

Abstract
Mutations in ABCA4 cause Stargardt disease and other blinding autosomal recessive retinal disorders. However, sequencing of the complete coding sequence in patients with clinical features of Stargardt disease sometimes fails to detect one or both mutations. For example, among 208 individuals with clear clinical evidence of ABCA4 disease ascertained at a single institution, 28 had only one disease-causing allele identified in the exons and splice junctions of the primary retinal transcript of the gene. Haplotype analysis of these 28 probands revealed 3 haplotypes shared among ten families, suggesting that 18 of the 28 missing alleles were rare enough to be present only once in the cohort. We hypothesized that mutations near rare alternate splice junctions in ABCA4 might cause disease by increasing the probability of mis-splicing at these sites. Next-generation sequencing of RNA extracted from human donor eyes revealed more than a dozen alternate exons that are occasionally incorporated into the ABCA4 transcript in normal human retina. We sequenced the genomic DNA containing 15 of these minor exons in the 28 one-allele subjects and observed five instances of two different variations in the splice signals of exon 36.1 that were not present in normal individuals (P < 10−6). Analysis of RNA obtained from the keratinocytes of patients with these mutations revealed the predicted alternate transcript. This study illustrates the utility of RNA sequence analysis of human donor tissue and patient-derived cell lines to identify mutations that would be undetectable by exome sequencing.
INTRODUCTION
Mutations in the gene encoding the ATP binding cassette transporter of the retina (ABCA4 NM_000350.2) cause a wide spectrum of recessive retinal diseases that range in severity from Stargardt disease to cone-rod dystrophy and retinitis pigmentosa depending upon the degree of residual transporter function in the encoded protein (1,2). The normal function of ABCA4 is to facilitate the transport of all-trans-retinal from the outer segment disk to the outer segment cytoplasm in the form of a mono-substituted phospholipid known as N-retinylidene-phosphatidylethanolamine (N-ret-PE) (3,4). When ABCA4 is defective, N-ret-PE tarries on the inner leaflet of the outer segment disk long enough for a second retinyl moiety to covalently bond to the nitrogen atom of the ethanolamine, irreversibly forming a toxic, insoluble, bis-retinoid known as A2PE. With relatively mild genotypes, the A2PE and its derivative A2E do not accumulate to injurious levels in the photoreceptor cells but instead accumulate in the underlying retinal pigment epithelium (RPE) as a result of the normal phagocytic turnover of the outer segments. There, the bisretinoids can (i) engorge the RPE causing the clinical findings known as a vermillion fundus or a masked choroid, (ii) accumulate beneath the RPE as yellowish deposits known as pisciform flecks, and/or (iii) cause the death of RPE cells. Cone photoreceptor cells are more sensitive to bisretinoid accumulation than rod photoreceptors so that ABCA4 genotypes of intermediate severity cause a cone selective photoreceptor loss that is recognized clinically and electrophysiologically as cone-rod dystrophy. The most severe genotypes cause a sufficient accumulation of bisretinoid in the rods that they also succumb, giving rise to a clinical and electrophysiological phenotype similar to typical retinitis Pigmentosa (2).
Viral-mediated gene replacement has been shown to rescue the retinal phenotype caused by Abca4 mutations in mice (5) and a phase 1 human clinical trial of such treatment is now underway (Clinical trial identifier: NCT01367444). Demonstrating the therapeutic efficacy of gene replacement in later phases will be facilitated by using the patients' genotype to both choose the optimal point in the disease course to administer the therapy as well as to balance the disease severity among individuals in the treatment and control groups. Schindler et al. (1) demonstrated that ABCA4 alleles contribute to an individual's phenotype in an additive fashion. They also calculated severity coefficients for 16 of the most common variations in the ABCA4 gene. This approach is most applicable to patients who have clinical features and genotypes that are both consistent with ABCA4 disease. However, genotype–phenotype correlations are rarely perfect even after decades of careful study. In any population of patients with apparently heritable macular disease, there will be people who appear clinically to have ABCA4 disease but who do not have two detectable disease-causing alleles. It would be unwise to enroll such subjects into clinical trials of invasive therapies such as gene replacement because if their disease is caused by some other gene or non-genetic phenocopy, the treatment would not help them and could harm them.
A number of disease-causing ABCA4 mutations must lie outside the coding sequences because some patients with classic clinical features exhibit at most one disease-causing variant when their coding regions are screened (1,6,7). Shotgun sequencing of the introns of these patients has not been very fruitful, in part because the gene is so large and in part because it harbors many repetitive elements. In the present study, we used sequencing of RNA extracted from normal human retina to identify sequences that are sufficiently close to canonical splice signals that the splicing machinery uses them for a detectable number of splicing events. We hypothesized that such sequences could act as mutation susceptibility sites that would make single nucleotide variations occurring within them much more likely to cause disease than similar variations elsewhere in the genomic sequence of the gene.
RESULTS
We reasoned that the individuals most likely to harbor a non-exomic disease-causing mutation in ABCA4 would be those who (i) exhibited multiple characteristic clinical features of ABCA4-related disease (see Materials and Methods) and (ii) harbored a single plausible disease-causing allele. Between 1997 and 2013, 208 probands were ascertained at the University of Iowa with characteristic clinical features of ABCA4-associated retinal disease. Sequencing of the ABCA4 coding sequences and canonical splice junctions in these individuals revealed zero (n = 3), one (n = 28) or two (n = 177) plausible disease-causing mutations. The 28 unrelated subjects with one detectable ABCA4 mutation served as the primary cohort for this study.
To estimate the number of different mutations that exist among the currently undetectable 8.2% of ABCA4 disease alleles (34/416) and to identify sub-regions of the gene that harbor them, we performed haplotype analysis of the 28 one-allele probands and their nuclear families. If a relatively small number of different mutations account for the majority of the currently undetectable disease alleles, we would expect to observe more haplotype sharing among the members of the one-allele cohort than among controls. Also, if some or all of these mutations are ancient, historical recombination events might limit the sharing to a small enough portion of the gene to materially aid in the identification of the mutations. To examine these possibilities, we first developed a multiplexed allele-specific assay for 60 tagged SNPs (Supplementary Material, Table S1) spanning the ABCA4 gene (see Materials and Methods) and used this assay to genotype the 28 members of the one-allele cohort, their family members and 18 control trios. We observed 3 shared haplotypes that were longer and more common in the individuals with 1 ABCA4 disease allele than in the 18 control trios (Fig. 1). Four members of the one-allele cohort share a haplotype (H1) spanning 28 kb centered on intron 31, three members share a haplotype (H2) spanning 114 kb centered on intron 16 and three members share a haplotype (H3) spanning 17 kb centered on intron 30. The remaining 18 members of the one-allele cohort exhibit haplotypic compositions that are indistinguishable from those of the 18 control trios. These data show that none of the currently undetectable disease-causing ABCA4 mutations in our patient population is likely to represent more than 15–20% of the total.
Figure 1.
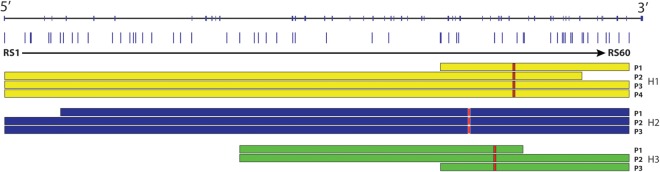
ABCA4 haplotype analysis. The positions of the 60-tagged SNPs used for the haplotype analysis are shown as small vertical lines beneath the schematic diagram of the genomic structure of the ABCA4 gene. The SNP numbers correspond to those in Supplementary Material, Table S1. The 5′ end of the gene is to the left. The contiguous SNPs shared by subjects with haplotypes H1, H2 and H3 are shown as horizontal bars.
We next considered a more focused possibility that cryptic splice sites created by intronic point mutations could account for a meaningful fraction of mutations in ABCA4. We hypothesized that the sites most susceptible to such mutations might be the sequences flanking the rare alternate splice junctions detectable in RNA extracted from normal retina. To test this hypothesis, we performed next-generation sequencing of RNA extracted from five different anatomic regions (nasal, temporal, superior, inferior and macular) of human donor retina (see Materials and Methods). Over 95% of the ABCA4 splice junctions observed in these sequencing data corresponded to the junctions present in the normal full-length retinal transcript of the gene. However, we also observed some minor splice variants (each less than 1% of the total) that were present in multiple regional samples (Fig. 2 and Supplementary Material, Fig. S1). To investigate whether sequence variations within these minor ABCA4 exons or their splice sites could be disease-causing, we sequenced the genomic DNA containing 15 of these alternate exons (Supplementary Material, Table S2) in the 28 members of the one-allele cohort. We observed five instances of two different sequence variations in the cohort that were present within the splice sites of a single minor exon (exon 36.1, Fig. 2). These two variants (V1 and V2, Table 1) were present in trans to known disease-causing ABCA4 mutations, are predicted to increase the strength of the splice signal in which they occur (Table 2) (8), were not present among 600 control alleles ascertained in the same clinic population, and were not present among 2184 alleles in the 1000 genomes database (9). The most frequent of these (V1) is on the allele previously recognized as bearing the haplotype H1 (Fig. 1). Fisher's exact test reveals that the number of exon 36.1 splice signal variants found among the undetected alleles of the one-allele cohort (5/28) is significantly greater (P < 10−6) than among the ABCA4 alleles of controls (0/600).
Figure 2.

RNA sequence analysis from normal human retina. Top: The genomic organization of ABCA4 is shown schematically with canonical exons in blue and the 15 most abundant alternate exons in pink. The 5′ end of the gene is to the left. RNA sequencing data supporting a specific splice junction is shown as a purple arc linking two exons. For 1 to 50 sequencing reads, the thickness of each arc is proportional to the number of reads supporting the given splice junction. Junctions supported by more than 50 reads are all shown at equal height. The positions of disease-causing variants V1–V7 are indicated with labeled asterisks. Middle: The portion of the splice junction map spanning canonical exons 36 and 37 is shown for samples obtained from five different regions of the retina (macula, superior, inferior, nasal and temporal). Two alternate exons, 36.1 and 36.2, each have sequence support for their splice acceptor and splice donor junctions in at least two of these retinal regions. Bottom: The sequence of alternate exon 36.1 and its flanking nucleotides is shown. Variant V1 (G>A, represented as R in the sequence) is 3 nucleotides upstream of the splice acceptor site, and variant V2 (C>A, represented as M in the sequence) is 4 nucleotides downstream of the splice donor site. Both variants increase the similarity of the splice junction sequence to a canonical splice sequence (Table 2).
Table 1.
Splice variants identified in the one allele ABCA4 patients
| Variant | Haplotype | Position | Genomic location | ABCA4 (NM_000350.2) | Primary cohort (n-28) | Validation cohort (n = 48) | Controls (n= 300) | 1000 genomes (n= 1092) |
|---|---|---|---|---|---|---|---|---|
| V1 | H1 | Exon 36.1–3 G>A | chr1:94,484,001 | c.5196+1137G>A | 4 | 4 | 0 | 0 |
| V2 | – | Exon 36.1+4 C>A | chr1:94,483,922 | c.5196+1216C>A | 1 | 0 | 0 | 0 |
| V3 | – | IVS36+1056 A>G | chr1:94,484,082 | c.5196+1056A>G | 1 | 0 | 0 | 0 |
| V4 | – | Exon 30.1 position 110 G>A | chr1:94,493,000 | c.4539+2001G>A | 1 | 0 | 0 | 0 |
| V5 | H2 | Exon 30.1 position 138 C>T | chr1:94,492,973 | c.4539+2028C>T | 3 | 4 | 1 | 0 |
| V6 | – | Val2114Val GTG>GTA | chr1:94,466,602 | c.6342G>A | 1 | 2 | 0 | 0 |
| V7 | H3 | IVS33+3 A>G | chr1:94,487,399 | c.4773+3A>G | 3 | 0 | 0 | 0 |
Table 2.
Predicted splice effect of variants identified in one allele patients (8)
| Canonical human splice sequence | Intron |
Acceptor |
Donor |
Intron |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −13 | −12 | −11 | −10 | −9 | −8 | −7 | −6 | −5 | −4 | −3 | −2 | −1 | 1 | 2 | 2 | 1 | +1 | +2 | +3 | +4 | +5 | |
| T | T | T | T | T | T | T | T | T | T | C | A | G | G | T | A | G | G | T | A | A | G | |
| 50.5% | 52.2% | 55.4% | 52.4% | 49.3% | 46.4% | 46.0% | 50.9% | 55.5% | 28.1% | 65.0% | 100.0% | 100.0% | 49.0% | 36.9% | 63.9% | 80.6% | 100.0% | 100.0% | 60.5% | 69.9% | 78.3% | |
| V1 chr1:94,483, 997–94,484,011 | C | T | G | T | C | T | A | C | A | C | G 0.2% A 5.6% |
A | G | G | A | |||||||
| V2 chr1:94,483, 921–94,483,927 | A | G | G | T | A | C 7.3% A 69.9% |
C | |||||||||||||||
| V3 chr1:94,484, 077–94,484,083 | T | A 9.8% G 80.6% |
G | T | A | A | G | |||||||||||||||
| V4 chr1:94,493, 000–94,493,014 | T | G | T | C | A | A | T | G | C | T | G | A | G | G | G 19.9% A 24.4% |
|||||||
| V5 chr1:94,492, 959–94,492,973 | C 28.0% T 50.5% |
C | T | C | C | A | G | C | A | T | C | A | G | G | A | |||||||
| V6 chr1:94,466, 600–94,466,606 | T | C | G | T | G 34.1% A 60.5% |
A | G | |||||||||||||||
| V7 chr1:94,487, 397–94,487,403 | G | G | G | T | A 60.5% G 34.1% |
T | G | |||||||||||||||
Another sequence variant (V3, Table 1) was incidentally discovered in one member of the one-allele cohort because of its proximity to V1. Like V1 and V2, this mutation lies in trans to the patient's known disease-causing mutation and was not observed in more than 600 control alleles ascertained at the University of Iowa or in the 1000 genomes database. Unlike V1 and V2, which each strengthen a detectably active minor splice signal, V3 creates a new splice signal within the intron (Table 2).
To test the functional effects of these three mutations, we took advantage of the fact that ABCA4 is expressed at low levels in cultured keratinocytes. We established primary keratinocyte cultures from at least one individual affected with each of these mutations. After passaging these cultures three times, RNA was extracted and used as template in an RT-PCR reaction spanning the mutation and at least one canonical splice junction. The resulting PCR products were separated with agarose electrophoresis, the bands were cut out of the gels and the DNA in these bands was subjected to sequencing. Figure 3 shows the result of this experiment for these three mutations. In each case, we observed that the mutation dramatically increases splicing at the alternate splice site when compared with the splicing pattern of RNA extracted from control keratinocytes.
Figure 3.

Functional confirmation of ABCA4 variants in patient-derived cell lines. RT-PCR analysis of ABCA4 in RNA extracted from human control retina (lane 1), human keratinocytes isolated from an unaffected individual (lane 2) and human keratinocytes isolated from a patient with ABCA4 associated retinal degeneration (lane 3). Two intronic splice site mutations (V1, A; and V2, B) in IVS 36 of the ABCA4 gene result in the introduction of an alternate exon (36.1, Fig. 2). An intronic splice site mutation within IVS 36 results in the introduction of a 177 bp segment of IVS 36 (V3, C). A synonymous codon change (Val2114Val) in exon 46 creates a premature donor splice site that results in deletion of the last 47 bases of exon 46 from the transcript (V6, D).
We observed one additional mutation (V4, Table 1) that appears to strengthen a possible splice site (8) (Table 2) within a minor exon observed in our RNA sequencing experiment (exon 31.1, Fig. 2). This mutation was observed in trans to the known disease-causing ABCA4 mutation in one member of the one-allele cohort, and was not observed in 600 control alleles ascertained in Iowa or in the 1000 genomes database.
In parallel to the minor splice variant experiments described above, we also performed next-generation sequencing of the 168 kb genomic region containing the entire ABCA4 gene (HG19 chr1:94 448 410–94 616 987) in nine members of the one-allele cohort using the Haloplex genomic fragment capture strategy (see Materials and Methods). In total, this experiment yielded 10 million uniquely mapped reads. These reads were aligned to the reference human genomic sequence using BWA (10). Departures from the reference were identified with GATK (11). Across all nine samples, more than 3800 sequence variations were detected. These variants were prioritized using the GATK variation quality score (greater than 50), and population frequency (less than 1% in all available population databases) resulting in an average of 10 high-quality variants per sample (38 instances of 30 different variations). Of these, only 12 instances of 8 variations were confirmed to exist by Sanger sequencing and found to lie on the ABCA4 allele opposite the individual's previously known disease-causing mutation. Five of these eight were present in the control subjects at a frequency that is too common for an allele that causes a rare Mendelian disease. One of the remaining variants (V3, Table 1) was also detected in the minor splice variant experiments described above. One novel variant (V5, Table 1) was observed in the three members of the one-allele cohort with haplotype H2 (Fig. 1). This variant strengthens an acceptor splice signal (8) within a minor exon that was observed in the original RNA sequencing data (exon 31.1, Fig. 2) but which was rare enough that it was not included in the minor splice variant hypothesis. Variant V5 was also observed in 1of the 600 control alleles ascertained in Iowa but was not observed among 2184 alleles in the 1000 genomes database. The one remaining rare variant in the Haloplex data is a C to T variation in IVS 3 that is present on the same allele as V5 in two of the three individuals with haplotype H2. It is also present in the same normal individual that harbors V5. Since V5 is present in all three affected individuals with the H2 haplotype, it seems more likely to be disease-causing than the IVS 3 variant.
We next asked whether any previously recognized variations in ABCA4 might also be acting via increased splicing of a rare alternate exon. We did this by comparing our RNA sequencing data to the list of all variants that we have previously observed in patients suspected to have ABCA4-mediated retinal disease (Supplementary Material, Table S3). We noted that a synonymous codon variant in exon 46 (V2114 V) strengthens the donor sequence of a rare alternate exon that was present in our RNA sequencing data from normal human retina. Despite its absence among the 6500 exomes summarized on the exome variant server (12), we had previously considered this variant (V6, Table 1) to be non-disease-causing because of its lack of predicted effect on the ABCA4 protein. However, similar to V1–V3, we were able to demonstrate altered splicing of ABCA4 at the predicted splice junction in the cultured keratinocytes of a subject harboring this mutation (Fig. 3). While reviewing the list of variants previously judged to be non-disease-causing, we also noted that three members of the one-allele cohort harbored an A to G mutation in the +3 position of the splice donor sequence of IVS33. Although in our experience variations at the +3 position are rarely disease-causing, this variation (V7, Table 1) is predicted to weaken the IVS 33 splice signal (8) and was not observed in any of 600 control alleles ascertained in Iowa or among the 2184 alleles in the 1000 genomes database. This variant was found in the 3 members of the primary cohort with haplotype H3 (Fig. 1) and was also identified by Duno et al. (13) in 1 of their 161 patients with Stargardt disease.
Although we did not observe any plausible disease-causing mutations in the 3′UTR or the proximal promoter of ABCA4 in the Haloplex sequencing experiment, only 9 of the 28 members of the one-allele cohort were included in this experiment. We therefore used Sanger sequencing to reevaluate the entire 3′UTR as well as 1000 base pairs of genomic DNA upstream from the transcription start site in all 28 members of the one-allele cohort. No plausible disease-causing variants were observed in these individuals.
The 7 variations summarized in Table 1 are collectively responsible for 14 of the 28 (50%) previously undetectable ABCA4 mutations in the one-allele cohort. Thus, we have now detected two plausible disease-causing mutations in 191/208 individuals (91.8%) in the Iowa cohort of patients suspected to have ABCA4 disease and 98.6% of these individuals have at least one of their disease alleles identified.
We next wanted to see what fraction of a group of patients with similar clinical features ascertained in other geographical areas would harbor these same variants. This validation cohort consisted of 48 unrelated individuals seen by physicians outside the University of Iowa who had both the clinical diagnosis of Stargardt disease and a single plausible disease-causing variation in ABCA4. Ten of these 48 individuals (20.8%) were found to harbor 1 of the 7 variants from the Iowa one-allele cohort (Table 1). Relatives were available from 5 of the 10 to demonstrate that these variants are in trans to their known disease-causing mutation (Supplementary Material, Fig. S2). This frequency was significantly greater (P < 10−10) than that seen in controls (1/600).
DISCUSSION
ABCA4-associated retinal disease is one of the most common causes of inherited retinal disease in children and young adults and it is encouraging that clinical trials of viral-mediated gene replacement are now underway (Clinical trial identifier: NCT01367444). However, the advent of such therapy has heightened the need for sensitive and specific genetic testing for this disease because for invasive mechanism-specific treatments like gene replacement, it is essential to know the cause of the patients' disease with certainty. ABCA4 is particularly challenging for molecular diagnosticians because the gene is extremely polymorphic with hundreds of alleles (1,6,14–16) that vary in pathogenic severity from benign to non-functional in small degrees (1). This allelic diversity is compounded by the fact that ABCA4 genotypes of varying severity cause a wide range of phenotypes, and each of these phenotypes overlaps those caused by mutations in a number of other genes. An additional difficulty is that ABCA4 disease alleles are recessive and are found in the heterozygous state in about 1 in 50 individuals in the general population. The final challenge, and the subject of this study, is that like many genes, a clinically meaningful fraction of disease-causing mutations in ABCA4 lie outside its coding sequences (1,6,7).
One can initially suspect the existence of non-exomic autosomal recessive mutations in a number of ways. Linkage analysis of multiplex families can map the location of the disease-causing mutations to the locus of a known disease-causing gene, while sequencing of the coding regions of the gene reveals no (17) or only one (18) plausible disease-causing variant. Alternatively, one can study a large cohort of unrelated individuals who share classic clinical features of recessive disease and observe that a subset of them manifest only one or no disease-causing variants in the coding sequences of the expected disease-causing gene. Since most human phenotypes are genetically heterogeneous and some also have a number of non-genetic phenocopies, the most likely individuals to harbor a non-exomic autosomal recessive mutation are members of multiplex families with evidence of linkage to a specific locus and isolated individuals with a single disease-causing mutation and convincing clinical features of the disease in question. In the present study, we investigated 28 unrelated individuals with multiple clinical features suggestive of ABCA4-associated retinal disease, but only a single plausible disease-causing mutation identified after Sanger sequencing of the complete coding sequence and splice junctions of the ABCA4 gene. Haplotype analysis of these 28 individuals revealed that only three of the previously undetectable mutations were likely to be present in more than one member of the cohort and that the most common of these accounted for only 4 of the 416 alleles in the original cohort of 208 patients. The haplotype analysis also suggests that 18 of the 28 undetectable alleles in the one-allele cohort seem to be present only once in more than 400 alleles. The large fraction (65%) of ABCA4 mutations with allele frequencies of 1/400 or less (Fig. 4) limit the utility of allele specific testing methods and increase the dependence on patient derived cell lines for functional demonstration of their pathogenicity.
Figure 4.

Allelic diversity of ABCA4. This figure shows the number of different disease-causing ABCA4 variants (y-axis) that occur at each frequency (x-axis) in a population of 404 patients with clinical features of ABCA4 disease ascertained by one of the authors. Variants seen eight or more times in the cohort are specifically labeled. Of the 258 different plausible disease-causing variants seen in this cohort, 168 (65%) were observed only once.
The sensitivity of a genetic test for an autosomal recessive disease is clinically important for two related reasons. First, each increment of additional sensitivity increases the fraction of affected individuals with two discoverable alleles, increasing the accuracy of counseling for these individuals and increasing the likelihood that mechanism-specific therapy will be useful for them. Second, each increment of additional sensitivity also increases the likelihood that observation of a single disease-causing variation in an individual is irrelevant to their disease. The ability to exclude a gene as the cause of disease in a given patient and move on to other diagnostic possibilities can be very important for diseases that are as genetically heterogeneous as the photoreceptor degenerations.
It is noteworthy that new mutations and new classes of mutations continue to be identified in the ABCA4 gene 16 years after its first association with retinal disease. Although this is due in part to improvements in sequencing technology, it is also due to our improved understanding of the detailed phenotype of the disease. The careful definition of a gene-specific-phenotype will become even more important as clinical DNA sequencing begins to be used to routinely query the entire genome. For a sequence variant to have true utility for clinical decision making, the pre-test probability that the region of the genome harboring it is involved in the patient's disease will have to be sufficient to overcome the very large amount of non-disease-causing variability in the genome. For example, in the current study, there were two dimensions of pre-sequencing focus that enabled a statistically significant result: (i) the clinical definition of ABCA4 disease that was employed when choosing the primary screening cohort and (ii) the relatively small portion of the ABCA4 gene that was screened in the experiment (15 segments ranging in size from 178 to 635 base pairs, each containing a minor exon detected by RNA sequencing).
Two additional findings in this study are also noteworthy with respect to personalized genomic medicine. First, despite extensive efforts to identify the second allele in the 28 members of the primary cohort, a second allele was not identified in 50% of these probands. Second, the novel mutations identified in the primary cohort account for an even smaller fraction of the missing alleles in the validation cohort (19.6%). One possible explanation for these observations is that ABCA4 is not the disease-causing gene for some of these patients. Another possibility is that mutations quite distant from the coding sequence could be involved. For example, Sagai and co-workers (19,20) showed that mutations up to 1 Mb from the coding sequence of a gene can affect expression of the gene sufficiently to cause disease. Most large genes contain a number of repetitive elements that are difficult to assess with any sequencing strategy, and ABCA4 is no exception. For example, there are 18 Alu repeats within the introns of ABCA4 that have not been thoroughly examined by any of the sequencing experiments that have been conducted to date. The difference in the findings between the primary and validation cohorts is likely due to the fact that the majority of mutations in ABCA4 are quite rare, present in fewer than 1/200 disease-causing alleles (Fig. 4). Thus, groups of patients with fewer than 50 members that are ascertained in different geographical areas are likely to harbor variants that are unique to each group. This, coupled with the fact that the validation cohort was sequenced much less extensively than the primary cohort is likely the explanation for the difference in sensitivity between these groups. This also suggests that for many disease-associated genes, genomic sequencing will be required to achieve mutation detection rates greater than 95%.
MATERIALS AND METHODS
Human subjects
All subjects provided written informed consent for this research study, which was approved by the Institutional Review Boards of the participating centers and adhered to the tenets set forth in the Declaration of Helsinki. The individuals in both the primary and validation cohorts had one plausible disease-causing mutation detected in ABCA4 after assessing the entire coding sequence and canonical retinal splice junctions with automated bidirectional Sanger sequencing using an ABI 3730 sequencer (Life Technologies, Carlsbad, CA, USA). In addition, each member of the primary cohort reported normal visual acuity in early childhood, a family history compatible with autosomal recessive inheritance, and exhibited five or more of the following features of ABCA4-associated retinal disease: decreased visual acuity before age 20, decreased visual acuity as the first visual symptom, symmetrical fundus findings, pisciform flecks, beaten metal macular atrophy, bulls-eye maculopathy, peripapillary sparing, vermillion fundus, masked choroid on fluorescein angiography, nummular pigment overlying extensive macular atrophy, central outer retinal atrophy on optical coherence tomography and central scotomas on Goldmann perimetry. Each member of the validation cohort had a clinical diagnosis of Stargardt disease made by their referring ophthalmologist before any molecular testing was performed.
DNA extraction
Blood samples were obtained from all subjects. DNA was extracted by following the manufacturer’s specifications for whole blood DNA extraction using Gentra Systems' Autopure LS instrument (AutoGen Inc., Holliston, MA, USA).
Haplotype analysis
Sixty tagged SNPs spanning the ABCA4 gene were selected from the International HapMap Project (http://hapmap.ncbi.nlm.nih.gov) (Supplementary Material, Table S1). The selected SNPs were required to be compatible with TaqMan SNP genotyping assays and were informative for HapMap haplotype assignment. Allele-specific genotyping was performed on the missing alleles of each of the 28 probands, their relatives and 18 control trios using the TaqMan SNP genotyping assays (Life Technologies) in a high-throughput micro-fluidic system (Fluidigm, San Fransisco, CA, USA) as per the manufacturer's instructions. Haplotypes of the missing ABCA4 alleles were identified by subtracting the genotype of the known allele.
Next-generation DNA sequencing
Capture of the genomic region containing ABCA4 (chr1:94 448 410–94 616 987) was performed using a Haloplex custom capture kit following the manufacturer's instructions (Agilent Technologies Inc., Santa Clara, CA, USA). Nine samples were barcoded, pooled and sequenced according to the manufacturer’s instructions on one paired end 100 bp lane of an Illumina HiSeq sequencer (Illumina, San Diego, CA, USA) at University of Iowa's DNA Core Facility.
Human donor tissue
With the consent of the donor's family, the eyes of a 91-year-old Caucasian female with no history of eye disease were obtained immediately after death from the Iowa Lions Eye Bank. Retinal punches (4 mm diameter) were collected from the macula (centered on the fovea centralis) as well as from the temporal, superior, nasal and inferior quadrants ∼15 mm from the macula. All punches were collected within 6 h of death.
Patient derived cells
Keratinocytes were isolated from 3 mm punch biopsies obtained from patients with clinically diagnosed stargardt disease, who harbored one plausible disease-causing ABCA4 mutation, following informed consent. Isolation was performed as described previously (21). Briefly, the epidermis was carefully dissected free from the underlying dermal tissue, placed in a flat bottom 1 ml tube containing 500 µl of 1 mg/ml dispase and incubated for 16 h at 4°C. Following incubation, the epidermis was carefully pealed free from the remaining dermal layer and placed in 0.25% tryspin/EDTA and incubated for 30 min at 37°C. Following incubation tissue was triturated with a polished pasture pipet to liberate cells. Cells were pelleted via centrifugation and cultured in low calcium epilife media (Gibco, M-EPI-500-CA).
RNA isolation and RT-PCR
Total RNA was extracted from human donor retina or cultured human keratinocytes using the RNeasy Mini-kit (Qiagen, Valencia, CA, USA) following the provided instructions. Briefly, cells were lysed, homogenized and diluted in 70% ethanol to adjust binding conditions. Samples were spun using RNeasy spin columns, washed and RNA was eluted with RNase-free water. One microgram of RNA was reverse transcribed into cDNA using the random hexamer (Invitrogen, Carlsbad, CA, USA) priming method. All PCR reactions were performed in a 50 µl reaction containing 1 × PCR buffer, 1.5 mm MgCl2, 0.2 mm dNTPs, 100 ng of DNA, 1.0 U of Platnium Taq (Invitrogen) and 20 pmol of each gene-specific primer (Integrated DNA Technologies, Coralville, IA, USA). All cycling profiles incorporated an initial denaturation temperature of 94°C for 10 min through 35 amplification cycles (30 s at 94°C, 30 s at annealing temperature of each primer and 1 min at 68°C) and a final extension at 68°C for 5 min. PCR products were separated by gel electrophoresis using 2% agarose e-gels (Invitrogen).
RNA sequencing
Isolated RNA was sent to the Hudson Alpha Institute (Huntsville, AL, USA) for paired-end sequencing by Illumina HiSeq (Illumina). Reads were aligned to the hg19 human reference genome with TopHat, using RefSeq gene transcript models for initial transcriptome alignment. Cufflinks was used for generating transcript assemblies and abundance estimation. Reference annotation-based transcriptome assembly was implemented using the RefSeq gene models. Junction information was visualized using the Integrative Genomics Viewer for alternate exon identification (Broad Institute, Cambridge, MA, USA).
Selection of 15 genomic regions for analysis in the primary cohort
Alternate exons in the ABCA4 RNAseq data were selected for sequencing in the primary cohort if they were present in at least 10 sequencing reads in two or more of the five retinal regions. Fifteen alternate exons met these criteria (genomic coordinates provided in the Supplementary Material, Table S2). Seven of these 15 alternate exons were present in all 5 retinal regions with greater than 10 reads. Three of the seven had reads connecting them to other exons in both directions. These 15 regions were sequenced in all 28 members of the primary cohort and 300 unaffected control individuals using automated DNA sequencing with dye termination chemistry on an ABI 3730 sequencer (Life Technologies).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by Elmer & Sylvia Sramek Charitable Foundation, Chicago, IL; Carver Endowment for Molecular Ophthalmology, Iowa City, IA; Howard Hughes Medical Institute, Chevy Chase,MD to E.M.S.; National Institutes of Health, Directors New Innovator Award (1-DP2-OD007483-01) to B.A.T.; National Institute of Health (EY-017451 to R.F.M., EY-016822 to E.M.S., EY-013203 to A.V.C.); and The Foundation Fighting Blindness, Owings Mills, MD. Funding to pay the Open Access publication charges for this article was provided by The Howard Hughes Medical Institute.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to the patients and their families for participating in this study.
REFERENCES
- 1.Schindler E.I., Nylen E.L., Ko A.C., Affatigato L.M., Heggen A.C., Wang K., Sheffield V.C., Stone E.M. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum. Mol. Genet. 2010;19:3693–3701. doi: 10.1093/hmg/ddq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheffield V.C., Stone E.M. Genomics and the eye. N. Engl. J. Med. 2011;364:1932–1942. doi: 10.1056/NEJMra1012354. [DOI] [PubMed] [Google Scholar]
- 3.Weng J., Mata N.L., Azarian S.M., Tzekov R.T., Birch D.G., Travis G.H. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 4.Quazi F., Lenevich S., Molday R.S. ABCA4 Is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012;3:925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong J., Kim S.R., Binley K., Pata I., Doi K., Mannik J., Zernant-Rajang J., Kan O., Iqball S., Naylor S., et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene. Ther. 2008;15:1311–1320. doi: 10.1038/gt.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster A.R., Heon E., Lotery A.J., Vandenburgh K., Casavant T.L., Oh K.T., Beck G., Fishman G.A., Lam B.L., Levin A., et al. An analysis of allelic variation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci. 2001;42:1179–1189. [PubMed] [Google Scholar]
- 7.Zernant J., Schubert C., Im K.M., Burke T., Brown C.M., Fishman G.A., Tsang S.H., Gouras P., Dean M., Allikmets R. Analysis of the ABCA4 gene by next-generation sequencing. Invest. Ophthalmol. Vis Sci. 2011;52:8479–8487. doi: 10.1167/iovs.11-8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abril J.F., Castelo R., Guigo R. Comparison of splice sites in mammals and chicken. Genome Res. 2005;15:111–119. doi: 10.1101/gr.3108805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duno M., Schwartz M., Larsen P.L., Rosenberg T. Phenotypic and genetic spectrum of Danish patients with ABCA4-related retinopathy. Ophthalmic Genet. 2012;33:225–231. doi: 10.3109/13816810.2011.643441. [DOI] [PubMed] [Google Scholar]
- 14.Lewis R.A., Shroyer N.F., Singh N., Allikmets R., Hutchinson A., Li Y., Lupski J.R., Leppert M., Dean M. Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in stargardt disease. Am. J. Hum. Genet. 1999;64:422–434. doi: 10.1086/302251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cideciyan A.V., Swider M., Aleman T.S., Tsybovsky Y., Schwartz S.B., Windsor E.A., Roman A.J., Sumaroka A., Steinberg J.D., Jacobson S.G., et al. ABCA4 Disease progression and a proposed strategy for gene therapy. Hum. Mol. Genet. 2009;18:931–941. doi: 10.1093/hmg/ddn421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Downs K., Zacks D.N., Caruso R., Karoukis A.J., Branham K., Yashar B.M., Haimann M.H., Trzupek K., Meltzer M., Blain D., et al. Molecular testing for hereditary retinal disease as part of clinical care. Arch. Ophthalmol. 2007;125:252–258. doi: 10.1001/archopht.125.2.252. [DOI] [PubMed] [Google Scholar]
- 17.den Hollander A.I., Koenekoop R.K., Yzer S., Lopez I., Arends M.L., Voesenek K.E., Zonneveld M.N., Strom T.M., Meitinger T., Brunner H.G., et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vache C., Besnard T., le Berre P., Garcia-Garcia G., Baux D., Larrieu L., Abadie C., Blanchet C., Bolz H.J., Millan J., et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Hum. Mutat. 2012;33:104–108. doi: 10.1002/humu.21634. [DOI] [PubMed] [Google Scholar]
- 19.Sagai T., Hosoya M., Mizushina Y., Tamura M., Shiroishi T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 2005;132:797–803. doi: 10.1242/dev.01613. [DOI] [PubMed] [Google Scholar]
- 20.Amano T., Sagai T., Tanabe H., Mizushina Y., Nakazawa H., Shiroishi T. Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev. Cell. 2009;16:47–57. doi: 10.1016/j.devcel.2008.11.011. [DOI] [PubMed] [Google Scholar]
- 21.Bickenbach J.R. Isolation, characterization, and culture of epithelial stem cells. Methods Mol. Biol. 2005;289:97–102. doi: 10.1385/1-59259-830-7:097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.