation of VTA DAergic neurons. These receptors are predominantly expressed in DAergic neurons and known to be critical for nicotine reinforcement, providing a potential common therapeutic molecular target to reduce nicotine and alcohol co-abuse.
1. Introduction
Nicotine and alcohol are the predominant co-abused drugs in the world. A significant proportion of alcoholics smoke and the majority of smokers (~60 %) binge drink or consume significant amounts of alcohol [1, 2] suggesting a shared mechanism of action between ethanol and nicotine, the addictive component of cigarette smoke. In the mesocorticolimbic “reward” circuitry of the brain, both drugs stimulate dopaminergic (DAergic) neurons in the ventral tegmental area (VTA), to elicit drug reinforcement [3-6].
Neuronal nicotinic acetylcholine receptors (nAChRs) are ligand-gated cation channels that, are activated by the endogenous neurotransmitter, ACh [7], as well as the tertiary alkaloid, nicotine. Twelve vertebrate genes encoding neuronal nAChR subunits have been identified (α2-α10, β2-β4) with five subunits coassembling to form a functional receptor [7]. High affinity nAChRs are heteromeric and consist of two or three α subunits coassembled with two or three β subunits. Low affinity receptors are mostly homomeric, predominantly consisting of α7 subunits. Several nAChR sub-types contribute to nicotine-mediated activation of DAergic neurons and reward/reinforcement including α4β2* (* indicates other unidentified subunits may coassemble with the indicated subunits), α6β2*, and α4α6β2* [8-13]. Of the critical VTA nAChR subunits, α6 is predominantly expressed in DAergic neurons [14, 15].
While ethanol is not a direct agonist of nAChRs, ethanol may induce an increase in ACh release from cholinergic neuron inputs into the VTA, thereby driving an increase in DAergic neuron activity via cholinergic signaling through nAChRs [16]. Interestingly, VTA infusion of α-conotoxin MII, a nAChR antagonist selective for α6, α3, or β3* nAChRs reduces ethanol induced NAc DA release, ethanol consumption and reinforcement in rodents [17, 18]. In addition, VTA DAergic neurons activated by acute ethanol express greater α6 nAChR subunit transcripts compared to DAergic neurons not activated by ethanol [19]. Interestingly, polymorphisms in CHRNA6, the gene encoding the α6 subunit has been associated with alcohol consumption in a representative sample of patients in the United States [20]. More recently, it has been shown that BAC transgenic mice expressing mutant α6 nAChR subunits that render α6* nAChRs hypersensitive to ACh consume more ethanol and are more sensitive to ethanol reward[21]. While these data implicate a role for α6* nAChRs in ethanol consumption and reinforcement, a direct involvement of this nAChR subtype in ethanol-induced activation of VTA DAergic neurons has not been demonstrated.
We sought to test the hypothesis that α6* nAChRs may be involved ethanol-induced activation of VTA DAergic neurons. Using a combination of pharmacology, genetics, and electrophysiology in a mouse midbrain slice preparation, we demonstrate that nAChRs containing the α6 subunit contribute to ethanol-mediated activation of VTA DAergic neurons.
2. Materials and Methods
Animals
α6 knockout (KO) homozygous mice and wild-type (WT) litter-mates (6-10 weeks of age) were used in all experiments. The α6 KO line has been back-crossed to the C57BL/6J strain at least nine generations. WT and KO animals were derived from heterozygous crosses. The genetic engineering of the α6 KO line has been described previously [14]. Animals were housed four/cage until the start of each experiment. Animals were kept on a standard 12-h light/dark cycle with lights on at 7:00 AM and off at 7:00 PM. Mice had access to food and water ad libitum. All experiments were conducted in accordance with the guidelines for care and use of laboratory animals provided by the National Research Council [22], as well as with an approved animal protocol from the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School.
2.1 Slice preparation
Mice were anesthetized by intraperitoneal (i.p.) injection of sodium pentobarbital (200 mg/kg, Vortech Pharmaceuticals, Dearborn, MI) and then decapitated. Brains were quickly removed and placed in an oxygenated ice-cold high sucrose artificial cerebrospinal fluid (SACSF) containing kynurenic acid (1 mM, Sigma, St. Louis, MO). Sagittal brain slices containing VTA and lateral dorsal tegmental brain regions (~180 μm) were made using a Leica VT1200 vibratome. The brain slices were incubated in oxygenated EBSS solution supplemented with glutathione (1.5 mg/ml, Sigma), N-ω-nitro-L-arginine methyl ester hydrochloride (2.2 mg/ml, Sigma), pyruvic acid (11 mg/ml, Sigma) and kynurenic acid (1 mM, Sigma) for 45 minutes at 34°C. Slices were transferred into oxygenated artificial cerebrospinal fluid (ACSF) at room temperature (24°C) for recording. SACSF solution contains (in mM, all chemicals purchased from Sigma): 250 sucrose, 2.5 KCl, 1.2 NaH2PO4•H2O, 1.2 MgCl2•6H2O, 2.4 CaCl2•2H2O, 26 NaHCO3, 11 D-Glucose. ACSF solution contains (in mM): 125 NaCl, 2.5 KCl, 1.2 NaH2PO4•H2O, 1.2 MgCl2•6H2O, 2.4 CaCl2•2H2O, 26 NaHCO3, 11 D-Glucose.
2.2 Electrophysiological recordings
Individual slices were transferred to a recording chamber continually superfused with oxygenated ACSF (30–32 °C) at a flow rate of ~ 2 ml/min. Cells were visualized using infrared differential interference contrast (IR–DIC) imaging on an Olympus BX-50WI microscope. Electrophysiological recordings were recorded using a Multiclamp 700B patch-clamp amplifier (Axon Instruments, Foster City, CA). Action potentials (APs) were obtained in the cell-attached configuration and gap-free acquisition mode in Clampex (Axon Instruments).
Ih currents in whole-cell configuration were elicited every 20 s by stepping from -60 mV to a test potential of -120 mV. APs and currents were filtered at 1 kHz using the amplifier's four-pole, low-pass Bessel filter, digitized at 10 kHz with an Axon Digidata 1440A interface and stored on a personal computer. The junction potential between the patch pipette and bath ACSF was nullified just prior to obtaining a seal on the neuronal membrane. At the end of recording, the cytoplasm was sucked into the recording pipette and the contents in the pipette were expelled into a microfuge-tube for single-cell PCR experiments to verify tyrosine hydroxylase (TH) expression as previously described [13].
Pipette solution contained (in mM, all chemicals purchased from Sigma): 121 KCl, 4 MgCl2•6H2O, 11 EGTA, 1 CaCl2•2H2O, 10 HEPES, 0.2 GTP and 4 mM ATP. Pipette solution was made with DEPC-treated distilled water to prevent RNA degradation.
ACSF was used for bath solution. Ethanol was added into ACSF. Ethanol, ACh, and α-conotoxin MII[E11A] were applied to each slice by gravity superfusion. α-conotoxin MII[E11A] was synthesized as previously described [23]. APs were recorded in the presence of a cocktail of inhibitors (Sigma) including atropine (1 μM) to block muscarinic receptors, bicuculline (20 μM) to block GABAA receptors, and CNQX (10 μM) to block AMPA receptors.
2.3 Data analysis
AP spikes were detected using a threshold detection protocol contained within pClampfit (pClamp v10.2, Axon Inst., Molecular Devices). Average fold change in AP frequency are presented as means ± standard errors of means (SEM). Paired t-tests were used to analyze differences between AP frequency at baseline (1 minute prior to drug application) and after 10 min application of drug. Unpaired t-tests were used to analyze data between genotypes or drug treatment unless otherwise indicated. Results were considered significant at p < 0.05.
3. Results
To test the involvement of α6* nAChRs in ethanol-mediated activation of VTA DAergic neurons, we recorded from VTA DAergic neurons in midbrain slices from mice that did not express CHRNA6, the gene encoding the α6 subunit (α6 KO mice) and their wild-type (WT) littermates. Slices containing the VTA were cut in the sagittal plane (Fig. 1A). DAergic neurons typically displayed a relative slow firing rate (1-5 hz, Fig. 1B) and a slow inward current upon hyperpolarization (Fig. 1C). DAergic neuron identity was verified after recording by detection of tyrosine hydroxylase (TH) mRNA as determine by single neuron RT-PCR (Fig. 1D). To test the effects of ethanol on DAergic neuron activity, AP frequency was monitored in cell-attached mode at baseline, during application of an intoxicating concentration of ethanol (50 or 100 mM), and after washout. Because the focus of our experiments was to uncover the contribution of nAChR activation in response to ethanol, recordings were made in the presence of a cocktail of inhibitors to block, muscarinic receptor, AMPA receptor, and GABAA receptor activity (see methods). No significant difference in baseline firing frequency of DAergic neurons was observed between WT and α6 KO animals (5.0 ± 1.8 and 4.0 ± 1.0 Hz, respectively). In WT VTA DAergic neurons, ten minute bath application of both ethanol concentrations produced an increase in AP frequency (~33 % and 35 % increase from baseline, p < 0.001, respectively, Fig 2A, D) that was completely reversed upon wash out. In contrast, ethanol did not significantly increase VTA DAergic neuron activity above baseline in α6 KO mice (Fig. 2B, D) in response to either concentration of ethanol. To simulate a more in vivo situation, responses to 100 mM ethanol were recorded in WT and α6 KO DAergic neurons in the absence of the inhibitor cocktail. In WT slices, 10 min bath application of 100 mM ethanol increased DAergic neuron AP frequency 1.42 ± 0.10 fold compared to baseline (n = 8). In contrast 100 mM ethanol increased AP frequency significantly less in α6 KO VTA DAergic neurons, (1.03 ± 0.057 fold, n = 7) compared to WT (p < 0.01, data not shown).
Figure 1.
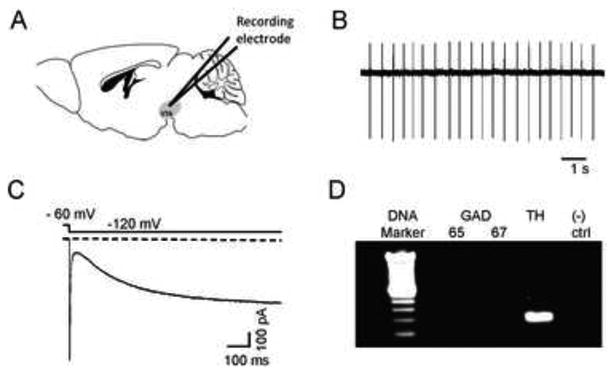
Characteristics of DAergic neurons in VTA sagittal slices. Illustration of a mouse sagittal section containing the VTA (shaded region). B) Representative cell-attached recording from a putative DAergic neuron. DAergic neurons had characteristic low baseline firing frequencies (1-5 Hz) and as shown in C), expressed the hyperpolarizing activated cation current, Ih. Currents were elicited by a hyperpolarizing step from a holding potential of -60 mV to -120 mV as indicated. D) At the end of each recording, the content of each neuron was aspirated into the patch pipette and TH expression was verified by single-cell real-time PCR. A representative DNA agarose gel is shown illustrating a typical result for a DAergic neuron. Only neurons that clearly expressed TH and not GAD 65/67 were included in the analysis. As a negative control for the PCR, a sample containing no RNA was used (“(-) ctrl”).
Figure 2.
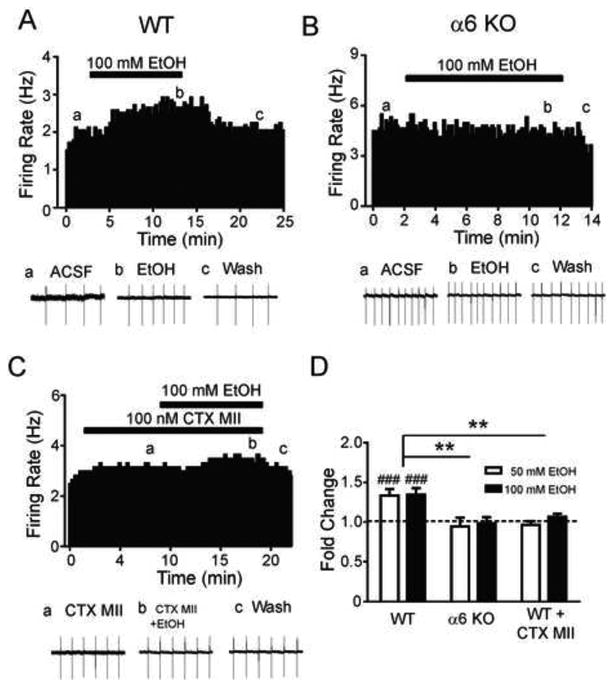
A contribution of a6* nAChRs to ethanol activation of VTA DAergic neurons. Representative action potential firing frequency histogram from an A) WT or B) α6 KO VTA DAergic neuron before, during, and after 10 min bath application of 100 mM ethanol (EtOH). C) Representative action potential firing frequency histogram from a WT VTA DAergic neuron before, during, and after 10 min bath application of 100 mM ethanol in the presence of a-CTX-MII[E11A]. Action potentials were recorded in cell-attached mode. Representative action potential traces (bottom of each panel, a, b, c) are shown from the corresponding times on the histograms. D) Fold-change in average firing frequency at baseline (1 min. prior to alcohol application, dotted line) compared to last min of ethanol application for each genotype/condition. ###p < 0.001 compared to baseline using a paired t-test; **p < 0.01, unpaired t-test. n = 5-8 neurons/condition.
To further test the hypothesis that activation of α6* nAChRs was necessary for the observed ethanol-mediated increase in VTA DAergic neuron activity, we bath applied 100 nM α-conotoxin MII[E11A] (α-CTX-MII[E11A]), an α6* nAChR subtype-selective antagonist [23], prior to and during application of ethanol (Fig. 2C, D) in WT slices. In the presence of α-CTX-MII[E11A], both 50 mM and 100 mM ethanol failed to significantly increase DAergic neuron AP frequency above baseline (Fig. 2C, D). Together these data suggest that activation of nAChRs containing the α6 subunit is necessary for the observed effects of ethanol on VTA DAergic neurons in midbrain slices.
Previously, we demonstrated that ethanol potentiates the DAergic neuron response to ACh in midbrain slices[24]. To test the contribution of α6* nAChRs to this effect we recorded WT and α6 KO VTA DAergic neuron activity in response to bath application of 300 μM ACh alone or in combination with 50 mM ethanol. When applied alone, ACh increased AP frequency of VTA DAergic neurons in WT and α6 KO slices (~20 % and 12 %, respectively, Fig. 3A, B, F). Two-way repeated measures ANOVA revealed a main effect of ACh on AP frequency over time (F18,180 = 3.96, p < 0.001, Fig. 4A) but not an effect of genotype. Co-application of ACh and ethanol in WT slices robustly increased DAergic neuron activity and this increase was significantly greater than ACh alone (p < 0.05, Fig. 3C, F). In contrast, VTA DAergic neuron activity in response to co-application of ACh and ethanol in α6 KO slices was significantly decreased compared to WT after co-application (p < 0.001, Fig. 3D, F). In addition, two-way repeated measures ANOVA revealed a main effect of ACh plus ethanol on AP frequency over time (F13,156 = 4.63, p < 0.001) an effect of genotype (F1,12 =16.05, p <0.001), and a significant time × genotype interaction (F13, 156 = 5.60, p < 0.001, Fig. 4B). Interestingly, DAergic neuron activity in response to ACh and ethanol was also significantly reduced in α6 KO slices compared to ACh alone (Fig. 3F). Finally, we also recorded the effect of ACh and ethanol on WT VTA DAergic neuron activity in the presence of α-CTX-MII[E11A].
Figure 3.
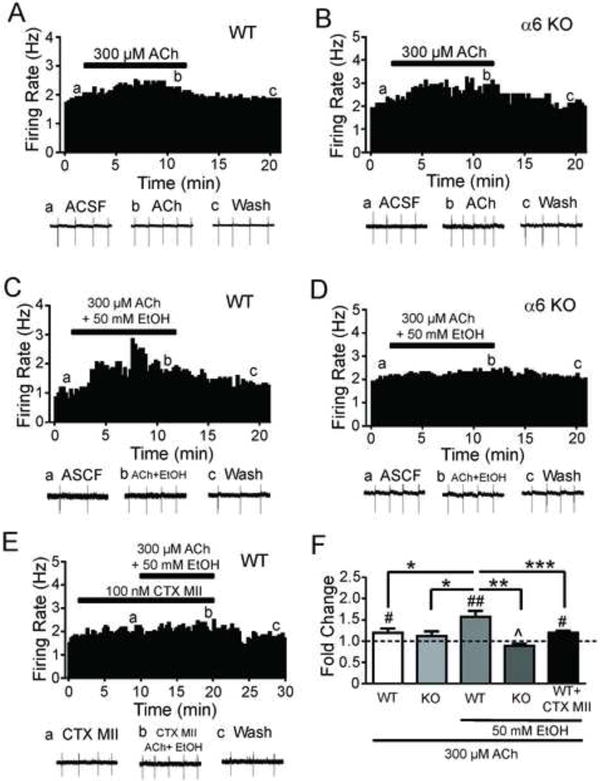
α6* nAChR expression and activation is necessary for alcohol potentiation of DAergic neuron responses to ACh. Representative action potential firing frequency histogram from an A) WT or B) α6 KO VTA DAergic neuron before, during, and after 10-min bath application of 300 μM ACh. Representative action potential traces (bottom of each panel, a, b, c) are shown from the corresponding times on the histograms. Representative action potential firing frequency histogram from a C) WT or D) α6 KO VTA DAergic neuron before, during, and after 10-min bath co-application of 300 μM ACh and 50 mM ethanol. E) Representative action potential firing frequency histogram from a WT VTA DAergic neuron before, during, and after 10 min bath co-application of 300 μM ACh and 50 mM ethanol in the presence of 100 nM α-CTX-MII[E11A]. F) Fold-change in average DAergic neuron firing frequency in response to 300 μM ACh alone in WT (n = 8) or α6 KO slices (n = 6), in the presence of 50 mM ethanol (n = 12 and 11, respectively), or in the presence of 50 mM ethanol and α-CTX-MII[E11A] in WT slices (n = 8). #p < 0.05, ##p<0.01 compared to baseline as in 1D. *p < 0.05, **p < 0.01, ***p < 0.001 response to ethanol compared between treatments/genotypes. ^p < 0.05 compared to ACh in α6 KO DAergic neurons.
Figure 4.

Time course of ACh and ACh + ethanol modulation of DAergic neuron activity. The effects of A) ACh alone or B) with ethanol on average normalized AP frequency in WT and α6 KO VTA DAergic neurons under each condition are shown (from Figure 3). The effect of ACh with ethanol in the presence of α-CTX-MII[E11A] in WT VTA DAergic neurons is also shown (red circles). ACh ± ethanol was applied at the times indicated by the bar. Recording times between groups were aligned based on time of ethanol application to facilitate comparison.
DAergic neuron activity in response to ACh and ethanol was reduced by pre-application of α-CTX-MII[EllA] compared to activity in response to ACh and ethanol alone (Fig. 3E, F). Two-way repeated measures ANOVA also revealed a main effect of ACh plus ethanol on AP frequency over time (F13, 221 = 16.1, p < 0.001) an effect of α-CTX-MII[EllA] treatment (F1,17 =15.83, p <0.001), and a significant time × α-CTX-MII[EllA] treatment interaction (F13, 156 = 4.30, p < 0.001, Fig. 4B). Together, these data indicate that α6* nAChR activation is necessary for ethanol potentiation of ACh response in VTA DAergic neurons.
4. Discussion
Previous studies indicate that cholinergic signaling through neuronal nAChRs may play a role in ethanol reinforcement and ethanol-induced DA release in NAc [25]. For example, systemic or VTA infusion of the non-specific nicotinic antagonist mecamylamine has been shown to reduce ethanol consumption, reinforcement, and ethanol-mediated NAc DA release in rodents and also subjective euphoric responses to ethanol in humans [26-30].
Here we show that expression and activation of nAChRs containing the α6 subunit are necessary for ethanol activation of VTA DAergic neurons in mouse midbrain slices. Ethanol did not significantly increase DAergic neuron firing frequency in α6 KO slices and the increase in DAergic neuron activation observed in WT slices was significantly reduced by an α6* nAChR selective antagonist. One caveat to the results from the present study is that all experiments were done in midbrain slices where critical inputs necessary for ethanol action may be severed. In addition, drugs of abuse including ethanol induce burst firing of VTA DAergic neurons which is difficult to detect in slice preparations [31]. Finally, because the focus of our experiments was on cholinergic signaling through nAChRs in DAergic neurons, glutamatergic, GABAergic, and muscarinic signaling was blocked in the majority of experiments. This could influence DAergic neuron activity in response to ethanol [32-34]. Because of these methodological limitations, in vivo electrophysiology and single unit recordings preferably in awake behaving animals will need to be done to verify a role for α6* nAChR in ethanol activation of the VTA.
α6* nAChRs have recently emerged as unique mediators of nicotine reinforcement and VTA DAergic neuron activation [10-12]. They are almost exclusively expressed in midbrain DAergic neurons [14, 15, 35] but may also be expressed in VTA GABAergic presynaptic terminals [36]. Previous studies have shown that infusion of the non-mutated version of α-conotoxin MII, a nAChR antagonist selective for α3β2*, β3*, and α6* subtypes (as opposed to α-conotoxin MII[E11A] used in the present study that is selective for α6* nAChRs), reduces ethanol consumption and reinforcement in rats and NAc DA release in mice [17, 18, 37]. Our data suggest that behavioral effects of α-conotoxin MII infusion in the VTA are most likely influenced by direct blockade of ethanol-mediated DAergic neuron activation through α6* nAChRs. If α6* nAChRs are critical for ethanol-mediated activation of VTA DAergic neurons, then one would expect that α6 KO mice would exhibit reduced ethanol-mediated behaviors associated with reward such as consumption. However, a recent study found no difference in ethanol consumption or preference in a two bottle 24 hr access model of ethanol intake between α6 KO mice and WT littermates [38]. In contrast, Powers et al. determined that mice expressing α6* nAChRs hypersensitive to ACh consume more ethanol than WT littermates and condition a place-preference in response to sub-reward threshold doses of ethanol indicating a role for α6* nAChRs in ethanol reward behavior [21]. Because ethanol is a non-specific drug and interacts with multiple gene targets including those that modulate DAergic neuron activity [4, 39], caution is warranted in interpreting these apparent discrepant results. In the absence of α6* nAChR expression, compensatory mechanisms may occur such that ethanol reward is maintained [14, 40]. In addition, behavioral assays used to assess ethanol reward value such as conditioned place preference and two bottle choice tests may be influenced by ethanol effects on non-reward behaviors such as anxiety which may be altered in nAChR mouse models.
In the future, it will be critical to identify the relevant nAChR subunits that coassemble with α6 to form the nAChRs that mediate ethanol-induced activation of DAergic neurons. Previous studies indicate that the β3 subunit almost exclusively co-assembles with α6 subunits in DAergic neurons and the β2 subunit is required for functional α6* nAChRs [14, 41]. In addition, two distinct sub-types of α6* nAChRs have been identified: α4α6β2β3* and α6(non-α4)β2β3* [35, 42]. Both subtypes are expressed at the level of VTA DAergic neuron soma [12, 13, 43]. Interestingly, we have previously demonstrated that DAergic neurons more sensitive to ethanol-induced activation over-express transcripts encoding α4, α6, and β3 nAChR subunits compared to less alcohol sensitive DAergic neurons [19]. We have also shown that α4* nAChRs contribute to ethanol activation of VTA DAergic neurons to influence alcohol reward [24]. Combined with the findings from the results of Powers et al. discussed above, it is possible that α4α6β2β3 nAChRs may significantly contribute to ethanol-induced activation of VTA DAergic neurons. Based on these data, selective antagonists, partial agonist, or desensitizing agents that target α6* nAChR subtype might have therapeutic value to help reduce ethanol consumption or at least blunt acute ethanol-induced reinforcement. Several nAChR ligands that reduce ethanol intake in animal studies have been identified including cytisine, lobeline, sazetidine-A, CP-601932, PF-4575180, and varenicline [29, 44-46]. Of these compounds, at least cytisine and varenicline can interact with α6* nAChRs potentially contributing to their effects on ethanol consumption; whereas the ability of the other ligands to interact with α6* nAChRs has not been tested [47, 48].
Many questions remain regarding the mechanistic bases for ethanol effects on cholinergic signaling within the VTA that leads to activation of DAergic neurons and increased DA release in the NAc. It is hypothesized that ethanol consumption increases ACh concentrations in the VTA (Fig. 5)[16]. It is assumed that the increase in ACh mediated by ethanol originates from cholinergic projection from the lateral dorsal tegmentum or pedunculopontine nucleus but the actual origin of ACh and the mechanism by which ethanol increases ACh in the VTA is unknown. Regardless, if ethanol increases ACh concentrations in the VTA, then this could activate DAergic neurons via nAChRs. Ethanol also potentiates the response to ACh at α4 and α6* and/or α4α6* nAChRs expressed in DAergic neurons which, in turn, could drive activation of VTA DAergic neurons increasing DA release in NAc. Because α4* and α6* nAChRs are also robustly expressed in DAergic neuron terminals which receive ACh from tonically active cholinergic interneurons in striatum [11, 48-51], one would expect that ethanol should also potentiate DA release from DAergic presynaptic terminals but this has yet to be demonstrated. Finally, neuronal nAChRs are also robustly expressed in VTA GABAergic neurons in addition to DAergic neurons but the role of the receptors in ethanol effects in the VTA is unknown [52-54]. Interestingly, a recent study indicates that activation of β2* nAChRs in both VTA GABAergic and DAergic neurons is required for nicotine reinforcement raising the interesting possibility that ethanol action on GABAergic VTA neurons may also be critical for ethanol reward [55].
Figure 5.
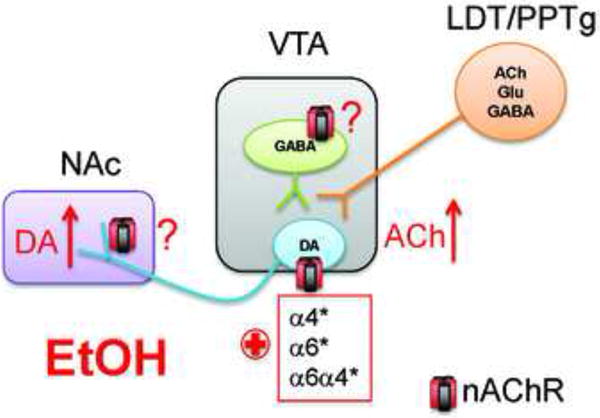
Ethanol actions in mesolimbic circuitry. In the VTA, ethanol stimulates DAergic neurons at least, in part, via nAChR activation. Ethanol increases ACh release (red arrow, presumably through cholinergic projection from the LDT/PPTg) which in turn activates nAChRs on DAergic neurons driving activity. Neuronal nAChRs mediating this effect contain α4 and/or α6 subunits. In addition, ethanol potentiates ACh activation at these high affinity nAChRs (red plus sign). The effect of ethanol on additional nAChRs in the mesolimbic reward circuitry including those expressed at DAergic neuron presynaptic terminals and in GABAergic neurons is unknown (red question marks). Ultimately activation of nAChRs in combination with other effects of ethanol in the VTA is hypothesized to increase DA release in NAc (red arrow). Prefrontal cortex (PFC); Lateral dorsal tegmentum (LDT); Pedunculopontine tegmentum (PPTg).
It is important to point out that other brain regions and circuits that modulate VTA function and that express distinct nAChR subtypes also may influence ethanol intake. Of particular interest is the habenular-interpeduncular (Hb-IPN) pathway that robustly expresses unique nAChR subtypes including α3β4* nAChRs [56]. α3β4* nAChR antagonists or partial agonists reduce ethanol consumption in rodent models suggesting that this subtype may also contribute to ethanol intake [45, 57]. As emerging evidence indicates that the Hb-IPN pathway is involved in drug withdrawal symptoms and aversion [58-60], it is possible that, during initial stages of alcoholism driven by reward [61], α6* nAChRs in the VTA are critical. However, with chronic ethanol exposure, neuroadaptations occur such that ethanol consumption is dominated by avoidance of negative, affective withdrawal symptoms which may be mediated by distinct nAChR subtypes in the Hb-IPN pathway (or other circuits). Future experiments should directly test the involvement of the Hb-IPN circuit in ethanol intake and modulation of VTA activity.
Our data indicate that α6* nAChR contribute to ethanol activation of VTA DAergic neurons and are required for ethanol potentiation of ACh responses at VTA DAergic neuron nAChRs in midbrain slices. Because α6* nAChRs also are involved in nicotine reinforcement, these data point toward a common molecular link between ethanol and nicotine-mediated activation of VTA DAergic neurons and suggest α6* nAChRs as therapeutic targets to reduce ethanol consumption.
Acknowledgments
This study was supported by the National Institute on Alcohol Abuse and Alcoholism award number R01AA017656 (ART), the National Institutes of Mental Health and General Medical Sciences R01MH53631 and PO1 GM48677 (JMM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
References
- 1.Batel P, Pessione F, Maitre C, Rueff B. Relationship between alcohol and tobacco dependencies among alcoholics who smoke. Addiction. 1995;90:977–80. doi: 10.1046/j.1360-0443.1995.90797711.x. [DOI] [PubMed] [Google Scholar]
- 2.Hurt RD, Offord KP, Croghan IT, Gomez-Dahl L, Kottke TE, Morse RM, et al. Mortality following inpatient addictions treatment. Role of tobacco use in a community-based cohort. JAMA. 1996;275:1097–103. doi: 10.1001/jama.275.14.1097. [DOI] [PubMed] [Google Scholar]
- 3.Rodd ZA, Melendez RI, Bell RL, Kuc KA, Zhang Y, Murphy JM, et al. Intracranial self-administration of ethanol within the ventral tegmental area of male Wistar rats: evidence for involvement of dopamine neurons. J Neurosci. 2004;24:1050–7. doi: 10.1523/JNEUROSCI.1319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95:619–26. doi: 10.1152/jn.00682.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, et al. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–4. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–4. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 7.Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, et al. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–7. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- 9.Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, et al. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–32. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 10.Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–27. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Exley R, Maubourguet N, David V, Eddine R, Evrard A, Pons S, et al. Distinct contributions of nicotinic acetylcholine receptor subunit {alpha}4 and subunit {alpha}6 to the reinforcing effects of nicotine. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1103000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gotti C, Guiducci S, Tedesco V, Corbioli S, Zanetti L, Moretti M, et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci. 2010;30:5311–25. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao-Shea R, Liu L, Soll LG, Improgo MR, Meyers EE, McIntosh JM, et al. Nicotine-mediated activation of dopaminergic neurons in distinct regions of the ventral tegmental area. Neuropsychopharmacology. 2011;36:1021–32. doi: 10.1038/npp.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, et al. Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22:1208–17. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha6* nicotinic acetylcholine receptors. Neuron. 2008;60:123–36. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larsson A, Edstrom L, Svensson L, Soderpalm B, Engel JA. Voluntary ethanol intake increases extracellular acetylcholine levels in the ventral tegmental area in the rat. Alcohol Alcohol. 2005;40:349–58. doi: 10.1093/alcalc/agh180. [DOI] [PubMed] [Google Scholar]
- 17.Larsson A, Jerlhag E, Svensson L, Soderpalm B, Engel JA. Is an alpha-conotoxin MII-sensitive mechanism involved in the neurochemical, stimulatory, and rewarding effects of ethanol? Alcohol. 2004;34:239–50. doi: 10.1016/j.alcohol.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Kuzmin A, Jerlhag E, Liljequist S, Engel J. Effects of subunit selective nACh receptors on operant ethanol self-administration and relapse-like ethanol-drinking behavior. Psychopharmacology (Berl) 2009;203:99–108. doi: 10.1007/s00213-008-1375-5. [DOI] [PubMed] [Google Scholar]
- 19.Hendrickson LM, Zhao-Shea R, Pang X, Gardner PD, Tapper AR. Activation of alpha4* nAChRs is necessary and sufficient for varenicline-induced reduction of alcohol consumption. J Neurosci. 2010;30:10169–76. doi: 10.1523/JNEUROSCI.2601-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoft NR, Corley RP, McQueen MB, Huizinga D, Menard S, Ehringer MA. SNPs in CHRNA6 and CHRNB3 are associated with alcohol consumption in a nationally representative sample. Genes Brain Behav. 2009;8:631–7. doi: 10.1111/j.1601-183X.2009.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powers MS, Broderick HJ, Drenan RM, Chester JA. Nicotinic Acetylcholine Receptors Containing alpha6 Subunits Contribute to Alcohol Reward-Related Behaviors. Genes Brain Behav. 2013 doi: 10.1111/gbb.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.National Research Council. Guide for the care and use of laboratory animals. Washington, D.C.: National Academy Press; 1996. [Google Scholar]
- 23.McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, et al. Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–52. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- 24.Liu L, Hendrickson LM, Guildford MJ, Zhao-Shea R, Gardner PD, Tapper AR. Nicotinic Acetylcholine Receptors Containing the alpha4 Subunit Modulate Alcohol Reward. Biol Psychiatry. 2013;73:738–46. doi: 10.1016/j.biopsych.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendrickson LM, Guildford MJ, Tapper AR. Neuronal nicotinic acetylcholine receptors: common molecular substrates of nicotine and alcohol dependence. Frontiers in psychiatry / Frontiers Research Foundation. 2013;4:29. doi: 10.3389/fpsyt.2013.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blomqvist O, Engel JA, Nissbrandt H, Soderpalm B. The mesolimbic dopamine-activating properties of ethanol are antagonized by mecamylamine. Eur J Pharmacol. 1993;249:207–13. doi: 10.1016/0014-2999(93)90434-j. [DOI] [PubMed] [Google Scholar]
- 27.Blomqvist O, Ericson M, Engel JA, Soderpalm B. Accumbal dopamine overflow after ethanol: localization of the antagonizing effect of mecamylamine. Eur J Pharmacol. 1997;334:149–56. doi: 10.1016/s0014-2999(97)01220-x. [DOI] [PubMed] [Google Scholar]
- 28.Ericson M, Blomqvist O, Engel JA, Soderpalm B. Voluntary ethanol intake in the rat and the associated accumbal dopamine overflow are blocked by ventral tegmental mecamylamine. Eur J Pharmacol. 1998;358:189–96. doi: 10.1016/s0014-2999(98)00602-5. [DOI] [PubMed] [Google Scholar]
- 29.Hendrickson LM, Zhao-Shea R, Tapper AR. Modulation of ethanol drinking-in-the-dark by mecamylamine and nicotinic acetylcholine receptor agonists in C57BL/6J mice. Psychopharmacology (Berl) 2009 doi: 10.1007/s00213-009-1488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chi H, de Wit H. Mecamylamine attenuates the subjective stimulant-like effects of alcohol in social drinkers. Alcohol Clin Exp Res. 2003;27:780–6. doi: 10.1097/01.ALC.0000065435.12068.24. [DOI] [PubMed] [Google Scholar]
- 31.Lodge DJ, Grace AA. The laterodorsal tegmentum is essential for burst firing of ventral tegmental area dopamine neurons. Proc Natl Acad Sci U S A. 2006;103:5167–72. doi: 10.1073/pnas.0510715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao C, Shao XM, Olive MF, Griffin WC, 3rd, Li KY, Krnjevic K, et al. Ethanol facilitates glutamatergic transmission to dopamine neurons in the ventral tegmental area. Neuropsychopharmacology. 2009;34:307–18. doi: 10.1038/npp.2008.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao C, Ye JH. Ethanol dually modulates GABAergic synaptic transmission onto dopaminergic neurons in ventral tegmental area: role of mu-opioid receptors. Neuroscience. 2008;153:240–8. doi: 10.1016/j.neuroscience.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theile JW, Morikawa H, Gonzales RA, Morrisett RA. GABAergic transmission modulates ethanol excitation of ventral tegmental area dopamine neurons. Neuroscience. 2011;172:94–103. doi: 10.1016/j.neuroscience.2010.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knockout mice. J Neurosci. 2003;23:7820–9. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang K, Buhlman L, Khan GM, Nichols RA, Jin G, McIntosh JM, et al. Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J Neurosci. 2011;31:2537–48. doi: 10.1523/JNEUROSCI.3003-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jerlhag E, Grotli M, Luthman K, Svensson L, Engel JA. Role of the subunit composition of central nicotinic acetylcholine receptors for the stimulatory and dopamine-enhancing effects of ethanol. Alcohol Alcohol. 2006;41:486–93. doi: 10.1093/alcalc/agl049. [DOI] [PubMed] [Google Scholar]
- 38.Kamens HM, Hoft NR, Cox RJ, Miyamoto JH, Ehringer MA. The alpha6 nicotinic acetylcholine receptor subunit influences ethanol-induced sedation. Alcohol. 2012;46:463–71. doi: 10.1016/j.alcohol.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morikawa H, Morrisett RA. Ethanol action on dopaminergic neurons in the ventral tegmental area: interaction with intrinsic ion channels and neurotransmitter inputs. Int Rev Neurobiol. 2010;91:235–88. doi: 10.1016/S0074-7742(10)91008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drago J, McColl CD, Horne MK, Finkelstein DI, Ross SA. Neuronal nicotinic receptors: insights gained from gene knockout and knockin mutant mice. Cell Mol Life Sci. 2003;60:1267–80. doi: 10.1007/s00018-003-2259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui C, Booker TK, Allen RS, Grady SR, Whiteaker P, Marks MJ, et al. The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci. 2003;23:11045–53. doi: 10.1523/JNEUROSCI.23-35-11045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–35. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 43.Liu L, Zhao-Shea R, McIntosh JM, Gardner PD, Tapper AR. Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing alpha4 and alpha6 subunits. Mol Pharmacol. 2012;81:541–8. doi: 10.1124/mol.111.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sajja RK, Rahman S. Lobeline and cytisine reduce voluntary ethanol drinking behavior in male C57BL/6J mice. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:257–64. doi: 10.1016/j.pnpbp.2010.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Chatterjee S, Steensland P, Simms JA, Holgate J, Coe JW, Hurst RS, et al. Partial agonists of the alpha3beta4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuropsychopharmacology. 2011;36:603–15. doi: 10.1038/npp.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steensland P, Simms JA, Holgate J, Richards JK, Bartlett SE. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, selectively decreases ethanol consumption and seeking. Proc Natl Acad Sci U S A. 2007;104:12518–23. doi: 10.1073/pnas.0705368104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bordia T, Hrachova M, Chin M, McIntosh JM, Quik M. Varenicline is a potent partial agonist at alpha6beta2* nicotinic acetylcholine receptors in rat and monkey striatum. J Pharmacol Exp Ther. 2012;342:327–34. doi: 10.1124/jpet.112.194852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, Grady SR. Pharmacology of alpha-conotoxin MII-sensitive subtypes of nicotinic acetylcholine receptors isolated by breeding of null mutant mice. Mol Pharmacol. 2007;71:1563–71. doi: 10.1124/mol.106.031492. [DOI] [PubMed] [Google Scholar]
- 49.Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–9. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- 50.Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 51.Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, et al. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74:1235–46. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–63. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci. 2003;23:3176–85. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–18. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tolu S, Eddine R, Marti F, David V, Graupner M, Pons S, et al. Co-activation of VTA DA and GABA neurons mediates nicotine reinforcement. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.83. [DOI] [PubMed] [Google Scholar]
- 56.Grady SR, Moretti M, Zoli M, Marks MJ, Zanardi A, Pucci L, et al. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the alpha3beta4* and alpha3beta3beta4* subtypes mediate acetylcholine release. J Neurosci. 2009;29:2272–82. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rezvani AH, Overstreet DH, Yang Y, Maisonneuve IM, Bandarage UK, Kuehne ME, et al. Attenuation of alcohol consumption by a novel nontoxic ibogaine analogue (18-methoxycoronaridine) in alcohol-preferring rats. Pharmacol Biochem Behav. 1997;58:615–9. doi: 10.1016/s0091-3057(97)10003-x. [DOI] [PubMed] [Google Scholar]
- 58.Salas R, Sturm R, Boulter J, De Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci. 2009;29:3014–8. doi: 10.1523/JNEUROSCI.4934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, et al. Aversion to nicotine is regulated by the balanced activity of beta4 and alpha5 nicotinic receptor subunits in the medial habenula. Neuron. 2011;70:522–35. doi: 10.1016/j.neuron.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 60.Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koob GF. Theoretical Frameworks and Mechanistic Aspects of Alcohol Addiction: Alcohol Addiction as a Reward Deficit Disorder. Curr Top Behav Neurosci. 2013;13:3–30. doi: 10.1007/7854_2011_129. [DOI] [PMC free article] [PubMed] [Google Scholar]