

Abstract
Mutations in Vps33 isoforms cause pigment dilution in mice (Vps33a, buff) and Drosophila (car) and the neurogenic arthrogryposis, renal dysfunction and cholestasis syndrome in humans (ARC1, VPS33B). The later disease is also caused by mutations in VIPAS39, (Vps33b interacting protein, apical-basolateral polarity regulator, SPE-39 homolog; ARC2), a protein that interacts with the HOmotypic fusion and Protein Sorting (HOPS) complex, a tether necessary for endosome–lysosome traffic. These syndromes offer insight into fundamental endosome traffic processes unique to metazoans. However, the molecular and cellular mechanisms underlying these mutant phenotypes remain poorly understood. Here we investigate interactions of wild-type and disease-causing mutations in VIPAS39/SPE-39 and Vps33b by yeast two hybrid, immunoprecipitation and quantitative fluorescent microscopy. We find that although few mutations prevent interaction between VIPAS39/SPE-39 and Vps33b, some mutants fragment VIPAS39/SPE-39-positive endosomes, but all mutants alter the subcellular localization of Vps33b to VIPAS39/SPE-39-positive endosomes. Our data suggest that the ARC syndrome may result through impaired VIPAS39/SPE-39 and Vps33b-dependent endosomal maturation or fusion.
INTRODUCTION
Trafficking of cargoes through the endocytic pathway requires endosome maturation, and this process frequently culminates with endosome–lysosome fusion (1–5). These endocytic trafficking mechanisms are defective in a wide variety of inherited human diseases that include neurodegeneration and pigment dilution syndromes such as Hermansky–Pudlak, Griscelli, and Chediak–Higashi syndromes (6–14). The complexity of these processes is reflected by genome-wide siRNA down-regulation experiments, where ∼20% of the human transcriptome is required for normal endosome function/morphology in cultured cells (15). Endosome maturation is one of the mechanisms that determine cargo progression along the endocytic route (2–5). This maturation process is characterized by the swapping of regulatory rab GTPases on the membrane of an endosome. Rab exchange is in turn necessary for the recruitment of cytosolic factors known as tethers, which bring two membranous organelles in close proximity in a step upstream of the pairing of soluble NSF attachment protein receptors (SNAREs) (2–5). Molecules that participate in the endosome maturation mechanism are also affected in Charcot–Marie–Tooth disease (rab7), Hermansky–Pudlak-like syndrome in mice (Vps33a, buff) and flies (Vps33a, carnation) and arthrogryposis, renal dysfunction and cholestasis syndromes 1 and 2 (Vps33b and VIPAS39/SPE-39, respectively) (16–22).
Much of what we know about this endosome maturation and fusion mechanisms has been pioneered by studies in Saccharomyces cerevisiae (23). Fusion along early and late stages of the endosomal route requires two tethers, the HOmotypic fusion and Protein Sorting (HOPS) complex and the class C core vacuole/endosome tethering (CORVET) complex. These tethers are heterohexameric complexes responsible for forward and retrograde fusion events between early and late endosome compartments, respectively (2,24–26). The HOPS and CORVET complexes share the class C Vps proteins Vps11, Vps16, Vps18 and Vps33, which bind to Vps39 and Vps41 to form the HOPS complex or Vps8 and Vps3 to constitute the CORVET complex (24,27,28). Although the molecular details of how tethers operate in yeast are well understood, the picture in metazoans is less well characterized and far more complex. The complexity of metazoan Vps class C tethers can be appreciated just by simply counting the number of isoforms for some of its subunits. Human Vps16, 39 and 41 possess at least two predicted splicing isoforms each, whereas the genomes of nematodes, flies and vertebrates have two genes encoding orthologs of S. cerevisiae Vps33, Vps33a and Vps33b (29). The function of these two genes is not redundant because, for instance, Drosophila Vps33b cannot rescue Vps33a-associated phenotypes, and mammalian Vps33a and b have non-overlapping phenotypes (19,22,30). Metazoan-specific molecules further emphasize the complexity of metazoan Vps class C tethers. This is the case for Vps33b and VIPAS39/SPE-39, two molecules whose genetic deficiencies produce similar phenotypes and constitute the focus of the present work (21,22).
VIPAS39/SPE-39 was previously referred to as C14orf133 (HGNC: 20347), Vps16b or VIPAR (21,31,32). SPE-39 was first identified in a Caenorhabditis elegans screen for genes required for the morphogenesis of the fibrous body membranous organelle, a lysosome-related organelle, assembled during spermatogenesis and required for fertilization (33). This was the first indication of a role for SPE-39 in endosome trafficking. Later, it was shown that VIPAS39/SPE-39 functions in other worm tissues, including phagocytic (macrophage-like), coelomocytes and oocytes. The spermatocyte phenotype is phenocopied by RNAi of the worm Vps33b ortholog (34). The phenotypic similarities between worm Vps33b and SPE-39 deficiencies extend to Drosophila and humans (21,22,32,34,35). Genetic defects in human Vps33b or VIPAS39/SPE-39 lead to the arthrogryposis, renal dysfunction and cholestasis syndromes type 1 and 2 (ARC1 and 2), respectively (OMIM 208085, 608552, 613404, 613401). This syndrome is characterized by neurogenic joint alterations, kidney and liver dysfunction, diverse neurodevelopmental pathology ranging from spinal cord neuropathology to lissencephaly, ichtyosis and facial dysmorphias (21,22,29,36,37). The similar phenotypes shown by either Vps33b or SPE-39 loss of function is probably due to the direct interaction of these two proteins (21).
Here we tested the hypothesis that altered Vps33b–VIPAS39/SPE-39 interaction is a major cause of vesicular trafficking abnormalities by analyzing a series of human mutations in Vps33b and VIPAS39/SPE-39. We identified the subunit of the HOPS complex to which VIPAS39/SPE-39 binds and defined regions necessary for Vps33b binding to VIPAS39/SPE-39. We focused on Vps33b and VIPAS39/SPE-39 ARC causative mutations as well as buff- and carnation-like Vps33b defects. Most missense ARC, buff- and carnation-like defects cluster in a Vps33b region shared by Vps33 isoforms from nematodes to chordates. This Vps33b region is required for VIPAS39/SPE-39 binding, yet this region is not necessary for Vps33b binding to either the late endosomal SNARE syntaxin 7, the Vps class C core or the HOPS complex. Critically, none of these binding activities of Vps33b are by themselves predictors of the pathogenic character of ARC, buff- and carnation-like mutations. Instead, the subcellular localization of Vps33b to the VIPAS39/SPE-39-positive endosome is the only common phenotype among all mutations in Vps33b. Our data suggest a model where defective VIPAS39/SPE-39 and Vps33b-dependent endosomal maturation and/or fusion contribute to the pathogenesis of the ARC syndrome. We propose that VIPAS39/SPE-39 provides specificity to conserved endo-lysosomal tethers so that they can engage in fusion reactions among the diverse endosomal compartments that distinguish metazoans from simpler organisms, such as yeast.
RESULTS
We analyzed the biochemical and cellular phenotypes of autosomal recessive missense and nonsense mutations in the gene encoding Vps33b. We focused on genetic defects that, in humans, generate the neurogenic arthrogryposis, renal dysfunction and cholestasis (ARC) syndrome, as well as mutations in its paralog Vp33a (the gene affected in mouse buff and Drosophila carnation mutants). We hypothesized that these mutations would disrupt critical and evolutionary conserved molecular interactions of Vps33b.
VIPAS39/SPE-39 binds Vps33b
The limited homology of VIPAS39/SPE-39 with Vps16 and the phenocopying observed in human Vps33b and VIPAS39/SPE-39 mutants suggests two alternative models for VIPAS39/SPE-39 interactions within the Vps class C complex (Fig. 1A) (34). These models make precise predictions about the interaction of VIPAS39/SPE-39 and Vps11, which are founded in the biochemical and structural analyses of the HOPS complex in S. cerevisiae (27,28,38). We tested these models by performing yeast two-hybrid (Y2H) analyses asking which one of the Vps core class C complex and/or HOPS subunits bind VIPAS39/SPE-39. VIPAS39/SPE-39 robustly bound to Vps33b as indicated by cell growth of a triple-auxotrophic yeast strain (Fig. 1B, lane 4; C, lane 4). In contrast, VIPAS39/SPE-39 failed to interact with Vps11 or 18 as predicted by model 1 (Fig. 1B, lanes 1 and 3). The functionality of Vps11 in this assay was asserted by its interaction with Vps39 (Fig. 1B and C, lanes 7 and 5, respectively) (28,38). The Vps33b association with VIPAS39/SPE-39 is not a reflection of promiscuous binding of Vps33b in Y2H as demonstrated by the lack of cell growth in strains carrying Vps33b in combination with itself and the HOPS subunits Vps39 and 41 (Fig. 1C, lanes 1–3). These data favor model 2, where VIPAS39/SPE-39 associates with the class C core complex as a Vps33b-interacting protein rather than as a Vps16-like molecule, as previously proposed by others (32,35).
Figure 1.
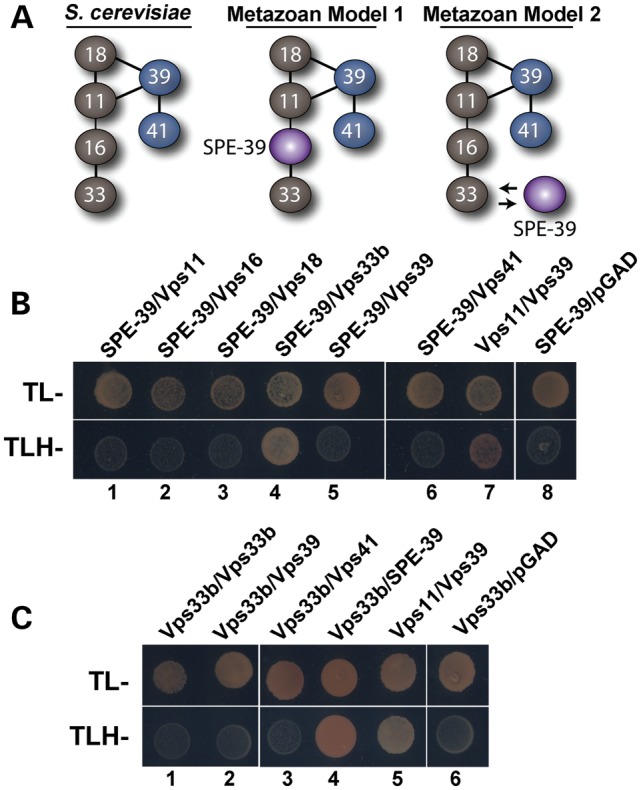
Direct interaction between VIPAS39/SPE-39 and Vps33b. (A) The yeast HOPS complex (left) and two possible models for how VIPAS39/SPE-39 interacts with other proteins in the metazoan HOPS complex. (B and C) Y2H assays testing possible interaction between Vps11, 16, 18, 39, 41, with VIPAS39/SPE-39 and Vps33b, respectively. Yeasts were grown in media deficient in tryptophan and leucine (TL−) to confirm that the bait and prey vectors were present in cells, and in interaction media deficient in tryptophan, leucine and histidine (TLH-) to detect interaction between the two proteins of interest. VIPAS39/SPE-39 binds to Vps33b; Vps11–Vps39 interaction was used as positive control and empty pGAD or pGB vector as negative control (B and C). Difference between (B) and (C) is that (B) has VIPAS39/SPE-39 as the bait and (C) has Vps33b as the bait.
Sequence alignment of human Vps33b reveals amino acids 221–260 as a region with high primary sequence conservation among the two mammalian Vps33 isoforms, as well as Vps33 polypeptides from Danio rerio, Drosophila and C. elegans (Fig. 2A and B). This region concentrates mutations affecting the phenotype of metazoan organisms spanning three phyla (Fig. 2A) (19,20,39). Furthermore, it is the place where the sec1-36 and sec1-76 alleles map in the S. cerevisiae Sec1 primary sequence, the founding member of the Sec1-Munc18 (SM) family proteins to which Vps33b belongs (40,41). We studied the association between Vps33b and VIPAS39/SPE-39 by analyzing how Vps33 mutations implicated in human ARC syndrome, mouse buff and Drosophila carnation affect interaction of these two proteins (Fig. 2D) (36,39,42,43). We first used Y2H to determine whether Vps33 mutations known to cause a phenotype in vivo affected association between Vps33 and VIPAS39/SPE-39. The ARC mutation L30P perturbs Vps33b–VIPAS39/SPE-39 association in vivo and this was faithfully recapitulated by the Y2H analysis (Fig. 2D, row 1) (21). Point mutations in the Vps33b 221–260 region fell in two categories: some disrupted VIPAS39/SPE-39 binding, such as a carnation-like mutation introduced into human Vps33b (Fig. 2D, row 3, G249V). In contrast, all other Vps33b missense mutant proteins were able to bind VIPAS39/SPE-39, irrespective of their pathogenic character. These include the ARC Vps33b D234H and S243F mutations (Fig. 2D, rows 2 and 7) and the buff-like mutation introduced into Vps33b (Fig. 2D, row 4). The same two binding behaviors to Vps33b were also identified in two VIPAS39/SPE-39 ARC pathogenic mutants (21). A nonsense defect truncating VIPAS39/SPE-39 at position 220 lost binding to Vps33b (Fig. 2D, row 8), yet a truncation at position 425 retained VIPAS39/SPE-39 binding to Vps33b (Fig. 2D, row 9).
Figure 2.
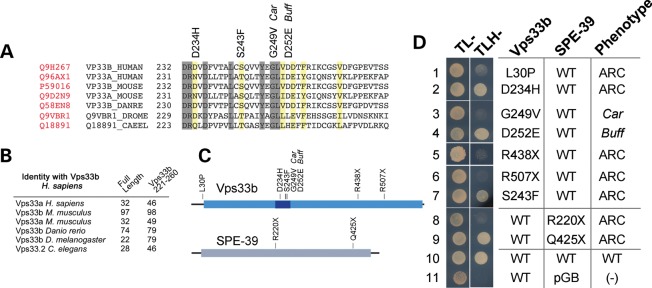
Effects of pathogenetic Vps33 mutations on Vps33b–VIPAS39/SPE-39 interaction. (A) Sequence alignment of human Vps33b from amino acids 172–271 with mammalian Vps33 isoforms, Vps33 polypeptides from D. rerio (DANRE), Drosophila melanogaster (DROME) and C. elegans (CAEEL). Identical residues are highlighted with gray boxes. Yellow boxes represent conserved residues. Uniprot numbers for each gene are in red. (B) The table depicts percent identity of human Vps33b with the sequences presented in (A), either with full-length proteins or with the Vps33b residues 221–260. (C) Schematic representation of Vps33b and VIPAS39/SPE-39 mutations tested in (D). The light blue box indicates the Vps33b SEC-1 domain. The dark blue box indicates the Vps33b residues 221–260. The gray box in VIPAS39/SPE-39 represents the Golgin A5 domain (Pfam PF09787) (D) Y2H evaluation of ARC, Car and Buff mutations on Vps33b–VIPAS39/SPE-39 interaction. Vps33b mutations L30P, G249V, R438X, R507X and VIPAS39/SPE-39 mutations R220X prevent Vps33b–SPE39 interaction (rows 1, 3, 5, 6, 8), whereas Vps33b mutations D234H, S243F, D252E and VIPAS39/SPE-39 mutation Q425X maintain the interaction (lanes 2, 4, 7, 9). Wild-type Vps33b and VIPAS39/SPE-39 (row 10) and wild-type Vps33b with empty vector pGBKT7 (row 11) were used as controls.
The Vps33b–VIPAS39/SPE-39 interaction phenotypes associated with pathogenic mutations in these two proteins were also analyzed by transfection of mutant constructs in mammalian cells. We transiently transfected HEK cells either with HA-tagged Vps33b wild-type, Vps33b carrying ARC-causing missense mutations, or the mouse or Drosophila Vps33a mutations engineered in Vps33b. Wild-type Vps33b co-immunoprecipitated with VIPAS39/SPE-39 in the absence and presence of the crosslinker DSP (Fig. 3A–C compare lanes 9 and 10; D, lanes 11 and 12). We introduced in vivo DSP crosslinking as a way to stabilize interactions potentially weakened by Vps33b pathogenic mutations (37,44–47). The specificity of the VIPAS39/SPE-39 co-precipitation with Vps33b-HA was determined by a two-pronged approach. First, VIPAS39/SPE-39 was absent from HA precipitations performed from non-transfected cell extracts (Fig. 3A–C, lanes 7 and 8; D, lanes 9 and 10). Moreover, Vps33b-HA precipitates were free of transferrin receptor, an abundant membrane protein localized to endocytic compartments where VIPAS39/SPE-39 resides (Fig. 3A) (34). Missense mutations expressed in HEK293 cells recapitulated phenotypes observed in Y2H analysis (Fig. 2). Thus, only the ARC L30P and Carnation-like G249V mutations prevented the interaction between Vps33b and VIPAS39/SPE-39, even in the presence of DSP (Fig. 3A, lanes 11 and 12; D, lanes 13 and 14). In contrast, and as predicted from Y2H, ARC mutations D234H (Fig. 3C, lanes 11 and 12), S243F (Fig. 3B, lanes 11 and 12) and Buff-like D252E (Fig. 3D, lanes 15 and 16) did not significantly affect Vps33b–VIPAS39/SPE-39 interaction. These results indicate that pathogenic mutations in Vps33b exert their effect by two distinct mechanisms that either affect or are independent of binding between Vp33b and VIPAS39/SPE-39.
Figure 3.

Effects of ARC mutations on Vps33b–VIPAS39/SPE-39 interaction in HEK cells. HEK cells were transiently transfected with either wild-type (WT) or mutant Vps33b tagged with HA. Cell extracts from untransfected HEK cells and cells expressing recombinant Vps33b were incubated at 4°C with (even lanes) or without (odd lanes) crosslinker, DSP. (A–D) Cell extracts were treated with magnetic beads that were decorated with rabbit polyclonal antibody directed against the HA epitope (lanes 7–12, A–C; lanes 9–16, D). The resulting immune complexes were resolved by SDS–PAGE and their composition was assessed by immunoblot with antibodies against HA, VIPAS39/SPE-39 and transferrin receptor (TrfR). Inputs represent 5% of the total immunoprecipitated proteins. VIPAS39/SPE-39 was co-immunoprecipitated with Vps33b WT, Vps33b D234H, S243F, D252E (lanes 9–12, A–C; lanes 11,12, 15, 16, D), but was not detected when Vps33b L30P and G249V were immunoprecipitated (lanes 11 and 12, B; 13 and 14, D). The negative control was HA and VIPAS39/SPE-39 mock immunoprecipitation from untransfected cells. Detection of the transferrin receptor, an abundant endosomal protein, was used as gel-loading control (lanes 1–6, A) as well as to confirm immunoprecipitation specificity (lanes 7–12).
We explored how Vps33b incorporation into HOPS complexes was affected by pathogenic mutations, irrespective of whether they affected VIPAS39/SPE-39 binding. We focused on the Vps33b ARC mutants L30P and D234H because, respectively, they either did or did not affect VIPAS39/SPE-39 binding (Figs 2 and 3). We tested how these Vps33b mutations affected their ability to incorporate into HOPS complexes, choosing the class C core complex subunit Vps18-Myc and the HOPS-specific subunit Vps41-Myc as markers (34,47). We co-expressed wild-type, L30P or D234H Vps33b-HA together with either Vps18-Myc or Vps41-Myc in HEK cells and tested whether Vps33b-HA co-immunoprecipitated with recombinant Vps18 or Vps41. The specificity of interactions was controlled as before in cells where Vps33b-HA was omitted (Fig. 4A and B, lane 5, respectively) or probing immunoprecipitated complexes for the presence of transferrin receptor (Fig. 4A). Vps33b-HA, Vps33b L30P-HA and D234H-HA were found to co-immunoprecipitate with Vps41-Myc and Vps18-Myc (Fig. 4A and B, lanes 6, 7 and 8, respectively), indicating that the integration of Vps33b into HOPS complexes was not affected by these ARC mutations.
Figure 4.

ARC mutations affecting Vps33b and VIPAS39/SPE-39 incorporate into the HOPS complex. (A and B) Untransfected HEK cells and HEK cells expressing Vps41-Myc (A) or Vps18-Myc (B) were transfected with Vps33b L30P-HA and D234H-HA. Extracts of cells treated with the crosslinker DSP were incubated with magnetic beads decorated with a monoclonal antibody directed against HA. HA and Myc epitopes were detected in the input lanes and from immunoprecipitations of Vps33b WT, L30P and D234H (lanes 6 and 7, A and B). (A) Transferrin receptor (TrfR) was used as a gel-loading control. Controls for specificity include TfrR, HA and Myc on western blots of gels that resolved immunoprecipitations from non-transfected cells (A and B, lanes 5–8).
Structural modeling of the pathogenic ARC mutations in Vps33b
Vps33b pathogenic mutations did not consistently affect binding to VIPAS39/SPE-39. Moreover, mutations did not hinder incorporation of Vps33b into HOPS complexes (Figs 2 and 3). However, most Vps33b mutations causing phenotypes in vertebrates and invertebrates clustered in the evolutionary conserved region encoded by the amino acids 221–260 of human Vps33b (Fig. 2B), suggesting that this domain is necessary for Vps33b function. These observations led us to make two structural predictions about Vps33b and its pathogenic mutations. First, our findings suggested that residues mutated in the domain 221–260 reside in domains not accessible to VIPAS39/SPE-39. Second, they predicted that the overall folding of Vps33b is not affected by Vps33b mutations, as deduced from their integration into HOPS complexes.
We modeled the structure of human Vps33b and we performed constrained molecular docking between VIPAS39/SPE-39 and Vps33b. The Vps33b region 221–260 corresponds to two small alpha-helical domains connected by a flexible loop (Fig. 5). This region was buried deep in the structure of Vps33b in a connecting region between two Vps33b lobes equivalent to domains 1–2 and 3a–3b in n-sec1 (48,49) (Fig. 5, gray ovals in Vps33b, upper left). Modeling of the Vps33b pathogenic mutations showed significant effect neither on the secondary structure of the Vps33b 221–260 region nor on the overall tertiary structure of Vps33b. This observation is in rapport with the incorporation into HOPS complexes of Vps33b mutants affecting this region (Fig. 4). Furthermore, the Vps33b 221–260 domain is not predicted to form a binding interface for VIPAS39/SPE-39 as determined by constrained molecular docking between VIPAS39/SPE-39 and Vps33b.
Figure 5.

Structural modeling of Vps33b, Syntaxin 7 and VIPAS39/SPE-39. Ribbon diagrams depict Vps33b and its complex with the SNARE domain of syntaxin 7 and VIPAS39/SPE-39. Gray ovals in Vps33b, upper left, denote two lobes in Vps33b equivalent to domains 1 and 2 and 3a–3b in n-sec1 (48,49). Residues marked red in Vps33b highlight the 221–260 region as well as the ARC Vps33b mutation L30P. Green depicts residue G249 affected by the car mutation in Drosophila Vps33a.
If the conserved Vps33b 221–260 region is necessary for Vps33b function, then pathogenic mutations in this domain should consistently impair Vps33b–VIPAS39/SPE-39 binding. This prediction is at odds with the differential effect of Vps33b G249V, which impairs binding, and D234H and D252E mutations, which do not. We tested the functional relevance of the 221–260 domain, which we inferred from its evolutionary conservation and the clustering of pathogenic mutations (Fig. 2A–C). We reasoned that missense mutations in this region encompassing residues adjacent to those affected by single pathogenetic mutations should prevent Vps33b–VIPAS39/SPE-39 interaction, as suggested by the Vps33b G249V mutation. We selected triple-alanine mutants that did not affect the predicted folding of Vps33b, yet encompassed the D234H and D252E residues. We generated three triple-alanine mutations covering residues D232-234A, D235-237A and V251-253A, which encompass D234H and D252E (Fig. 6A). These three Vps33b mutants impaired Vps33b–VIPAS39/hSPE39 interaction in Y2H assay (Fig. 6B, rows 1–3). These results indicate a crucial role of Vps33b domain 221–260 in facilitating its binding to VIPAS39/SPE-39 by an indirect mechanism.
Figure 6.
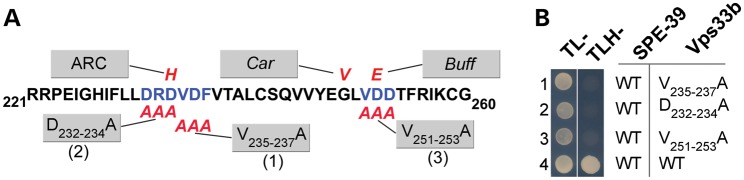
Vps33b domain 221–260 is necessary for binding with VIPAS39/SPE-39. (A) Schematic representation of the region 221–260, which clusters ARC, Car, Buff mutations affecting human, Drosophila and mouse, respectively. ARC, Car, Buff and triple-alanine mutations are represented in red. (B) Y2H assay testing VIPAS39/SPE-39 interaction with three different Vps33b triple-alanine mutations (rows 1–3) and wild-type Vps33b (row 4).
Pathogenic ARC mutations do not impair Vps33b–SNARE interactions
We focused on the Vps33b–SNARE interaction because SNARE binding rather than VIPAS39/SPE-39 association could be a common pathogenic mechanism for all Vps33b pathogenic mutations. Vps33b belongs to the SM protein family capable of binding SNAREs and defined by the presence of the Sec-1 domain (Fig. 2B, dark blue box) (50). We modeled the structure of human Vps33b complexed with the syntaxin 7 SNARE domain (Fig. 5). Vps33b and syntaxin 7 structures were derived from the crystal structure coordinates of their homologs n-sec1 bound to syntaxin 1 (49). Syntaxin 7-predicted binding interfaces were away from the Vps33b domain 221–260 (Fig. 5). The Vps33b L30P mutant was the closest residue to a binding interface between Vps33b and syntaxin 7, yet the predicted conformational changes induced by the L30P mutant did not result in alterations of syntaxin 7-binding surface in Vps33b or its overall structure. We experimentally tested these structural predictions determining the binding of Vps33b to endosomal syntaxins that recognize Drosophila Vps33 isoforms: syntaxin 7, 13 and 16 (30). We deleted their transmembrane domains in order to perform Y2H assays. None of the syntaxins bound to Vps33b (Fig. 7A, rows 1, 2 and 4) compared with a control Vps33b–VIPAS39/SPE-39 (Fig. 7A, row 5). Syntaxins adopt a closed conformation, with their N-terminus flipping over their C-terminus preventing the exposure of their SNARE domain (48,51–53). We tested the binding of Vps33b to an endosomal syntaxin SNARE domain by deletion of its N-terminus. Vps33b binds to syntaxin 7 in Drosophila (30), and syntaxin 7 subcellular distribution is affected in VIPAS39/SPE-39 deficiency (34); hence, we focused on this endosomal SNARE and tested the binding of Vps33b to syntaxin 7 and its SNARE domain. Whereas Vps33b did not bind to syntaxin 7 (Fig. 7A, row 1; B, row 3), Vps33b bound to the syntaxin 7 SNARE domain (Fig. 7B, lane 1; C, row 1). None of the missense mutations affected the binding of Vps33b to the syntaxin 7 SNARE domain (Fig. 6C, rows 2 and 3, 5 and 6). These results indicate that Vps33b ARC missense mutations globally compromise neither Vps33b folding nor syntaxin 7 recognition.
Figure 7.
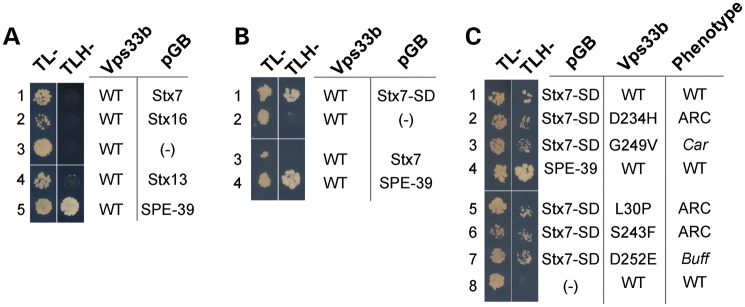
Mutant Vps33b retains binding to Syntaxin7 SNARE domain. Y2H assays were performed to investigate the interaction of either Vps33b or Vps33b containing ARC mutations with SNARE proteins. Interaction between Vps33b and VIPAS39/SPE-39 was used as a positive control and the empty vector pGB as a negative control. (A) Vps33b did not bind to syntaxin 7, 13 and 16 in the Y2H assay under triple dropout conditions (TLH-) (rows 1, 4, 2, respectively). (B) Vps33b interacted with the SNARE domain of syntaxin 7 but not the full-length syntaxin 7 (rows 1 and 3). (C) Buff, Car and ARC mutations in Vps33b did not prevent its interaction with the SNARE domain of syntaxin 7 (rows 2, 3, 5, 6, 7).
Pathogenic ARC mutations alter Vps33b subcellular distribution on the VIPAS39/SPE-39-positive endosome
Mammalian HOPS complex subunits have been described in Golgi compartments, early–late endosomes (54–59) and the VIPAS39/SPE-39-positive endosome, which is characterized by the presence of rab 5, 7 and 11 and exists in HEK293 cells as one or two copies per cell (34). We hypothesized that Vps33b pathogenic mutations would alter the morphology or co-localization of Vps33b-HA with these compartments. We performed quantitative high-resolution Delta deconvolution microscopy of cells expressing recombinant Vps33-HA with endogenous markers of late endosomes (syntaxin 7), early endosomes (EEA1), Golgi compartment (GM130) and mitochondria as control (Fig. 8). We quantified co-localization using Pearson's coefficient, which quantifies how variations in two channels conform to each other irrespective of the gain (60,61). The highest co-localization was observed between wild-type Vps33b-HA and syntaxin 7 with Pearson's coefficient of ∼0.4, whereas Golgi and early endosome co-localizations coefficients were half of the syntaxin 7 value (Fig. 8, compare A with B and C). None of the Vps33b-HA mutants modified these co-localization values. In all these cases, we determined the specificity of co-localization by rotating one channel with respect to the other by 15°, which dropped Pearson's coefficients to zero. We further tested the discriminatory power of our quantitative imaging analysis calculating Pearson's coefficient between recombinant forms of Vps33b-HA and the mitochondrial dye Mitotracker without or with 15° rotation, which were all negative or zero values, respectively (Fig. 8D).
Figure 8.

Subcellular distribution of recombinant Vps33b. HEK293 cells were transfected with wild-type and mutant forms of Vps33b tagged with HA. Cells were double-labeled with HA antibodies and one of the following: syntaxin 7 (Stx7, A), EEA1 (B), GM130 (C) and Mitotracker (D). Cells were imaged by Delta deconvolution microscopy. Co-localization was determined using Pearson's co-localization coefficient at 0° or after a 15° channel rotation in 20–28 cells per condition (A), 12 cells per condition (B and D) and 18 cells per condition (C). No statistical differences were detected among groups at 0° in (A)–(D), Kruskal–Wallis test. Bar 5 μm and inserts are 3-fold enlargement of the indicated area.
VIPAS39/SPE-39 defines a unique endocytic compartment that exists in HEK293 cells as one or two copies per cell (34). The integrity of this VIPAS39/SPE-39 compartment is required for cargo delivery along the endocytic route and the normal distribution of late endosomal SNAREs (34). The VIPAS39/SPE-39 endosome robustly co-localizes with endogenous or recombinant forms of wild-type Vps33b (34,43), hence we used co-localization with the VIPAS39/SPE-39 endosome as an indirect read out of Vps33b function. We characterized the effects of Vps33b mutations on Vps33b subcellular localization using quantitative and qualitative morphometric parameters between Vps33b-HA and endogenous VIPAS39/SPE-39 (Figs 9 and 10). The overall distribution of all Vps33b-HA-positive puncta was similar for wild-type and mutant Vps33b-HA (Fig. 9). In fact, analysis of Vps33b puncta frequencies, intensities (not shown) and size per cell were indistinguishable among wild-type and mutants (Fig. 10A). The morphology of the VIPAS39/SPE-39 endosome was not altered by the expression of wild-type or some mutant forms of Vps33b-HA (Figs 9 and 10). In contrast, the Vps33b-HA L30P mutation induced fragmentation of the VIPAS39/SPE-39 endosome (Fig. 9D and H), as indicated by increased VIPAS39/SPE-39 puncta frequencies per cell and puncta size reductions (Fig. 10B). The fragmentation of the VIPAS39/SPE-39 endosome resulted in a significant drop in Vps33b-HA co-localization with VIPAS39/SPE-39 with the L30P allele (Fig. 10C).
Figure 9.
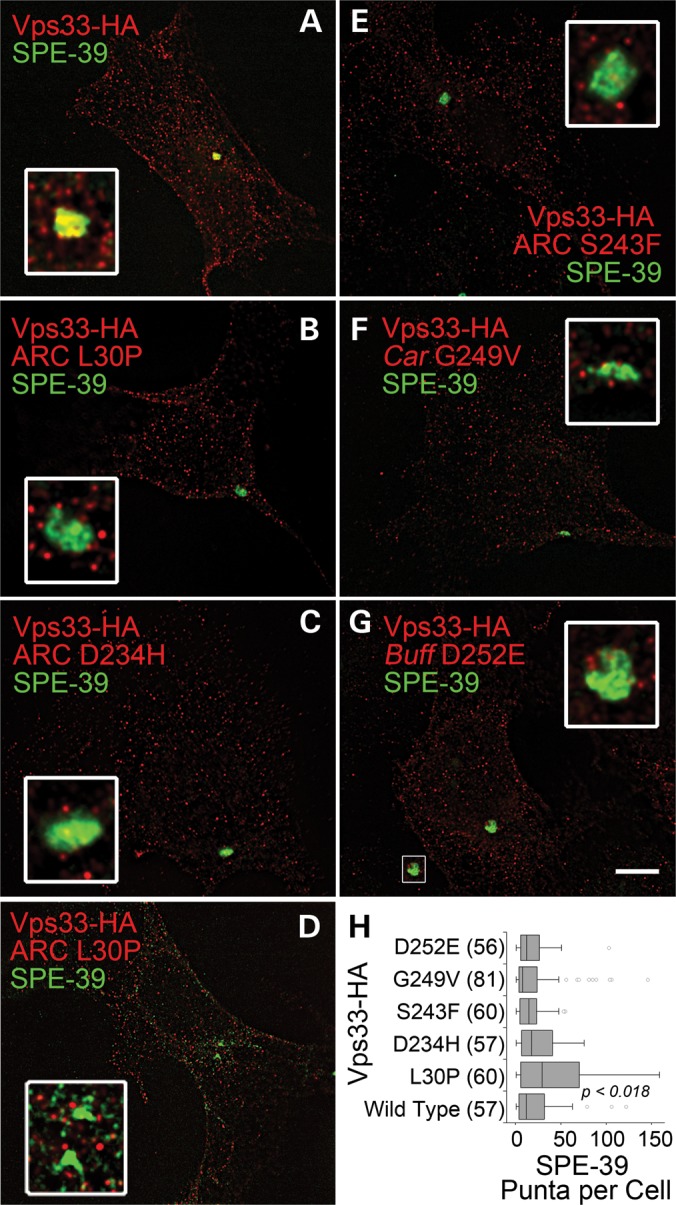
Subcellular distribution of recombinant Vps33b and endogenous VIPAS39/SPE-39. HEK293 cells were transfected with wild-type and mutant forms of Vps33b tagged with HA. Cells were imaged by Delta deconvolution microscopy. (B) and (D) depict cells expressing Vps33b-HA L30P mutant that differ in the fragmentation of the VIPAS39/SPE-39 endosome. Inserts are 4-fold enlargements of the VIPAS39/SPE-39 endosomal organelle. Bar 5 μm. (H) A quantification of the number of VIPAS39/SPE-39 endosomes per cell after expression of Vps33-HA. Numbers in parentheses are the number of cells obtained from three independent experiments. P-values were obtained after the Kruskal–Wallis test followed by the Wilcoxon–Mann–Whitney U-test.
Figure 10.

Quantitative analysis of recombinant Vps33b and endogenous VIPAS39/SPE-39 co-localization. (A and B) Probability plots of puncta counts for Vps33-Ha wild-type and mutant and endogenous VIPAS39/SPE-39. Inserts represent probability plots of puncta area measured in pixels. P-values were obtained with the Kolmogorov–Smirnov test and refer to comparisons between Vps33b-HA wild-type and L30P. (C) Co-localization between wild-type and mutant Vps33b-HA with endogenous VIPAS39/SPE-39. Co-localization was determined using Pearson's co-localization coefficient at 0° or after a 15° channel rotation in 18–25 cells per condition. P-values were obtained after the Kruskal–Wallis test followed by the Wilcoxon–Mann–Whitney U-test. (D) Patterns of co-localization between Vps33-HA and VIPAS39/SPE-39. (A and A′) Overlapping pattern. (B and B′) Juxtaposed pattern. (C and C′) No co-localization. (A′–C′) Imaris-generated isosurfaces of the images depicted in (A–C). (E) Frequency distribution of the patterns described in (C). P-values (Fisher's exact probability test for a 2 × 3 table).
We observed that the distribution of wild-type Vps33b-HA in the VIPAS39/SPE-39-positive endosome could be qualitatively described as three discrete patterns, here designated A–C (Fig. 10D). Pattern A is characterized by significant pixels overlapping between Vps33-HA and VIPAS39/SPE-39 endogenous signals, B corresponds to juxtaposed pixels minimally overlapping and C is characterized by Vps33b-HA puncta adjacent to VIPAS39/SPE-39 without pixel overlap. We scored these patterns in wild-type and all Vps33b mutants and determined their percentile distribution (Fig. 10E). The distribution of all mutants was significantly different from wild-type Vps33b-HA. All mutants have a decreased number of A-type VIPAS39/SPE-39 endosomes at the expense of an increased in those endosomes with the juxtaposed pattern B (Fig. 9D). These results demonstrate that all pathogenic mutations in Vps33b converge on a common cellular phenotype characterized by a defective overlap of VIPAS39/SPE-39- and Vps33b-positive compartments. Moreover, this phenotype is independent of the binding between VIPAS39/SPE-39 and Vps33b.
DISCUSSION
We used human mutations in Vps33b to define regions necessary for Vps33b binding to VIPAS39/SPE-39 as well as endosomal phenotypes common to Vps33b pathogenic mutations. We focused on Vps33b, the only class C Vps protein for which there are known human mutations, because of the genetic evidence indicating that Vps33b and VIPAS39/SPE-39 interact (21,34–36,43). Mutation of either Vps33b or VIPAS39/SPE-39 in C. elegans, Drosophila and humans produces, in each one of these species, nearly identical phenotypes. These mutations lead to the multi-systemic ARC1 and two syndromes in humans (OMIM 208085, 608552, 613404, 613401). We expanded the portfolio of ARC pathogenic mutations, creating buff- and carnation-like Vps33b defects (19,20). We predicted that these Vps33a defects would generate biochemical and cellular phenotypes when engineered into the Vps33b primary structure due to the high sequence similarity between these human Vps33 isoforms. Most missense ARC, buff- and carnation-like defects are within a Vps33b domain found in all Vps33 isoforms from nematodes to chordates. This Vps33b region is required for VIPAS39/SPE-39 binding as exemplified by loss of binding to the Vps33b G249V allele and the triple-alanine mutants in the 221–260 domain of Vps33b. However, this domain is not necessary for either the binding of Vps33b to syntaxin 7 or Vps33b incorporation into the Vps class C core and HOPS complexes. None of these binding activities of Vps33b are by themselves predictors of the pathogenic character of ARC, buff- and carnation-like mutations. Moreover, localization to Golgi, early and late endosomes does not discriminate between wild-type and mutant forms of Vps33b. Instead, the subcellular localization of Vps33b to the VIPAS39/SPE-39-positive endosome is a common phenotype among all mutations in Vps33b. This phenotype is characterized by a decrease in the overlapping pattern of recombinant mutated forms of Vps33b with the VIPAS39/SPE-39-positive endosome.
VIPAS39/SPE-39 is also referred to as Vps16b due to a region of homology between VIPAS39/SPE-39 and the C-terminus of Vps16a (32). This observation has been the basis for a model where VIPAS39/SPE-39 would replace Vps16a from class C Vps complexes (Fig. 1A, model 1). We previously demonstrated that Vps16a co-immunoprecipitates, co-sediments and co-localizes with VIPAS39/SPE-39 and Vps33b, suggesting that VIPAS39/SPE-39 is not replacing Vps16a from class C Vps complexes (Fig. 1A, model 2) (34). Our present data support model 2 since VIPAS39/SPE-39 binds Vps33b in Y2H but not Vps11 (Fig. 1A, model 2), a binding profile that differs from that predicted for a Vps16a-like molecule binding Vps33b and Vps11 (Fig. 1A, model 1). A critical test to distinguish between these models rests on the prediction of model 1 that Vps33b mutations disrupting its binding to VIPAS39/SPE-39 should prevent other class C or HOPS complex subunits to co-isolate with mutant Vps33b (Fig. 1A, model 1). This prediction contrasts with our findings, where Vps33b mutants that prevent VIPAS39/SPE-39 binding are still capable of precipitating subunits of the class C complex, Vps18 and the HOPS complex, Vps41 (Fig. 5). Thus, our data favor model 2, where Vps33b binds both VIPAS39/SPE-39 and Vps16a.
Although all Vps33b mutations tested share endosomal phenotypes (Figs 9 and 10), their molecular binding properties to SNAREs and VIPAS39/SPE-39 do not offer a unitary molecular mechanism to explain their pathogenic nature. The structural modeling of Vps33b complexed with syntaxin 7 and VIPAS39/SPE-39 provides preliminary insight into mechanisms by which these Vps33b pathogenic mutations may operate. First, Vps33b mutations do not seem to globally alter the secondary and tertiary structures of Vps33b, a prediction consistent with the binding of these Vps33b mutants to syntaxin 7 and their incorporation into HOPS complexes, and their normal co-localization with Golgi, early and late endosome markers. Second, the Vps33b 221–260 region is buried in the structure of Vps33b. The primary sequence integrity of Vps33b 221–260 is necessary for the binding of VIPAS39/SPE-39. This Vps33b domain is segregated from putative Vps33b SNARE and VIPAS39/SPE-39 surface-binding interfaces. Thus, plausible models for the mechanism affected by pathogenic mutations are (i) propagated structural changes in surfaces required for the binding of Vps33b to VIPAS39/SPE-39 or (ii) impaired transition of Vps33b among conformations required for its biological activity. We favor this last model because Vps33b 221–260 domain resides on the region of Vps33b that could act as a hinge. Moreover, an impaired transition of Vps33b among conformations is compatible with the puzzling observation that small increases in a residue size such as in Vps33b car(G249V) and the Vps33b D252E mutant differ so dramatically in their binding to VIPAS39/SPE-39 yet display similar endosome phenotypes. This interpretation of our data is consistent with observations in yeast sec1, where mutations in the 221–260-equivalent domain generate cellular phenotypes (40,41).
The major difference in the alignment between syntaxin 7 and syntaxin 1a (3C98 B chain) is a deletion of the N-terminal 30 residues, including a number of charged residues which are disordered in the crystal structure (48). Nonetheless, the N-terminus of the model still interacts with the same domain of Vps33b as seen in the interaction of syntaxin a1 with Munc18a (48,49). The details of the interaction are undoubtedly different as indicated by the absence of Y2H interaction between Vps33b and syntaxin 7 (1–238) but a positive interaction with Syntaxin 7 SNARE domain (165–227, Fig. 7). These Y2H results and our structural interpretation are in rapport with elegant biochemical studies in yeast Vps33b demonstrating that Vps33b binding is restricted to the SNARE domain, yet endosomal SNARE N-termini bind to Vps11, 16 and 18 (40). Intact SNARE binding to Vps33b mutants as well as putative additional binding surfaces in mammalian Vps11, 16 and 18 would explain the normal co-localization of all mutants to syntaxin 7-positive endosomes observed in our studies.
Down-regulation of VIPAS39/SPE-39 impairs delivery of cargoes between early and late endosomal compartments in human cells, proteins taken up by fluid-face endocytosis in nematode cells and bacteria in Drosophila hemocyte phagosomes (34,35). The precise nature of the VIPAS39/SPE-39 compartment in humans is still incompletely understood. However, the VIPAS39/SPE-39-positive endosome is characterized by its content of transferrin receptor, rab5, 11 and predominantly rab7b, suggesting it is an endosome maturation intermediary (21,34,43). Deconvolution microscopy revealed that Vps33b localizes with the VIPAS39/SPE-39 endosome in at least two discernible patterns referred to as overlapped and juxtaposed (patterns A and B, respectively, Fig. 10C). We interpret these patterns as possible stages in the maturation of the VIPAS39/SPE-39 endosome necessary for tethering incoming Vps33b-positive organelles. This hypothesis predicts that mutant forms of Vps33b should modify the relative proportions of these patterns. In fact, expression of Vps33b pathogenic mutations increases juxtaposed pattern of Vps33b with the VIPAS39/SPE-39 endosome (pattern B, Fig. 10D) with a concomitant decrease in the overlap between mutant Vps33b and VIPAS39/SPE-39 (pattern A, Fig. 10D). This model could explain a striking feature of the VIPAS39/SPE-39 endosome with its low copy number in HEK293 cells despite Vps33b being distributed in multiple organelles in these cells. Since Vps33b and VIPAS39/SPE-39 mutations phenocopy each other in humans, C. elegans and Drosophila, we deem it unlikely that VIPAS39/SPE-39 defines a subset of independently functioning Vps class C complexes containing Vps33b constitutively bound to VIPAS39/SPE-39. Rather, we favor a model where Vps33b is upstream of VIPAS39/SPE-39, with Vps33b organelles funneling into a VIPAS39/SPE-39 endosome. Thus, in a tethering reaction, VIPAS39/SPE-39 would mark an ‘acceptor’ organelle for an incoming Vps33b-only positive organelle. We propose that molecules, such as VIPAS39/SPE-39, form protein complexes that are conserved and define novel compartments and endosomal transport routes in metazoans.
MATERIALS AND METHODS
Y2H analyses
Y2H Gold strain, the DNA-binding domain and activation domain vectors, pGBKT7 and pGADT7, respectively, came from the Matchmaker Two-Hybrid System 3 (Clontech, Mountain View, CA, USA). Double dropout supplement -Leu/-Trp DO media plates were used to select double-transformed yeast with pGBKT7 and pGADT7 constructs. Protein interaction was tested using triple dropout supplement -Leu/-Trp/-His DO plates plus 2 mm 3-aminotriazole.
Yeast were transformed using 0.1 m lithium acetate (LiAc). Briefly, we inoculated a single colony into 2 ml of YPD for 24 h at 30°C. About 0.5–1 ml of this overnight culture was inoculated into 50 ml of YPD, cultured at 30°C until (optical density) O.D595 = 0.6. Next, the cells were pelleted by centrifugation at 750 g for 2 min. The pellet was resuspended in 5 ml of 0.1 m LiAc and incubated at room temperature for 30 min. Cells were pelleted by centrifugation at 750 g for 5 min and resuspended in 1 ml of LiAc. Next, we mixed 10 μl of carrier DNA (salmon testis, 10 mg/ml), 10 μl of sample DNA, 100 μl of competent cells and 230 μl of 50% polyethylene glycol 3350. This mixture was vortexed, incubated for 1 h at room temperature, spread on selection plate and incubated at 30°C for at least 72 h.
Plasmids and recombinant DNA
For all plasmids, the cDNA of interest was amplified by PCR and cloned into the TOPO vector pcDNA 3.1 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's directions. The insert was subsequently cut out, and the resulting fragment was subcloned into pGBKT7 or pGADT7 and confirmed to be error-free by DNA sequencing.
Vps and VIPAS/SPE-39 Y2H plasmids
Vps11, 16, 18, 33b, 39, 41 and VIPAS39/SPE-39 were inserted into pGBKT7 and pGADT7 from plasmids encoding human Vps33b and VIPAS39/SPE-39, C-terminal murine vps11-HA, vps16-HA, vps18-myc, N-terminal GFP-murine vps18, GFP-vps39 and C-terminal human vps41-myc. Vps11, 16 and vps39 plasmids were kind gifts from Dr Robert Piper, and vps18-myc was provided by Dr Liping Wang.
Vps11 was amplified by forward primer 5′cacccatatgatggcggcctacctgcag3′ and reverse primer 5′gtcgacctaagttcccctcctgga3′. ClaI and XhoI restriction enzymes were used to cut Topo vector. Primers forward 5′cacccatatgatggactgttac3′ and reverse 5′gaattctcacttcttctgggc3′ were used to amplify vps16, and these primers, respectively, introduced NdeI and EcoRI sites. Vps18 was amplified by the forward primer 5′cacccatatgatggcgtccatc3′ and the reverse primer 5′ccgggctatagccagctga3′, and these primers, respectively, introduced novel NdeI or XmaI sites. The forward primer 5′caccgaattcatggcttttccc3′ and the reverse primer 5′ggatcctcaggctttcacctcac3′ were used to amplify vps33b, and these primers, respectively, introduced novel EcoRI and BamHI restriction sites. Vps39 was amplified by the forward primer 5′cacccatatgatgcatgacgct3′ and the reverse primer 5′ggatccctaggtgtcggctga3′, and these primers, respectively, introduced novel NdeI and BamHI sites. Vps41 was amplified by the forward primer 5′cacccatatgatggcggaagca3′ and the reverse primer 5′ggatccctattttttcatctccaaaattgcacttcc3′, and these primers, respectively, introduced novel NdeI and BamHI sites. The forward primer 5′cacccatatgatgaatcggac3′ and the reverse primer 5′ggatccttaattcttccatcg3′ were used to amplify VIPAS/SPE-39, and these primers, respectively, introduced novel NdeI and BamHI sites.
Vps33b and syntaxin Y2H plasmids
Human cDNAs of Syntaxin 7 (MHS1010-74354), 12 (MHS1010-73802) and 16 (MHS1010-97227865, Open Biosystems, Lafayette, CO, USA) were amplified by PCR. Primers were designed to amplify syntaxins without their transmembrane domain and N-terminal end. EcoRI–BamHI restriction enzymes were used to cut Topo vector and subclone syntaxins into the pGBKT7 Y2H plasmid.
Syntaxin 7 (amino acids 1–238), 12 (amino acids 1–248) and 16 (amino acids 1–301) were amplified with the forward primer 5′caccgaattcatgtcttacact3′ and reverse primer 5′ggatcctcacagggtttttct3′, forward primer 5′caccgaattcatgtcatacg3′ and reverse primer 5′ggatcctcagcgagatttttt3′, and forward primer 5′caccgaattcatggccaccagg3′ and reverse primer 5′ggatcctcacttccgattcttctt3′, respectively.
The SNARE domain of Syntaxin 7 (amino acids 165–227) was amplified using the forward primer 5′caccgaattcatgctccgtctt3′ and the reverse primer 5′ggatcctcatgcccttgacag3′.
ARC mutations
Vps33b and VIPAS39/SPE-39 nonsense and missense mutations identified in ARC patients were created from the wild-type sequence by site-directed mutagenesis using the following primers:
Vps33-Leu30Pro: forward 5′agctcatctatctgccggagcagcttcctgg3′ and reverse 5′ccaggaagctgctccggcagatagatgagct3′; Vps33-Asp234His: forward 5′tttctcttggacagacatgtggactttgtga3′ and reverse 5′tcacaaagtccacatgtctgtccaagagaaa3′; Vps33-Ser243Phe: forward 5′tgacagcactttgcttccaagtggtttatga3′ and reverse 5′tcataaaccacttggaagcaaagtgctgtca3′; Vps33-Gly249Val (Drosophila Car mutation): forward 5′aagtggtttatgaggtcctagtagatgacac3′ and reverse 5′gtgtcatctactaggacctcataaaccactt3′; Vps33-Asp252Glu (mouse Buff mutation): forward 5′tgagggcctagtagaggacaccttccgcatc3′ and reverse 5′gatgcggaaggtgtcctctactaggccctca3′; Vps33b-Arg438X: forward 5′ttctccaatctgtgaagagctgggct3′ and reverse 5′agcccagctcttcacagattggagaa3′; Vps33b-Arg507X: forward 5′gatctgaaagtgccctgagacatggcttacg3′ and reverse 5′cgtaagccatgtctcagggcactttcagatc3′; Spe39-Arg220X: forward 5′agagctggaggtgtgacaggttgccct3′ and reverse 5′agggcaacctgtcacacctccagctct3′; spe39-Arg425X: forward 5′ccctgtgcagatattataggagtatgtcaatct3′ and reverse 5′agattgacatactcctataatatctgcacaggg3′.
Vps33b triple-alanine mutations were designed by site mutagenesis using the following primers:
D232-234A: forward 5′tatctttctcttggccgcagctgtggactttgtgaca3′ and reverse 5′tgtcacaaagtccacagctgcggccaagagaaagat3′; V235-237A: forward 5′tttctcttggacagagatgcagcagcagtgacagcactttgctcc3′ and reverse 5′ggagcaaagtgctgtcactgctgctgcatctctgtccaagagaaa3′; S243-245A: forward 5′agcactttgcgcagcagcagtttatgagg3′ and reverse 5′cctcataaactgctgctgcgcaaagtgct3′; V251-253A: forward 5′tggtttatgagggcctagcagctgccaccttccgcatcaagtgtg3′ and reverse: 5′cacacttgatgcggaaggtggcagctgctaggccctcataaacca3′.
Antibodies
The following primary antibodies were used in this study: polyclonal anti-myc (A190105A) and anti-HA (A190108A, Bethyl Laboratories, Montgomery, TX, USA), monoclonal anti-transferrin receptor (13-6800, Zymed Laboratories), monoclonal anti-SPE-39 (34), monoclonal anti syntaxin 7 (A gift of Dr Andrew A. Peden) and monoclonal anti-HA (MMS-101R, Covance). Monoclonal antibodies against EEA1 (610457) and GM130 (558712) were from BD Biosciences, San Jose, CA, USA. Secondary antibodies used for western blot were HRP-goat anti-mouse or anti-rabbit (626420 and G21234, Invitrogen). Secondary antibodies for immunofluorescence were Alexa Fluor 488 anti-rabbit, Alexa Fluor 488 anti-mouse, and Alexa Fluor 555 anti-rabbit (A11034, A11029 and A21429, Molecular Probes, Eugene, OR, USA).
Crosslinking and immunoprecipitation
Crosslinking and immunoprecipitation experiments were performed as described previously (37,45,46). Briefly, confluent HEK293T cells were washed twice with ice-cold PBS/1 mm MgCl2/0.1 mm CaCl2 followed by incubation with 1 mm DSP (Cat# 22585, Thermo Scientific, Rockford, IL, USA) or DMSO control in PBS/1 mm MgCl2/0.1 mm CaCl2 for 2 h on ice. Cells were incubated for 15 min with 25 mm Tris, pH 7.4, to quench DSP, then rinsed twice with ice-cold PBS/1 mm MgCl2/0.1 mm CaCl2. Cells were incubated for 30 min at 4°C with lysis buffer, Buffer A (150 mm NaCl, 10 mm HEPES, 1 mm EGTA, 0.1 mm MgCl2) + 0.5% Triton X-100. The lysate was centrifuged at 16 000g for 15 min, the supernatant diluted in Buffer A + 0.5% Triton X-100 to 1 µg/µl, then incubated with Dynal immuno-magnetic-precipitation beads (Dynal, Oslo, Norway). Beads and lysate were incubated at 4°C for 2 h, washed six times with Buffer A + 0.1% Triton X-100, resuspended with 30 µl with 4× SDS–PAGE sample buffer and incubated at 75°C for 5 min. Immunoprecipitated material was analyzed on an SDS–PAGE western blot.
Cell culture and transfection
HEK293T (ATCC, Manassas, VA, USA) cells were incubated at 37°C with 10% CO2 in DMEM (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Hyclone) and 100 μg/ml penicillin and streptomycin (Hyclone). HEK cells were transfected for 4 h in six-well plates with 1–2 µg of recombinant DNA using 0.25% Lipofectamine 2000 (Invitrogen) diluted in Opti-Mem (Gibco, Grand Island, NY, USA) and then incubated in culture media.
Immunofluorescent imaging
Cells plated on Matrigel (BD Bioscience) or poly-l-lysine-coated glass coverslips were washed two times with PBS/1 mm MgCl2/0.1 mm CaCl2 and then fixed with 4% paraformaldehyde in PBS for 20 min on ice. Cells were washed two times with PBS, then permeabilized and blocked with 0.02% saponin (Sigma, St Louis, MO, USA), 15% horse serum (Hyclone), 2% BSA and 1% fish skin gelatin (Sigma) in PBS for 30 min at room temperature. Cells were incubated with the primary antibodies described above for 30 min in 37°C bath, rinsed three times with block solution, incubated for 30 min at 37°C with the secondary antibodies and rinsed two times with block solution. The cells were then mounted on slides with gelvatol.
Fixed HEK cells expressing wild-type vps33b-HA or vps33b-HA carrying ARC mutations were imaged using deconvolution microscopy as described by Zlatic et al. (47). Slides were imaged on a 200M inverted microscope using 63×/1.4 and 100×/1.4 oil DIC objectives (Carl Zeiss) and a Sedat filter set. Images were collected using a scientific grade, cooled charge-coupled Cool-Snap HQ camera with ORCA-ER chip on a multiwavelength, wide-field, three-dimensional microscopy system using the Slidebook 4.0 OS X software (Intelligent Imaging Innovations, Denver, CO, USA). Out-of-focus light was removed with a constrained iterative deconvolution algorithm as described in Swedlow et al. (62). Metamorph software, version 6.1 (Universal Imaging, Sunnyvale, CA, USA), NIS-Elements AR 3.0, Imaris 6.3.1 (Bitplane, St Paul, MN, USA) and Image J 1.41 (NIH) software packages were used for image analysis.
Molecular modeling
Molecular models were derived using the automated model server Panther (http://bmcc3.cs.gsu.edu). The model for Syntaxin-7 was derived from the neuronal-sec1, syntaxin 1a complex (49). Profile–profile based alignment found 14 distinct homologous protein structures in the pdb, with the b-chain of entry 3C98 having the highest identity (18.8% identity) and structural similarity for a complete sequence homology. Distance restraints were derived via Delaunay triangulation (63) of the 14 distinct homologs and used to restrain the model to structural consensus positions. The model for Vps33b was determined in a similar manner; however, the sequence identity is lower (11.8%) and only eight homologous proteins were found. Distance restraints were derived from these eight molecules and used to restrain the model to its structural consensus. The model from 3C98 A-chain was chosen as it was already in a complex with the syntaxin molecule and thus eliminated one docking step in the modeling. The model for VIPAS39/SPE-39 was more difficult to derive. Profile–profile scans identified numerous fragments of overlap between the structure of human mitochondrial RNA-polymerase (3SPA) (64). However, the overall identity is low (5.5%); therefore, some aspects of this part of the model complex are tentative. The homology is best for the N-terminal 200 residues. The model for VIPAS39/SPE-39 was docked to the model for vps33b using the algorithm docking function in AMMP (65) using the non-bonded and electrostatic potential energy as a minimization target. The total charge of the individual protein molecules was renormalized to zero to account for missing counterions by subtracting the average charge from each atom as both proteins had a non-zero net negative charge would not be able to dock due to electrostatic repulsion. Distance restraints to minimize the distance between the regions 1–200 of VIPAS39/SPE-39 and vps33b (residues 234, 243, 249 and 252) were included.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by grants from the National Institutes of Health to V.F. (NS42599, GM077569) and S.W.L. (GM082932). S.A.Z. was supported by T32, the Graduate and Post-doctoral Training in Toxicology training grant (NIEHS, 1P50NS071669).
ACKNOWLEDGEMENTS
We are indebted to the Faundez laboratory members for their comments and to Drs Guy Benian (Mozart) and Hiroshi Qadota for their invaluable help with Y2H.
REFERENCES
- 1.van Meel E., Klumperman J. Imaging and imagination: understanding the endo-lysosomal system. Histochem. Cell Biol. 2008;129:253–266. doi: 10.1007/s00418-008-0384-0. doi:10.1007/s00418-008-0384-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brocker C., Engelbrecht-Vandre S., Ungermann C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. 2010;20:R943–R952. doi: 10.1016/j.cub.2010.09.015. doi:10.1016/j.cub.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 3.Poteryaev D., Datta S., Ackema K., Zerial M., Spang A. Identification of the switch in early-to-late endosome transition. Cell. 2010;141:497–508. doi: 10.1016/j.cell.2010.03.011. doi:10.1016/j.cell.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Rink J., Ghigo E., Kalaidzidis Y., Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. doi:10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 5.Huotari J., Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. doi:10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dell'angelica E.C. AP-3-dependent trafficking and disease: the first decade. Curr. Opin. Cell Biol. 2009;21:552–559. doi: 10.1016/j.ceb.2009.04.014. doi:10.1016/j.ceb.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Wei M.L. Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. doi:10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 8.Di Pietro S.M., Dell'Angelica E.C. The cell biology of Hermansky-Pudlak syndrome: recent advances. Traffic. 2005;6:525–533. doi: 10.1111/j.1600-0854.2005.00299.x. doi:10.1111/j.1600-0854.2005.00299.x. [DOI] [PubMed] [Google Scholar]
- 9.Li W., Rusiniak M.E., Chintala S., Gautam R., Novak E.K., Swank R.T. Murine Hermansky-Pudlak syndrome genes: regulators of lysosome-related organelles. Bioessays. 2004;26:616–628. doi: 10.1002/bies.20042. doi:10.1002/bies.20042. [DOI] [PubMed] [Google Scholar]
- 10.Huizing M., Helip-Wooley A., Westbroek W., Gunay-Aygun M., Gahl W.A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu. Rev. Gen. Hum. Genet. 2008;9:359–386. doi: 10.1146/annurev.genom.9.081307.164303. doi:10.1146/annurev.genom.9.081307.164303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Gele M., Dynoodt P., Lambert J. Griscelli syndrome: a model system to study vesicular trafficking. Pigment Cell Melanoma Res. 2009;22:268–282. doi: 10.1111/j.1755-148X.2009.00558.x. doi:10.1111/j.1755-148X.2009.00558.x. [DOI] [PubMed] [Google Scholar]
- 12.Filimonenko M., Stuffers S., Raiborg C., Yamamoto A., Malerod L., Fisher E.M., et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. doi:10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee J.A., Beigneux A., Ahmad S.T., Young S.G., Gao F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007;17:1561–1567. doi: 10.1016/j.cub.2007.07.029. doi:10.1016/j.cub.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 14.Urwin H., Authier A., Nielsen J.E., Metcalf D., Powell C., Froud K., et al. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum. Mol. Genet. 2010;19:2228–2238. doi: 10.1093/hmg/ddq100. doi:10.1093/hmg/ddq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collinet C., Stoter M., Bradshaw C.R., Samusik N., Rink J.C., Kenski D., et al. Systems survey of endocytosis by multiparametric image analysis. Nature. 2010;464:243–249. doi: 10.1038/nature08779. doi:10.1038/nature08779. [DOI] [PubMed] [Google Scholar]
- 16.Meggouh F., Bienfait H.M., Weterman M.A., de Visser M., Baas F. Charcot-Marie-Tooth disease due to a de novo mutation of the RAB7 gene. Neurology. 2006;67:1476–1478. doi: 10.1212/01.wnl.0000240068.21499.f5. doi:10.1212/01.wnl.0000240068.21499.f5. [DOI] [PubMed] [Google Scholar]
- 17.Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J.M., et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. doi:10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cogli L., Piro F., Bucci C. Rab7 and the CMT2B disease. Biochem. Soc. Trans. 2009;37:1027–1031. doi: 10.1042/BST0371027. doi:10.1042/BST0371027. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki T., Oiso N., Gautam R., Novak E.K., Panthier J.J., Suprabha P.G., et al. The mouse organellar biogenesis mutant buff results from a mutation in Vps33a, a homologue of yeast vps33 and Drosophila carnation. Proc. Natl Acad. Sci. USA. 2003;100:1146–1150. doi: 10.1073/pnas.0237292100. doi:10.1073/pnas.0237292100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sevrioukov E.A., He J.P., Moghrabi N., Sunio A., Kramer H. A role for the deep orange and carnation eye color genes in lysosomal delivery in Drosophila. Mol. Cell. 1999;4:479–486. doi: 10.1016/s1097-2765(00)80199-9. doi:10.1016/S1097-2765(00)80199-9. [DOI] [PubMed] [Google Scholar]
- 21.Cullinane A.R., Straatman-Iwanowska A., Zaucker A., Wakabayashi Y., Bruce C.K., Luo G., et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat. Genet. 2010;42:303–312. doi: 10.1038/ng.538. doi:10.1038/ng.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gissen P., Johnson C.A., Morgan N.V., Stapelbroek J.M., Forshew T., Cooper W.N., et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat. Genet. 2004;36:400–404. doi: 10.1038/ng1325. doi:10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 23.Balderhaar H.J., Ungermann C. CORVET and HOPS tethering complexes – coordinators of endosome and lysosome fusion. J. Cell Sci. 2013;126:1307–1316. doi: 10.1242/jcs.107805. doi:10.1242/jcs.107805. [DOI] [PubMed] [Google Scholar]
- 24.Peplowska K., Markgraf D.F., Ostrowicz C.W., Bange G., Ungermann C. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev. Cell. 2007;12:739–750. doi: 10.1016/j.devcel.2007.03.006. doi:10.1016/j.devcel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 25.Wickner W. Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu. Rev. Cell Dev. Biol. 2010;26:115–136. doi: 10.1146/annurev-cellbio-100109-104131. doi:10.1146/annurev-cellbio-100109-104131. [DOI] [PubMed] [Google Scholar]
- 26.Nickerson D.P., Brett C.L., Merz A.J. Vps-C complexes: gatekeepers of endolysosomal traffic. Curr. Opin. Cell Biol. 2009;21:543–551. doi: 10.1016/j.ceb.2009.05.007. doi:10.1016/j.ceb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brocker C., Kuhlee A., Gatsogiannis C., Balderhaar H.J., Honscher C., Engelbrecht-Vandre S., et al. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc. Natl Acad. Sci. USA. 2012;109:1991–1996. doi: 10.1073/pnas.1117797109. doi:10.1073/pnas.1117797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ostrowicz C.W., Brocker C., Ahnert F., Nordmann M., Lachmann J., Peplowska K., et al. Defined subunit arrangement and rab interactions are required for functionality of the HOPS tethering complex. Traffic. 2010;11:1334–1346. doi: 10.1111/j.1600-0854.2010.01097.x. doi:10.1111/j.1600-0854.2010.01097.x. [DOI] [PubMed] [Google Scholar]
- 29.Gissen P., Johnson C.A., Gentle D., Hurst L.D., Doherty A.J., O'Kane C.J., et al. Comparative evolutionary analysis of VPS33 homologues: genetic and functional insights. Hum. Mol. Genet. 2005;14:1261–1270. doi: 10.1093/hmg/ddi137. doi:10.1093/hmg/ddi137. [DOI] [PubMed] [Google Scholar]
- 30.Akbar M.A., Ray S., Kramer H. The SM protein Car/Vps33A regulates SNARE-mediated trafficking to lysosomes and lysosome-related organelles. Mol. Biol. Cell. 2009;20:1705–1714. doi: 10.1091/mbc.E08-03-0282. doi:10.1091/mbc.E08-03-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.L'Hernault S.W., Faundez V. On the endosomal function and gene nomenclature of human SPE-39. Nat. Genet. 2011;43:176. doi: 10.1038/ng0311-176. doi:10.1038/ng0311-176. [DOI] [PubMed] [Google Scholar]
- 32.Pulipparacharuvil S., Akbar M.A., Ray S., Sevrioukov E.A., Haberman A.S., Rohrer J., et al. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci. 2005;118:3663–3673. doi: 10.1242/jcs.02502. doi:10.1242/jcs.02502. [DOI] [PubMed] [Google Scholar]
- 33.Zhu G.D., L'Hernault S.W. The Caenorhabditis elegans spe-39 gene is required for intracellular membrane reorganization during spermatogenesis. Genetics. 2003;165:145–157. doi: 10.1093/genetics/165.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu G.D., Salazar G., Zlatic S.A., Fiza B., Doucette M.M., Heilman C.J., et al. SPE-39 family proteins interact with the HOPS complex and function in lysosomal delivery. Mol. Biol. Cell. 2009;20:1223–1240. doi: 10.1091/mbc.E08-07-0728. doi:10.1091/mbc.E08-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akbar M.A., Tracy C., Kahr W.H., Kramer H. The full-of-bacteria gene is required for phagosome maturation during immune defense in Drosophila. J. Cell Biol. 2011;192:383–390. doi: 10.1083/jcb.201008119. doi:10.1083/jcb.201008119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gissen P., Tee L., Johnson C.A., Genin E., Caliebe A., Chitayat D., et al. Clinical and molecular genetic features of ARC syndrome. Hum. Genet. 2006;120:396–409. doi: 10.1007/s00439-006-0232-z. doi:10.1007/s00439-006-0232-z. [DOI] [PubMed] [Google Scholar]
- 37.Gokhale A., Larimore J., Werner E., So L., Moreno-De-Luca A., Lese-Martin C., et al. Quantitative proteomic and genetic analyses of the schizophrenia susceptibility factor dysbindin identify novel roles of the biogenesis of lysosome-related organelles complex 1. J. Neurosci. 2012;32:3697–3711. doi: 10.1523/JNEUROSCI.5640-11.2012. doi:10.1523/JNEUROSCI.5640-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plemel R.L., Lobingier B.T., Brett C.L., Angers C.G., Nickerson D.P., Paulsel A., et al. Subunit organization and Rab interactions of Vps-C protein complexes that control endolysosomal membrane traffic. Mol. Biol. Cell. 2011;22:1353–1363. doi: 10.1091/mbc.E10-03-0260. doi:10.1091/mbc.E10-03-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hershkovitz D., Mandel H., Ishida-Yamamoto A., Chefetz I., Hino B., Luder A., et al. Defective lamellar granule secretion in arthrogryposis, renal dysfunction, and cholestasis syndrome caused by a mutation in VPS33B. Arch. Dermatol. 2008;144:334–340. doi: 10.1001/archderm.144.3.334. doi:10.1001/archderm.144.3.334. [DOI] [PubMed] [Google Scholar]
- 40.Lobingier B.T., Merz A.J. Sec1/Munc18 protein Vps33 binds to SNARE domains and the quaternary SNARE complex. Mol. Biol. Cell. 2012;23:4611–4622. doi: 10.1091/mbc.E12-05-0343. doi:10.1091/mbc.E12-05-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hashizume K., Cheng Y.S., Hutton J.L., Chiu C.H., Carr C.M. Yeast Sec1p functions before and after vesicle docking. Mol. Biol. Cell. 2009;20:4673–4685. doi: 10.1091/mbc.E09-02-0172. doi:10.1091/mbc.E09-02-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jang J.Y., Kim K.M., Kim G.H., Yu E., Lee J.J., Park Y.S., et al. Clinical characteristics and VPS33B mutations in patients with ARC syndrome. J. Pediatr. Gastroenterol. Nutr. 2009;48:348–354. doi: 10.1097/mpg.0b013e31817fcb3f. doi:10.1097/MPG.0b013e31817fcb3f. [DOI] [PubMed] [Google Scholar]
- 43.Smith H., Galmes R., Gogolina E., Straatman-Iwanowska A., Reay K., Banushi B., et al. Associations among genotype, clinical phenotype and intracellular localization of trafficking proteins in ARC syndrome. Hum. Mut. 2012;33:1656–1664. doi: 10.1002/humu.22155. doi:10.1002/humu.22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Craige B., Salazar G., Faundez V. Phosphatidylinositol-4-kinase type II alpha contains an AP-3 sorting motif and a kinase domain that are both required for endosome traffic. Mol. Biol. Cell. 2008;19:1415–1426. doi: 10.1091/mbc.E07-12-1239. doi:10.1091/mbc.E07-12-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salazar G., Zlatic S., Craige B., Peden A.A., Pohl J., Faundez V. Hermansky-Pudlak syndrome protein complexes associate with phosphatidylinositol 4-kinase type II alpha in neuronal and non-neuronal cells. J. Biol. Chem. 2009;284:1790–1802. doi: 10.1074/jbc.M805991200. doi:10.1074/jbc.M805991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zlatic S.A., Ryder P.V., Salazar G., Faundez V. Isolation of labile multi-protein complexes by in vivo controlled cellular cross-linking and immuno-magnetic affinity chromatography. J. Vis. Exp. 2010 doi: 10.3791/1855. 1855 pii 10.3791/1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zlatic S.A., Tornieri K., L'Hernault S.W., Faundez V. Clathrin-dependent mechanisms modulate the subcellular distribution of class C Vps/HOPS tether subunits in polarized and nonpolarized cells. Mol. Biol. Cell. 2011;22:1699–1715. doi: 10.1091/mbc.E10-10-0799. doi:10.1091/mbc.E10-10-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burkhardt P., Hattendorf D.A., Weis W.I., Fasshauer D. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J. 2008;27:923–933. doi: 10.1038/emboj.2008.37. doi:10.1038/emboj.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Misura K.M., Scheller R.H., Weis W.I. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. doi:10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 50.Sudhof T.C., Rothman J.E. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. doi:10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rathore S.S., Bend E.G., Yu H., Hammarlund M., Jorgensen E.M., Shen J. Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc. Natl Acad. Sci. USA. 2010;107:22399–22406. doi: 10.1073/pnas.1012997108. doi:10.1073/pnas.1012997108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carr C.M., Rizo J. At the junction of SNARE and SM protein function. Curr. Opin. Cell Biol. 2010;22:488–495. doi: 10.1016/j.ceb.2010.04.006. doi:10.1016/j.ceb.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber S.H., Rah J.C., Min S.W., Liu X., de Wit H., Dulubova I., et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. 2008;321:1507–1510. doi: 10.1126/science.1163174. doi:10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim B.Y., Kramer H., Yamamoto A., Kominami E., Kohsaka S., Akazawa C. Molecular characterization of mammalian homologues of class C Vps proteins that interact with syntaxin-7. J. Biol. Chem. 2001;276:29393–29402. doi: 10.1074/jbc.M101778200. doi:10.1074/jbc.M101778200. [DOI] [PubMed] [Google Scholar]
- 55.Pols M.S., van Meel E., Oorschot V., ten Brink C., Fukuda M., Swetha M.G., et al. Hvps41 and VAMP7 function in direct TGN to late endosome transport of lysosomal membrane proteins. Nat. Commun. 2013;4:1361. doi: 10.1038/ncomms2360. doi:10.1038/ncomms2360. [DOI] [PubMed] [Google Scholar]
- 56.Richardson S.C., Winistorfer S.C., Poupon V., Luzio J.P., Piper R.C. Mammalian late vacuole protein sorting orthologues participate in early endosomal fusion and interact with the cytoskeleton. Mol. Biol. Cell. 2004;15:1197–1210. doi: 10.1091/mbc.E03-06-0358. doi:10.1091/mbc.E03-06-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pols M.S., ten Brink C., Gosavi P., Oorschot V., Klumperman J. The HOPS proteins hVps41 and hVps39 are required for homotypic and heterotypic late endosome fusion. Traffic. 2013;14:219–232. doi: 10.1111/tra.12027. doi:10.1111/tra.12027. [DOI] [PubMed] [Google Scholar]
- 58.Swetha M.G., Sriram V., Krishnan K.S., Oorschot V.M., ten Brink C., Klumperman J., et al. Lysosomal membrane protein composition, acidic pH and sterol content are regulated via a light-dependent pathway in metazoan cells. Traffic. 2011;12:1037–1055. doi: 10.1111/j.1600-0854.2011.01214.x. doi:10.1111/j.1600-0854.2011.01214.x. [DOI] [PubMed] [Google Scholar]
- 59.Sriram V., Krishnan K.S., Mayor S. Deep-orange and carnation define distinct stages in late endosomal biogenesis in Drosophila melanogaster. J. Cell Biol. 2003;161:593–607. doi: 10.1083/jcb.200210166. doi:10.1083/jcb.200210166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adler J., Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A. 2010;77:733–742. doi: 10.1002/cyto.a.20896. doi:10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 61.French A.P., Mills S., Swarup R., Bennett M.J., Pridmore T.P. Colocalization of fluorescent markers in confocal microscope images of plant cells. Nat. Protoc. 2008;3:619–628. doi: 10.1038/nprot.2008.31. doi:10.1038/nprot.2008.31. [DOI] [PubMed] [Google Scholar]
- 62.Swedlow J.R., Sedat J.W., Agard D.A. Deconvolution in optical microscopy. In: Jansson P.A., editor. Deconvolution of Images and Spectra. Deconvolution of Images and Spectra. San Diego, CA: Academic Press; 1997. pp. 284–307. [Google Scholar]
- 63.Bose P., Xiaxia Y., Harrison R.W. Atlanta, GA, USA: 2011. pp. 338–344. Encoding protein structure with functions on graphs IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW) [Google Scholar]
- 64.Ringel R., Sologub M., Morozov Y.I., Litonin D., Cramer P., Temiakov D. Structure of human mitochondrial RNA polymerase. Nature. 2011;478:269–273. doi: 10.1038/nature10435. doi:10.1038/nature10435. [DOI] [PubMed] [Google Scholar]
- 65.Harrison R.W. In: Peter P., Dey M.N.A., Gatton T.M., editors. Montreal, Canada: 2003. pp. 77–81. Amortized fast multipole algorithm for molecular modeling Proceedings of the International Conference on Computer Science and Its Applications ICCSA. [Google Scholar]