

Abstract
Mutant matrilin-3 (V194D) forms non-native disulphide bonded aggregates in the rER of chondrocytes from cell and mouse models of multiple epiphyseal dysplasia (MED). Intracellular retention of mutant matrilin-3 causes endoplasmic reticulum (ER) stress and induces an unfolded protein response (UPR) including the upregulation of two genes recently implicated in ER stress: Armet and Creld2. Nothing is known about the role of Armet and Creld2 in human genetic diseases. In this study, we used a variety of cell and mouse models of chondrodysplasia to determine the genotype-specific expression profiles of Armet and Creld2. We also studied their interactions with various mutant proteins and investigated their potential roles as protein disulphide isomerases (PDIs). Armet and Creld2 were up-regulated in cell and/or mouse models of chondrodysplasias caused by mutations in Matn3 and Col10a1, but not Comp. Intriguingly, both Armet and Creld2 were also secreted into the ECM of these disease models following ER stress. Armet and Creld2 interacted with mutant matrilin-3, but not with COMP, thereby validating the genotype-specific expression. Substrate-trapping experiments confirmed Creld2 processed PDI-like activity, thus identifying a putative functional role. Finally, alanine substitution of the two terminal cysteine residues from the A-domain of V194D matrilin-3 prevented aggregation, promoted mutant protein secretion and reduced the levels of Armet and Creld2 in a cell culture model. We demonstrate that Armet and Creld2 are genotype-specific ER stress response proteins with substrate specificities, and that aggregation of mutant matrilin-3 is a key disease trigger in MED that could be exploited as a potential therapeutic target.
INTRODUCTION
The chondrodysplasias are a clinically and genetically heterogeneous group of skeletal diseases (chondrodysplasias) that encompass over 300 different phenotypes (1). Although the clinical presentation varies from mild to lethal they are often characterized by abnormal endochondral ossification that results in disproportionate short stature. Mutations within the genes encoding a variety of cartilage extracellular matrix (ECM) structural proteins can result in numerous chondrodysplasias. These include cartilage oligomeric matrix protein (COMP), matrilin-3 (MATN3) and type IX collagen, which result in pseudoachondroplasia (PSACH: OMIN 177170) and multiple epiphyseal dysplasia (MED: OMIN 132400, 600204, 607078 and 614135) (2); and type X collagen that causes metaphyseal chondrodysplasia type Schmid (MCDS: OMIN 156500) (3).
The majority of disease-associated mutations in these genes cause misfolding of the respective proteins and their subsequent retention within the endoplasmic reticulum (ER) (4,5). Mutant protein retention results in ER stress and can lead to an UPR, whereby ER-resident proteins that are involved in protein folding are up-regulated to aid in folding and protecting the cells from stress (6,7). Other consequences of the UPR include protein degradation via ER-associated degradation (ERAD), attenuation of protein synthesis and eventually apoptosis if the misfolded protein and ER stress persist (6,8,9). Mutant protein accumulation in the ER and its subsequent degradation have been recognized as a key pathological feature in a range of different diseases, including several neurological disorders and diabetes. More recently, ER stress has been demonstrated in several mouse models of human chondrodysplasias, including PSACH-MED (10–13) and MCDS (14–16).
Matrilin-3 is the third member of the matrilin family of modular proteins and consists of a single von-Willebrand factor A-like domain (A-domain), four EGF-like motifs and a C-terminal coiled-coil oligomerization domain (17). The A-domain is arranged into a classical Rossman fold and contains a single intra-chain disulphide bond. To date, all MED causing mutations in matrilin-3 are located within the single A-domain and primarily affect residues that comprise the central β-sheet (2,18–21). A murine model of MED with the Matn3 V194D mutation develops a progressive short-limb dysplasia resulting from decreased chondrocyte proliferation and dysregulated apoptosis. Mutant matrilin-3 is retained within the rER of chondrocytes, which leads to an UPR characterized by the upregulation of the ER-resident chaperones BiP and GRP94 (10). Microarray studies further confirmed the upregulation of numerous genes associated with ER stress and a conventional transcriptional UPR (12), including several members of the protein disulphide isomerase family A (PDIAs), specifically PDIA3, -4 and -6. The PDIAs are members of the thioredoxin superfamily of enzymes that can catalyse thiol-disulphide oxidation, reduction and isomerization and are critical for correct disulphide bond formation and/or rearrangement if incorrect (non-native) bonds are formed (22,23). There are at least 19 members of the PDIA family, and these multi-domain proteins contain at least one domain that is homologous to thioredoxin. Many of these thioredoxin domains contain a pair of active site cysteine residues (CXXC) that can shuttle between the disulphide and dithiol forms (22,24).
Interestingly, among the most highly up-regulated genes in Matn3 V194D chondrocytes were Armet (arginine-rich, mutated in early stage tumours) and Creld2 (cysteine-rich with EGF-like domains 2); two genes that have only recently been implicated in ER stress and UPR following a variety of physiological and pathological triggers (25–27).
Armet, also known as MANF (Mesencephalic Astrocyte derived Neurotrophic Factor), was identified as a gene up-regulated by various forms of ER stress in different cell lines and by cerebral ischaemia in rat (26–28). The expression of Armet was not only similar to that of the molecular chaperone BiP/GRP78, but the upregulation of Armet was also shown to be mediated by an ERSE-II element, which is frequently found in the promoters of ER chaperone genes (27). Furthermore, the over-expression of Armet in HeLa cells inhibited cell proliferation and ER stress-induced cell death (26). Interestingly, two important disease mechanisms in the Matn3 V194D mouse include a reduction in chondrocyte proliferation and dysregulated apoptosis (10,12), suggesting an influence of Armet in the initiation and/or progression of MED.
Creld2 was recognized as an ER stress-inducible gene through microarray analysis of Neuro2a cells treated with thapsigargin. Furthermore, it has also been shown to contain a typical ER stress response element (ERSE) in its promoter region, which is positively regulated by ATF6 (25). Creld2 has been reported to mediate the intracellular trafficking of nicotinic acetylcholine receptor (nAChR) α4 and β2 subunits and is believed to be involved in the folding and assembly of α4β2 nAChRs within the ER, perhaps indicating a more general role for Creld2 in protein folding and trafficking through the ER (29).
Despite these recent descriptions, the precise roles of Creld2 and Armet remain poorly understood in human biology and genetic diseases. This current study aimed at determining the genotype-specific expression profiles of Armet and Creld2, while also investigating their interactions with mutant proteins.
RESULTS
Armet and Creld2 are up-regulated in cell and murine models of MED caused by matrilin-3 V194D
To establish the relative levels of Armet and Creld2 proteins in Matn3 V194D mutant cartilage, sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting were performed on the intracellular proteins of chondrocytes isolated from the cartilage of new born and 5-day-old mice. Western blotting confirmed that the protein levels of both Armet and Creld2 were increased in the chondrocytes of Matn3 V194D mice compared with wild-type controls at birth and 5 days of age (Fig. 1A, 5 days and Supplementary Material, Fig. S1, new born). Densitometry established that Armet was significantly up-regulated ∼2-fold at birth (not shown) and ∼4-fold in 5-day-old mice (Fig. 1B; **P < 0.01). Similarly, Creld2 was up-regulated ∼2-fold in newborn (not shown) and ∼3-fold in 5-day-old mutant mice (Fig. 1B; **P < 0.01). These data verified a time-dependent increase in the protein levels of Armet and Creld2, which corresponded to the gradual accumulation of mutant matrilin-3 as previously reported (10,12).
Figure 1.
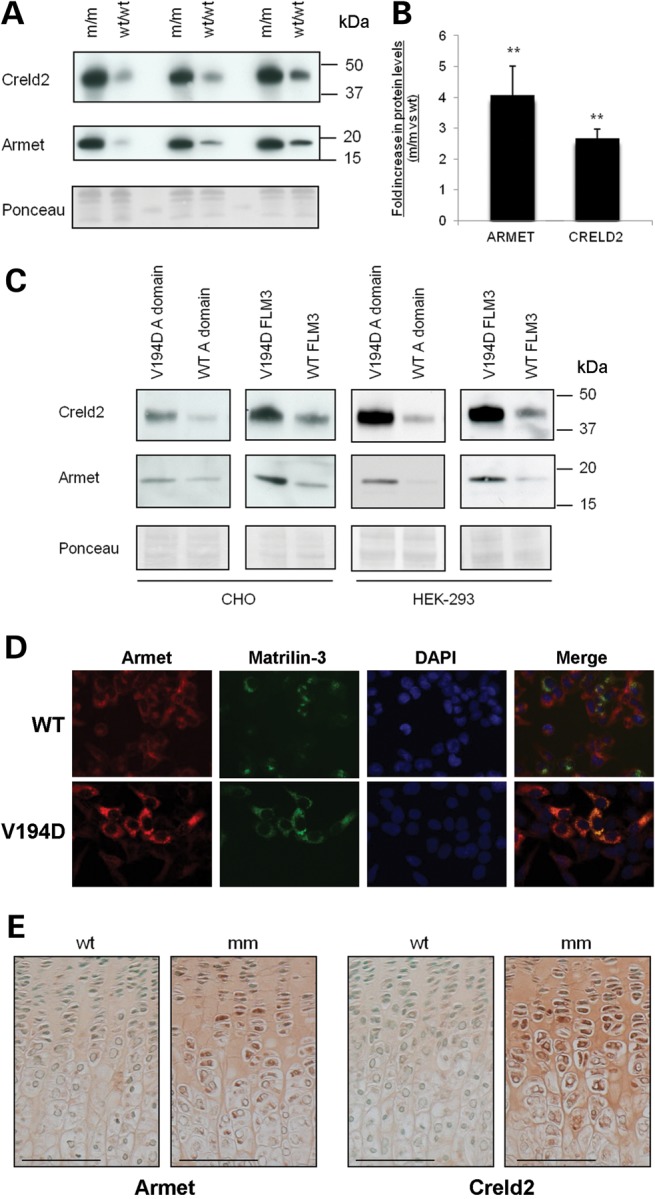
Armet and Creld2 are increased in V194D matrilin-3 mouse chondrocytes, cell culture models and growth plate cartilage. (A) Chondrocytes were isolated from the rib cartilage of 5-day-old Matn3 V194D [m/m] and wild-type (WT/WT) mice. Total protein from 1 × 105 cells was analysed by SDS–PAGE and western blotting using antibodies against Armet (∼18 kDa) and Creld2 (∼45 kDa). Equal protein loading was verified by Ponceau staining and three litters per genotype (∼5–10 mice pooled per litter) were analysed in three separate experiments. (B) Western blots were scanned and analysed by densitometry which demonstrated that there was a ∼4-fold increase in Armet and a ∼2-fold increase in Creld2 (independent t-test, **P < 0.01). (C) Cell lysate samples from CHO and HEK-293 cells expressing wild-type and V194D matrilin-3 were analysed by SDS–PAGE and western blotting and increased amounts of Armet and Creld2 were detectable in lysates from cells expressing the V194D mutation in both the full-length (FLM3) and single A-domain forms. Equal protein loading was verified by Ponceau staining. (D) Dual-labelling immunofluorescence microscopy confirmed that V194D matrilin-3 (green) and Armet (red) co-localized as an intracellular protein accretion (yellow/orange); DAPI was used to identify cell nuclei. (E) IHC using Armet and Creld2 antibodies on the tibia growth plates from 3-week-old wild-type (WT) and V194D mutant mice (mm). Chondrocytes in all zones of the mutant growth plate showed increased levels of intracellular staining for both Armet and Creld2. Interestingly, staining was also observed in the ECM of tibia growth plate cartilage. Scale bar is 100 µm; kDa = kilodaltons.
We have previously shown in cell culture models of MED that wild-type matrilin-3, either full-length protein or the single A-domain, is efficiently secreted into the culture media, whereas matrilin-3 containing MED mutations is retained intracellular (30). We therefore investigated whether the relative levels of Armet and Creld2 would also be increased in CHO and HEK-293 cell culture models expressing the p.V194D MED mutation. SDS–PAGE and western blotting demonstrated that both Armet and Creld2 were consistently increased in both the cell lines (Fig. 1C), thus confirming that the cell culture models of MED re-capitulated this characteristic pathological feature of the murine model and also demonstrating that the upregulation of Armet and Creld2, in response to mutant matrilin-3 expression, was not chondrocyte specific. Furthermore, immunofluorescent analyses confirmed that Armet (Fig. 1D) and Creld2 (not shown) were co-localized with mutant matrilin-3 in the ER.
Armet and Creld2 are increased and secreted proteins in the growth plates of V194D matrilin-3 mice
Immunohistochemistry (IHC) was used to confirm that Armet and Creld2 were up-regulated in the growth plates of mutant mice and also to establish their precise localization within this tissue. IHC of growth plates from Matn3 V194D mice confirmed that Armet was up-regulated and accumulated within chondrocytes of the growth plate (Fig. 1E) and around the secondary centre of ossification from 1 week of age (Supplementary Material, Fig. S2). This observation was similar to the pattern of mutant matrilin-3 retention as described previously (10,12). Surprisingly, Armet was also secreted into the ECM of growth plate cartilage of mutant mice but not wild-type controls. The increased expression of Armet and its secretion into the ECM was observed from birth (Fig. 1E and supplementary Material, Fig. S3). Similarly, IHC of growth plates from Matn3 V194D mice showed that Creld2 accumulated within chondrocytes and its secretion into the ECM of mutant cartilage was detectable from 1 week of age (Fig. 1E and Supplementary Material, Fig. S4). These IHC data therefore support the western blotting experiments, confirming that the relative levels of Armet and Creld2 are increased within the chondrocytes of mutant mice. The accumulation of both proteins appeared to increase over time and peaked at ∼1 week for Armet, while the relative levels of Creld2 remained comparatively high over the full-time period studied.
Armet and Creld2 are not increased in murine models of Comp-associated PSACH-MED but are increased in Col10a1 MCDS
Although our studies indicated that the upregulation of Armet and Creld2 in response to the expression of Matn3 V194D was not chondrocyte specific, we wished to determine whether their upregulation was gene product and/or mutation specific. We therefore used IHC and SDS–PAGE western blotting to study three additional mouse models of ER stress-induced chondrodysplasia; mild PSACH resulting from a mutation in the C-terminal globular domain of COMP (Comp T585M) (11), severe PSACH resulting from a common in-frame deletion in the type III repeat of COMP (Comp D469del) (13) and MCDS resulting from a NC1 mutation in type X collagen (Col10a1 N617 K) (14).
We first examined the growth plates from 1-week-old Comp T585M and Comp D469del mice by IHC and the relative levels of Armet and Creld2 remained unchanged in both mutant mice compared with their wild-type controls (Fig. 2A and Supplementary Material, Fig. S5). To confirm that there was no increase in Armet or Creld2 in the chondrocytes of these PSACH-MED models, SDS–PAGE and western blotting were performed on the intracellular proteins extracted from chondrocytes isolated from the cartilage of 5-day-old mice (Fig. 2B). Finally, we examined a mouse model of MCDS resulting from a mutation in the NC1 domain of type X collagen (Col10a1 N617 K), and demonstrated that the relative levels of both Armet and Creld2 were increased within the chondrocytes and ECM of the hypertrophic zone (Fig. 2C). Due to the restricted expression of type X collagen in hypertrophic chondrocytes, we were unable to confirm this observation by SDS–PAGE and western blotting, but the upregulation of Armet and Creld2 mRNA in Col10a1 N617K hypertrophic chondrocytes has recently been confirmed by microarray and in situ in a separate study (31).
Figure 2.
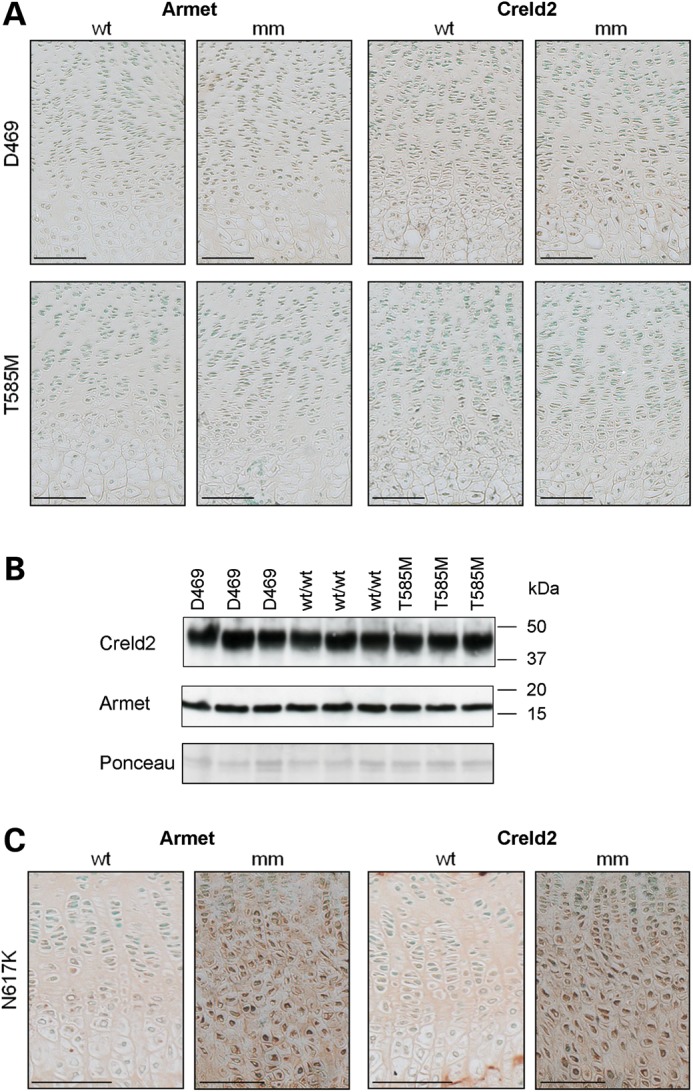
Creld2 and Armet are not up-regulated in mouse models of COMP-related PSACH-MED, but are increased in a model of MCDS. (A) IHC using Armet and Creld2 antibodies on tibia growth plates from 3-week-old Comp T585M (T585M), Comp D469del (D469) and matched wild-type (WT) mice. No increase in staining was observed for Armet or Creld2 in either mutant (mm) mouse model compared with wild-type (WT). (B) Chondrocytes were isolated from the cartilage of 5-day-old Comp T585M (T585M), Comp D469del (D469) and wild-type (WT) mice. Total protein from 1 × 105 cells was loaded per lane and analysed by SDS–PAGE and western blotting. No detectable differences in the levels of Creld2 and Armet were observed in cell extracts from mutant mice (T585M and D469) compared with wild-type (WT) controls. Equal protein loading was verified by Ponceau staining. (C) IHC on tibia growth plates from 3-week-old Col10a1 N617K (N617K) mice demonstrated both intracellular and ECM staining in the hypertrophic zone of growth plate cartilage. Scale bar is 100 µm; kDa = kilodaltons.
Overall, these data demonstrate that the upregulation of Armet and Creld2 is not specific to Matn3 V194D, but is also a response to Col10a1 N617K expression, whereas two mutant forms of COMP (T585M and D469del) do not induce increased expression.
Matrilin-3 interacts with Armet and Creld2 in a complex with other chaperones and foldases
We used co-immunoprecipitation to investigate whether both the full-length and A-domain forms of matrilin-3 could interact with Armet and Creld2, while ERp72/PDIA4, a well characterized PDI known to interact with various mutant forms of the matrilin-3 A-domain (30), was included as a control. These experiments confirmed that full-length mutant matrilin-3 interacted with Armet, Creld2 and ERp72/PDIA4; while the use of mutant A-domain alone refined the site of these interactions to this specific region (Fig. 3A).
Figure 3.

Armet and Creld2 interact in a complex with matrilin-3, but not with COMP. (A) Cell lysate proteins of HEK293 cells transfected with wild-type (WT) and V194D matrilin-3 expression constructs (FLAG-tagged full-length (FLM3) and the single A-domain) were immunoprecipitated with ANTI-FLAG affinity gel. SDS–PAGE and western blotting demonstrated that ERp72, Creld2 and Armet were co-precipitated with mutant matrilin-3 alone and that these interactions could be mediated by the A-domain (right panel). GAPDH confirmed equal loading and cell lysates from untransfected HEK 293 cells was used as a control. (B) Cell lysate proteins of HT1080 cells transfected with GFP alone or GFP-tagged wild-type COMP (WT COMP) and D469del mutant COMP (D469) expression constructs were immunoprecipitated with anti-GFP-sepharose beads. SDS–PAGE and western blotting did not identify any interactions between COMP and Armet or Creld2 (right panel). GAPDH confirmed equal loading and cell lysates from untransfected HT1080 cells was used as a control. Key: FLM3 = full-length matrilin-3; A-domain = A-domain alone comprising residues 77–263 of matrilin-3; kDa = kilodaltons.
Proteomic interrogation by liquid chromatography-mass spectrometry (LC-MS)/MS of the Flag-precipitated protein complexes revealed that a number of different chaperones and foldases were also present in both the wild-type and mutant matrilin-3 protein complexes (Table 1). These included BiP/GRP78, GRP94, PDI (PDIA1, -3, -4 and -6) and peptidylprolyl isomerases A and B; however, there were clearly more peptides detected for PDIA4, PDIA6, BiP/GRP78 and GRP94 in the mutant protein samples. Finally, the only protein that was almost entirely absent from wild-type matrilin-3 complexes, but was present in significant quantities in mutant protein complexes of both the A-domain and full-length matrilin-3, was hypoxia up-regulated protein 1 (HYOU1/ORP150/GRP170), which is a protein implicated in ER stress and several human diseases (32).
Table 1.
Summary of the results of spectral counting from FLAG and GFP-precipitated complexes of matrilin-3 and COMP, respectively
| Gene | Type | 293 | WT FLM3 | V194D FLM3 | WT A-domain | V194D A-domain | 1080 | WT COMP | D469del COMP |
|---|---|---|---|---|---|---|---|---|---|
| PDIA1 | PDI | 0/0 | 4/6 | 8/7 | 6/3 | 8/8 | 0/0 | 7/8 | 6/5 |
| PDIA3 | PDI | 0/0 | 3/0 | 4/0 | 3/0 | 6/7 | 0/0 | 0/0 | 0/0 |
| PDIA4 | PDI | 0/0 | 3/3 | 22/24 | 5/3 | 13/7 | 0/0 | 8/5 | 9/11 |
| PDIA6 | PDI | 0/4 | 9/4 | 11/10 | 8/5 | 10/10 | 0/0 | 0/0 | 0/0 |
| PPIA | PPI | 0/3 | 2/2 | 0/0 | 0/2 | 3/0 | 0/3 | 0/2 | 0/0 |
| PPIB | PPI | 0/0 | 4/3 | 4/7 | 5/0 | 5/5 | 0/0 | 3/3 | 7/5 |
| GRP78 | Chaperone | 3/8 | 14/16 | 50/46 | 14/8 | 52/43 | 0/10 | 26/20 | 27/29 |
| GRP94 | Chaperone | 0/4 | 9/3 | 32/20 | 9/6 | 22/19 | 4/2 | 19/23 | 21/15 |
| ORP150 | Chaperone | 0/0 | 0/0 | 10/12 | 2/0 | 10/10 | 0/0 | 0/0 | 4/2 |
| DNAJC3 | Co-Chaperone | 0/0 | 0/0 | 0/2 | 0/0 | 3/0 | 0/0 | 0/0 | 0/0 |
| DNAJC10 | Co-Chaperone | 0/0 | 0/0 | 2/3 | 0/0 | 2/3 | 0/0 | 0/0 | 0/0 |
| MATN3 | ECM protein | 0/7 | 15/12 | 26/28 | 37/9 | 24/15 | – | – | – |
| COMP | ECM protein | – | – | – | – | – | 0/0 | 26/18 | 32/12 |
Total protein pools following FLAG and GFP precipitation were analysed by LC-MS/MS and the data evaluated using Mascot against the UniProt human database and validated with Scaffold using peptide/protein confidence values of 0.95 and 0.99, respectively. Positively identified proteins were defined as those having a number of matched peptide spectra >2. Two biological replicates were used in all experiments and the number of matched spectra from each experiment is shown separated with a forward slash (/).
Key: 293 = untransfected HEK293 cells; 1080 = untransfected HT1080 cells; WT = wild type; V194D = mutant matrilin-3; D469del = mutant COMP; FLM3 = full-length matrilin-3; A-domain = matrilin-3 A-domain alone; PDI = protein disulphide isomerase; PPI = peptidylprolyl isomerase; ECM = extracellular matrix protein.
Armet and Creld2 are not present in a complex with COMP and other chaperones
Following co-immunoprecipitation, we did not detect any interactions between COMP (wild type or D469del) and either Armet or Creld2 (Fig. 3B). LC-MS/MS of the GFP-precipitated protein complexes confirmed that wild-type COMP formed complexes with a variety of chaperones and foldases including PDIAs (PDIA1 and -4), BiP/GRP78, GRP94 and peptidylprolyl isomerase B (PPIB) (Table 1). However, unlike Matn3 V194D there were no quantitative differences in the proteomic portrait of D469del mutant COMP compared with wild-type, with the possible exception of hypoxia up-regulated protein 1 (HYOU1), which was only detected in mutant COMP samples.
Increased expression of Armet and Creld2 is associated with a specific increase in PDIs in Matn3 and Col10a1 disease models
We have previously shown by microarray analysis that a UPR is initiated through the expression of Matn3 V194D (12). Interestingly, of the 20 most significantly up-regulated genes, three were members of the PDIA protein family, namely Pdia3/Erp57, Pdia4/Erp72 and Pdia6/P5, while PDI was shown to be up-regulated by western blotting. These expression data are therefore consistent with the proteomic profile of the co-precipitated mutant protein complexes.
In order to gain insight into why Armet and Creld2 were only increased in the murine models of Matn3-MED and Col10a1-MCDS, and not the Comp models of PSACH-MED, we compared the relative expression levels of Armet, Creld2 and members of the PDIA protein family in chondrocytes from the following mouse models; Matn3 V194D (12), Comp D469del (13), Comp T585M (K Pirog personal communication) and Col10a1 N617K (31) (Table 2). It is noteworthy that there was a consistent and significant upregulation of several PDIAs in the Matn3 V194D and Col10a1 N617K mutant mice, whereas there was no such upregulation in the Comp D469del and Comp T585M mice. These expression data and the co-immunoprecipitation studies (Fig. 3) led us to consider whether Armet and/or Creld2 possessed PDI-like activities.
Table 2.
PDIs are up-regulated in mouse models of Matn3 MED and Col10a1 MCDS but not Comp PSACH-MED
| Gene and/or protein name | V194D d5 | D469del d5 | T585M d5 | N617K NB |
|---|---|---|---|---|
| Creld2 | 5.77 | – | 1.30 | 7.66 |
| Armet/MANF | 4.29 | −1.33 | 1.24 | 2.99 |
| PDIA1/P4HB | 1.57 | – | −1.03 (ns) | 2.49 |
| PDIA2/PDIP | 1.42 (ns) | – | 1.02 (ns) | – |
| PDIA3/ERp57/GRP58 | 2.03 | 1.12 | 1.24 | 2.05 |
| PDIA4/ERp72 | 3.16 | 1.76 (ns) | 1.22 | 16.37 |
| PDIA5/PDIR | 1.34 | 1.07 | −1.09 (ns) | 1.62 (ns) |
| PDIA6/P5 | 3.18 | −1.09 | 1.27 | 4.97 |
Selected gene expression profiles of chondrocytes from 5-day-old (or new born for Col10a1) wild-type and the mutant mice genotypes Matn3 V194D (12), Comp D469del (13), Comp T585M (unpublished) and Col10a1 N617K (31) were compared. Gene expression levels were previously determined by microarray analysis as described in relevant papers. The relative expression levels of Armet, Creld2 and PDIAs1-6 are represented as fold change (mutant versus wild type), and all values are significant unless otherwise stated (ns = not significant). Significant upregulation was determined by PPLR values of >0.95.
Key: d5 = 5-day-old; NB = new born.
Creld2 possesses putative PDI-like activity whereas Armet does not
Armet has recently been proposed to be a putative PDIA due to the presence of a CXXC motif in the C-terminal domain (22,33), which is a common feature of thiol/disulphide oxidoreductases (22). Similarly, Creld2 has a number of CXXC motifs, which may indicate that it also possesses isomerase activity. In order to investigate this potential function, we generated a series of Armet and Creld2 substrate-trapping mutants similar to those previously generated for other PDIAs (e.g. ERp72, ERp57, ERp46 and PDI), which have all previously been validated and characterized in depth (34). Following sequence alignment comparisons between Creld2, Armet and PDI, we identified potential amino- and carboxyl-terminal CXXC motifs (Supplementary Material, Fig. S6). For each substrate-trapping construct, we used in vitro mutagenesis to convert the second cysteine of each selected CXXC motif into alanine, thereby generating eight constructs, which included wild-type and double N- and C-terminal mutants (Supplementary Material, Fig. S7). HT1080 cells were then transfected with wild-type or mutant (N-CXXA, C-CXXA and N/C-CXXA) constructs and selection with G418 allowed stable cell lines to be established. Once confluent, and following treatment with NEM to prevent post-lysis thiol exchange, the total cell lysate proteins were separated by SDS–PAGE and proteins were detected by western blotting with an anti-V5 antibody. We also included as a positive control in this experiment the previously characterized ERp72 substrate-trapping mutant as an example of the typical profile that would be expected from a confirmed PDI (34). In this definitive study, the authors showed unequivocally that substrate-trapping mutants of ERp57 (PDIA3), PDI (PDIA1), P5 (PDIA6), ERp18 (PDIA6), ERp72 (PDIA4) and ERp46 (PDIA15) all formed higher order mixed disulphides, whereas the repsective wild-type proteins did not (34). We therefore included in our experiments only the substrate-trapping mutant form of ERp72 (and not the wild-type protein) as a visual example for the formation of higher order mixed disulphides.
All the three substrate-trapping mutants of Armet (N-CXXA, C-CXXA and N/C-CXXA) had a similar profile to the wild type (under both reducing and non-reducing conditions), which suggested that no higher order mixed disulphides had been formed with substrate proteins (Fig. 4A), whereas the ERp72 substrate-trapping mutant clearly showed the formation of mixed disulphide complexes (Fig. 4B), which were consistent with the previously published profile (34). Similar to ERp72, the mutation of putative active sites in Creld2 allowed the formation of a number of higher order complexes containing Creld2-V5 (Fig. 4C, left panel). These complexes were disrupted following reduction by dithiothreitol (DTT), confirming that mixed disulphide bonds had formed (Fig. 4C, right panel). Furthermore, there was specificity in which CXXC motif was mediating the interaction(s). For example, only the N-CXXA and N/C-CXXA proteins formed high-molecular weight mixed disulphides, indicating that it was the amino-terminal CXXC motif that possessed the isomerase activity. To identify the individual substrates forming mixed disulphide complexes with Creld2, we performed LC-MS/MS after affinity isolation of the protein complexes using anti-V5 agarose beads (Fig. 4D and Table 3). This analysis, which was performed in HT1080 cells, identified laminin-5 β3 (LAMB3), collagen α1(VI) and α3(VI), thrombospondin-1 and integrin α-3 as potential substrates, while BiP/GRP78, GRP94 and PDIA1, -3 and -6 were also present within these complexes.
Figure 4.

Creld2 processes putative PDI-like activity, whereas Armet does not. Total cell lysate proteins from HT1080 cells stably transfected with Armet-V5, Creld2-V5 or ERp72-V5 substrate-trapping mutants were separated by SDS–PAGE and analysed by western blotting with an anti-V5 antibody. (A) There was no evidence of higher order mixed disulphides formed between putative substrate proteins and either wild-type Armet (WT) or the individual substrate-trapping mutants (N/C-CXXA, C-CXXA and N-CXXA). (B) In comparison, the control ERp72 substrate-trapping mutant (C-CXXA) demonstrated the formation of mixed disulphides with substrate proteins. (C) Both the N-CXXA and N/C-CXXA substrate-trapping mutants of Creld2 formed high-molecular weight mixed disulphides (left panel) that were resolved on reduction (right panel). In contrast, wild-type Creld2 (WT) and the C-CXXA trapping mutant did not form higher molecular weight aggregates with putative substrate proteins. (D) V5 co-immunoprecipitated proteins from the various Creld2 substrate-trapping cell lines were resolved by SDS–PAGE and viewed by silver staining or instant blue (insert). Total protein pools >50 kDa were excised from each lane of the instant blue gel for liquid chromatography-mass spectrometry/MS analysis. Key: N/C-CXXA = amino and carboxyl terminal double substrate-trapping mutant; C-CXXA = carboxyl terminal substrate-trapping mutant; N-CXXA = amino terminal substrate-trapping mutant; WT = wild- type Armet or Creld2; HT1080 = untransfected HT1080 cells; lysate = total protein lysate prior to V5 co-immunoprecipitation; R = reduced protein samples; NR = non-reduced protein samples; kDa = kilodaltons.
Table 3.
Creld2 is part of mixed disulphide complexes with structural proteins, chaperones and PDIs
| Protein | Type | HT1080 | Creld2 WT | Creld2 N-CXXA | Creld2 C-CXXA | Creld2 N/C-CXXA |
|---|---|---|---|---|---|---|
| BiP | Chaperone | 0/0 | 4/0/6 | 16/17/19 | 3/0/9 | 17/16/17 |
| GRP94 | Chaperone | 0/0 | 0/0/3 | 0/11/3 | 0/0/2 | 8/8/9 |
| HSP90β | Chaperone | 4/0 | 4/0/4 | 10/5/10 | 0/0/4 | 17/5/17 |
| HSPA8 | Chaperone | 0/0 | 6/0/3 | 7/13/9 | 2/0/4 | 14/5/14 |
| SERPINH1 | Chaperone | 0/0 | 0/0/0 | 2/6/2 | 0/0/0 | 4/3/3 |
| PDIA1 | PDI | 0/0 | 3/0/0 | 3/6/4 | 0/0/4 | 7/6/5 |
| PDIA3 | PDI | 0/0 | 0/0/0 | 3/6/5 | 0/0/7 | 12/11/8 |
| PDIA4 | PDI | 0/0 | 0/0/0 | 0/3/0 | 0/0/0 | 7/4/4 |
| LAMB3 | ECM protein | 0/0 | 0/0/0 | 11/15/22 | 2/0/14 | 14/13/17 |
| α1(VI) chain | ECM protein | 0/0 | 0/0/0 | 7/9/15 | 0/0/0 | 9/5/5 |
| α3(VI) chain | ECM protein | 0/0 | 0/0/0 | 3/9/22 | 0/0/14 | 19/15/12 |
| TSP1 | ECM protein | 0/0 | 0/0/0 | 14/11/23 | 0/0/5 | 6/0/7 |
| Integrin alpha-3 | Receptor | 0/0 | 0/0/0 | 6/4/14 | 0/0/0 | 9/6/10 |
| Pyruvate kinase isozymes M1/M2 | Glycolytic enzyme | 0/0 | 0/0/9 | 10/11/13 | 0/0/5 | 11/5/16 |
A summary of the proteins identified by spectral counting from V5-precipitated complexes of the Creld2 substrate-trapping mutants. Total protein pools were analysed by LC-MS/MS and the data analysed using Mascot against the UniProt human database and validated with Scaffold using peptide/protein confidence values of 0.95 and 0.99, respectively. Positively identified proteins were defined as those having a number of matched peptide spectra greater than two. Three biological replicates were used in all experiments and the number of spectra from each experiment is separated with a forward slash (/).
Key: N/C-CXXA = amino and carboxyl terminal double substrate-trapping mutant; C-CXXA = carboxyl terminal substrate-trapping mutant; N-CXXA = amino terminal substrate-trapping mutant; WT = wild-type Creld2; HT1080 = untransfected HT1080 cells; PDI = protein disulphide isomerase; PPI = peptidylprolyl isomerase; ECM = extracellular matrix.
N-CXXA Creld2 substrate-trapping mutant shows specificity for mutant matrilin-3
Since HT1080 cells do not express cartilage structural proteins, we wished to test directly whether Armet or Creld2 substrate-trapping mutants could capture mutant matrilin-3. As expected co-transfection of the Armet HT1080 substrate-trapping cell lines with WT and V194D matrilin-3 expression constructs (Fig. 5A; left panel) failed to identify any mixed disulphides (Fig. 5A; right panel), thereby confirming that Armet most likely formed non-covalent interactions with matrilin-3 protein-folding complexes (see Fig. 3A). In contrast, the N-CXXA and N/C-CXXA substrate-trapping mutants of Creld2 were able to capture full-length mutant matrilin-3 (Fig. 5B; left panel) that existed as heterogeneous higher order mixed disulphide complexes under non-reducing conditions (Fig. 5B; right panel). This finding was consistent with the previous experiment (Fig. 4C) and confirmed the location of the isomerase activity of Creld2 to the amino terminal CXXC motif. However, we also noted that the C-CXXA mutant was able to interact to a small extent with mutant matrilin-3, suggesting that there may be some limited activity at this CXXA site of Creld2.
Figure 5.

N-CXXA Creld2 substrate-trapping mutant shows specificity for mutant matrilin-3. Wild-type Armet-V5 and Creld2-V5 (WT) and the various substrate-trapping (N/C-CXXA, C-CXXA and N-CXXA) cell lines were co-transfected with wild-type (WT M3) or V194D mutant (V194D) matrilin-3 expression constructs. (A) Reducing SDS–PAGE and western blotting (anti-FLAG) on total cell lysate proteins confirmed the co-expression of WT and V194D matrilin-3 in all Armet cell lines (left panel: WT Armet and the various substrate-trapping lines). However, non-reducing SDS–PAGE and western blotting for Armet (anti-V5) did not detect any higher order mixed disulphides in all substrate-trapping and wild-type cell lines (right panel). (B) Co-immunoprecipitation with V5 followed by reducing SDS–PAGE and western blotting for Creld2 (anti-V5) and matrilin-3 (anti-FLAG) confirmed interactions between full-length V194D matrilin-3 and only the N/C-CXXA and N-CXXA substrate-trapping cell lines (left panel). When these samples were run under non-reducing conditions, the presence of matrilin-3 (anti-FLAG) containing higher order mixed disulphide complexes was demonstrated (right panel). Total cell lysate proteins from HT1080 cells (either untransfected or transfected with WT Creld2) were used controls. Key: WT M3 = wild-type matilin-3; V194D = V194D mutant matrilin-3; N/C-CXXA = amino and carboxyl terminal double substrate-trapping mutant; C-CXXA = carboxyl terminal substrate-trapping mutant; N-CXXA = amino terminal substrate-trapping mutant; WT = wild-type Armet or Creld2; HT1080 = untransfected HT1080 cells; kDa = kilodaltons.
Substitution of the two terminal cysteine residues from the A-domain of V194D matrilin-3 prevents aggregation, promotes mutant protein secretion and reduces the levels of Creld2 and Armet
The expression of V194D matrilin-3 consistently results in aggregation through non-native disulphide bonds and intracellular retention in cell and mouse models of MED (Fig. 6A and B, respectively) (12,30,35). Moreover, this aggregation is mediated through the mutant A-domains alone (Fig. 6A; right panel) and can be resolved to a single molecular form on reduction (21,30) (and not shown). We therefore tested the hypothesis that alanine substitution of the two terminal cysteine residues (Cys77 and Cys263) from the A-domain of V194D matrilin-3 would reduce mutant protein aggregation, promote the secretion of a proportion of V194D matrilin-3 and influence the expression of PDIs.
Figure 6.

Mutant matrilin-3 forms non-native disulphide bonded aggregates that can be resolved by deletion of the A-domain terminal cysteine residues. SDS–PAGE and western blot analysis confirmed the intracellular retention and aggregation over time (t = day 0 or day 3 following confluency after transfection) of V194D mutant matrilin-3 (V194D) in the cell lysate of (A) cellular models or (B) isolated mouse chondrocytes. (A; left panel) demonstrates that only wild-type (WT) matrilin-3 is secreted into the culture medium predominately as a tetramer, whereas as V194D mutant matrilin-3 (V194D) is retained initially as various oligomeric forms (t = 0), but these aggregate over time (t = 3) to form high-molecular weight aggregates. (A; right panel) confirms that aggregation is mediated through the A-domain by the presence of various oligomeric forms (1x to 5x). V194D and R121W are typical MATN3-MED mutations, whereas E252K is a known polymorphism and is comparable to wild type (WT). (C) In vitro substitution of the terminal cysteine residues of the A-domain (V194D_no cys) resolved mutant protein aggregation (V194D) and did not affect oligomerization of either the wild-type (WT_no cys) or mutant proteins when analysed by non-reducing SDS–PAGE and western blotting (anti-FLAG). (D) Substitution of the terminal cysteine residues with alanine promoted secretion of the mutant V194D protein (V194D-no cys) as visualized by reducing SDS–PAGE. Furthermore, (E) there was also selective reduction in the levels of ERp72, Creld2, and Armet, but not GRP78 or GRP94. Ponceau staining (not shown) and GAPDH were used as loading controls. Key: WT = wild-type matrilin-3; V194D = mutant matrilin-3; V194D_no cys & WT_no cys = matrilin-3 with cysteine residues 77 and 263 replaced with alanine (mutant and wild-type respectively); 293 = cell lysate from untransfected HEK293 cells; agg = non-native disulphide bonded aggregates of mutant matrilin-3; q = tetramer, t = trimer, d = dimer and m = monomers of matrilin-3; ns = non-specific band; kDa = kilodaltons. All gels show full-length matrilin-3 unless stated otherwise and the number of replicates in (E) is indicated.
The removal of both cysteine residues prevented the formation of V194D matrilin-3 higher order disulphide-bonded aggregates (Fig. 6C), but did not prevent the folding and secretion of wild-type matrilin-3 A-domain. Moreover, this genetic manipulation also promoted to some extent the secretion of V194D matrilin-3 (Fig. 6D) and selectively reduced the protein levels of ERp72, Creld2 and Armet, but not GRP78 and GRP94 (Fig. 6E).
DISCUSSION
This study demonstrates for the first time the genotype-specific upregulation and secretion of Armet and Creld2 in various cell and mouse models of genetic skeletal diseases. Moreover, we have demonstrated that both Armet and Creld2 have substrate specificity in their binding to structural proteins and that Creld2 possess PDI-like activity. This is the first study to identify a potential role for both Armet and Creld2 and to confirm that they are key markers in human ER stress-related diseases. Finally, we present data that the aggregation of mutant matrilin-3 through non-native disulphide bonds between the A-domains is a key disease trigger that promotes specialization of the UPR toolbox (36).
Perhaps surprisingly, we observed that both Armet and Creld2 were present within the cartilage ECM of mutant mouse growth plates, confirming that these proteins can be secreted under certain pathological conditions. Many ER resident proteins are prevented from being secreted via an ER retention motif (KDEL) at their extreme C-terminus, which binds with high affinity to KDEL receptors (KDEL-R) located in the intermediate compartment or in the cis-Golgi (37). Interestingly, both Armet and Creld2 contain putative ER retention motifs at their C-terminus (RTDL and REDL, respectively) (38), suggesting that these proteins might remain resident in the ER. However, while previous reports have shown that both Armet and Creld2 are primarily retained within the ER or Golgi, they can also be secreted into cell culture media under certain experimental conditions (25,26,39). For example, these proteins can be secreted if the putative ER retention signals are disrupted, either by deletion or having a peptide tag engineered at the C-terminus (26,27,39). In other experiments, the ER retention motif has not been disrupted but cells have been subjected to ER stress by chemical agents such as tunicamycin or by culturing cells in serum-free medium. Overall, these studies have suggested that Armet and Creld2 can be secreted in vitro during different ER stress conditions. Since Armet and Creld2 are only detectable at significant levels in the ECM of Matn3 V194D and Col10a1 N617K mutant growth plates and not in wild-type controls, our study demonstrates for the first time that both proteins are up-regulated and secreted during ER stress in gene-targeted disease models. This observation is consistent with the hypothesis that under normal physiological conditions, Armet and Creld2 are able to bind to the KDEL-R and remain within the ER (40); however, it is believed that KDEL-variants bind with lower affinity than KDEL itself (38). During ER stress, the relative levels of Armet and Creld2 are increased, along with other UPR-related proteins such as BiP and GRP94, while the relative expression of the three mammalian KDEL receptors (KDELR1–3) are not increased (41). Indeed, microarray analysis confirmed that the relative levels of Kdelr1, Kdelr2 and Kdelr3 in V194D mutant compared with control chondrocytes were −1.22, 1.43 and 1.28, respectively (not shown). Therefore, upon activation of the UPR proteins such as BiP, which contain perfect ER retention motifs that have higher affinity for the KDEL receptor, would compete with proteins which contain a motif that has a lower binding affinity, thereby allowing the latter proteins to escape the ER and ultimately be secreted. The interesting observation that Armet and Creld2 are secreted under conditions of increased ER stress suggests the possibility that they may be exploited as soluble extracellular biomarkers of ER stress-related diseases that are gene product and/or mutation specific.
Substrate-trapping experiments demonstrated that Creld2 possesses PDI-like activity and in addition to matrilin-3, we also identified laminin-5 β3, collagen VI and thrombospondin-1 (TSP1) as potential substrates. All three proteins have numerous intramolecular disulphide bonds. Indeed, the α1(VI) and α3(VI) chains contain up to three and seven VWA domains, respectively (42), which are very similar in structure to the one found in matrilin-3, while laminin-5 β3 has numerous intermolecular disulphide bonds in various domains and also forms disulphide bonded trimers with the α3 and γ2 chains. TSP1 is the archetypal member of the TSP protein family, which includes COMP (TSP5), but has additional domains not shared with COMP such as the type 1 repeats and procollagen homology domain. Mutations in the VWA domains of type VI collagen result in Bethlem and Ulrich muscular dystrophies (42), while mutations in LAMB3 cause junctional epidermolysis bullosa (both Herlitz and non-Herlitz type) (43). In both diseases, ER stress and an UPR have not been investigated as potential mechanisms, although mutations in type VI collagen have been shown to cause impaired secretion and mutant protein retention (44,45). The role of ER stress and UPR specialization in these connective tissues diseases, and in particular whether ARMET and Creld2 are up-regulated, warrants further investigation in order to delineate a range of different phenotypes that may share a common disease trigger.
The co-immunoprecipitation experiments were also consistent with the recently published interaction map of endoplasmic reticulum chaperones and foldases (36). For example, we noted that matrilin-3 formed ER multi-protein complexes with BiP/GRP78, GRP94, selected PDIAs and PPIs. Indeed, the PPIB (cyclophilin B) multi-protein composite containing PDIA1, PDIA4/ERp72, PDIA6/P5, BiP/GRP78 and GRP94 was found with matrilin-3 A-domain complexes (Table 1), while the calnexin cycle components (CANX and CALR) were absent, which supports the separation of these two systems in this disease model (36). Moreover, there were increased levels of these proteins in mutant matrilin-3 complexes, which was consistent with our previous gene expression studies (Table 2). Wild-type COMP also showed preference for the PPIB multi-protein complex, but in contrast there was no increase in these components with D469del mutant COMP, confirming our previous finding of a lack of transcriptional UPR with this mutation (13). Finally, the interesting observation that HYOU1 was almost exclusively associated with mutant V194D matrilin-3, and to a lesser extent D469del COMP, highlights an important role for this protein in binding to mutant protein substrates and its cytoprotective role warrants further investigation in MED disease models (32).
Overall, our data suggest that the upregulation of Armet and Creld2 with the PPIB ER-folding complex may not simply be a consequence of UPR activation but could be part of a specialized UPR that is tailored for certain misfolded proteins that expose an unpaired cysteine residue. Indeed, Armet has recently been suggested to facilitate the formation of cysteine bridges and protein folding in the ER during neurodegenerative diseases (33), while a C96Y mutation in insulin leads to the disruption of intramolecular disulphide bonds (46), and the expression of this mutation in pancreatic cell lines leads to induction of Armet and Creld2 along with various PDIAs (47).
The upregulation of various PDIAs in the Matn3 V194D mutant chondrocytes might be expected since mutant matrilin-3 forms misfolded aggregates, both in vivo and in vitro, which dissociate upon reduction, indicating that these complexes form via non-native disulphide bonds. Furthermore, mutant matrilin-3 A-domains have also been shown to directly interact with PDIA4/ERp72 in a cell culture model of MED (30). Similarly, although expected to form stable type X collagen homotrimers, certain mutant forms of type X collagen have been shown to form an unusual dimer, which also dissociates upon reduction (48). The formation of this mutant type X collagen dimer, via a non-native disulphide bond, is believed to result from a dramatic conformational change since the sulfhydryl group in the NC1 domain is not solvent-exposed in the correctly folded α1(X) trimer. Finally, several mutant forms of type X collagen have been shown to co-immunoprecipitate with PDI (49); however, additional experimentation is required to define the role of Armet and Creld2 in MCDS.
Conversely, there is no evidence to support non-native disulphide bond formation and protein aggregation due to mutations in COMP. For example, cell culture studies on COMP with p.D469del, p.D472Y or p.D475N mutations confirmed impaired secretion of the mutant protein, but also demonstrated that both retained and secreted mutant COMP forms pentameric molecules similar to the wild-type protein (50–52). Overall, these studies demonstrate that specific mutations in the type III repeat region of COMP do not affect its ability to form pentamers, suggesting that these mutations do not cause a dramatic conformational change of the protein that might leave unpaired cysteine residues.
We hypothesized that the formation of non-native disulphide bonded aggregates of mutant V194D matrilin-3 was the direct result of delayed or incomplete folding of the central β-sheet of the A-domain that results in prolonged exposure of the thiol groups of the two terminal cysteine residues (Cys77 and Cys263). This in turn promotes the formation of intermolecular disulphide bonds between matrilin-3 oligomers that cause mutant protein aggregation and render these complexes resistant to retro-translocation and ERAD. To test this hypothesis, we engineered expression constructs in which the two terminal cysteines were replaced by alanine residues. In the first instance, this proof-of-principle experiment demonstrated that removal of Cys77 and Cys263 did not disrupt to any great extent the folding and secretion of wild-type matrilin-3. This result in itself was not surprising since some vWFA domains and related integrin I-domains do not possess these terminal cysteine residues (53). In contrast, we saw a dramatic effect on the aggregation and secretion of mutant matrilin-3. For example, deletion of Cys77 and Cys263 completely abolished the formation of disulphide-bonded high-molecular weight aggregates and promoted the secretion of a proportion of V194D matrilin-3. Moreover, this resulted in a reduction of specific chaperones and foldases including Armet, Creld2 and ERp72, thus confirming their direct role in mediating disulphide bond formation in this disease model.
In conclusion, this study has discovered that the aggregation of mutant matrilin-3 is a key disease trigger in this form of MED, and that the prevention of this protein accretion, by either enhancing the folding with corrector-molecules (54,55) or preventing non-native disulphide bond formation by blocking the unpaired cysteine residues (56), offers novel therapeutic targets for further validation.
MATERIALS AND METHODS
Transgenic mice models of human genetic skeletal diseases
Matn3 V194D, Comp T585M mice, Comp D469del mice and Col10a1 N617K mice were generated as previously described (10,11,13,14). Mice homozygous for the respective mutations were compared with their wild-type counterparts for these studies.
Matrilin-3 and COMP cellular models of PSACH-MED
Complementary DNAs encoding full-length matrilin-3 (FLM3) and the A-domain alone (both wild type and p.V194D) had previously been cloned into pCMV-Tag4 (FLM3) and pSecTag2A (A-domain) vectors with C-terminal C-MYC or FLAG epitope tags, respectively, and used to establish stable CHO cell lines (30). These cDNAs were also subcloned into the expression vector pCEP4 and transfected into HEK 293 cells as described previously (21). Expression constructs encoding wild-type COMP and the D469del mutation had previously been cloned into pEGFP-N3 with a C-terminal GFP tag and transfected into HT1080 cells (13). Mutation of the terminal cysteines (Cys77 and Cys263) to alanine residues in the A-domain of full-length matrilin-3 expression constructs (WT and p.V194D) was performed by PCR-based in vitro mutagenesis similar to the approach previously used to introduce MED mutations (21).
CHO, HEK 293 and HT1080 cells were cultured as previously described with Dulbecco's Modified Eagle's medium supplemented with 10% FBS (Fisher Scientific), 2 mm l-glutamine (Sigma), 100 U/ml penicillin and 100 mg/ml streptomycin (Biowhittaker), non-essential amino acids and vitamins (Sigma) and antibiotics appropriate to the expression vectors (13,21,30). All cells were incubated at 37°C in humidified air containing 5% CO2. CHO and HEK 293 cells were lysed by incubation in RIPA lysis buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 1% sodium deoxycholate, 0.1% SDS) on ice for 15 min. The cell extract was removed, centrifuged at 13 600g for 5 min and the supernatant removed for analysis.
Isolation of total mouse chondrocyte protein
Chondrocytes were isolated from the rib cartilage of pooled litters of 5-day-old mice as described previously (12). Briefly, rib cages were dissected from wild-type (WT) and mutant (mm) mice and treated with 2 mg/ml type 1A collagenase (Sigma) for 1 h. The costal cartilage was then dissected from the rib cage and other connective tissues were removed. The clean cartilage was further treated with collagenase for 3 h, and the collagenase digest passed through a cell strainer (70 μm) and centrifuged for 5 min at 560g to pellet the released chondrocytes. The cell pellet was washed twice with PBS and resuspended in 5 ml PBS. Chondrocyte numbers were determined using a haemocytometer and aliquots of 1 × 105 cells prepared. Cell pellets were collected by centrifugation and resuspended in 5X SDS loading buffer.
SDS–PAGE and western blotting of mouse and cell line protein extract
Twenty micrograms of total cell lysate (CHO or HEK293) or mouse chondrocyte protein extract were separated by SDS–PAGE on 4–12% Bis-Tris gels (Invitrogen). Proteins were transferred to nitrocellulose membranes for western blot analysis. Ponceau staining and/or GAPDH were used to confirm the equal loading of protein prior to probing with antibodies specific to Armet, GAPDH (Abcam), Creld2, GRP78, GRP94, ERp72 (all Santa Cruz Biotechnology), matrilin-3 (R&D), COMP (Genetex) and FLAG (Sigma). Protein bands were visualized by enhanced chemiluminescence (Perkin-Elmer Inc) and quantified using AIDA densitometry software. For the Armet and Creld2 blots using chondrocyte protein extracts, the ‘cell protein aliquots’ from three different biological replicates were analysed in triplicate and quantified for each genotype. Analysis of cell lysates from CHO and HEK293 cells was performed in triplicate on one isolate.
Immunohistochemical analysis of growth plate cartilage
Limbs from male mice were prepared for IHC as described previously (10). Sections were blocked with goat serum (Dako Cytomation) in PBS/1% bovine serum albumin (BSA) and incubated for 1 h with primary antibodies specific to Armet or Creld2. Sections were then incubated with the appropriate secondary antibody (biotinylated anti-rabbit IgG, Dako Cytomation) in PBS/1% BSA, followed by incubation with ABCcomplex/HRP reagent (Vector laboratories Ltd.) for 30 min. Sections were developed using 3,3′-diaminobenzidine (Dako Cytomation), counterstained with methyl green, dehydrated in EtOH, cleared in xylene and mounted in Vectamount. The sections were imaged using a Zeiss Axiovision microscope.
Immunocytochemical analysis of MED cell culture models
Fixed cells were permeabilized with 0.2% Triton/PBS for 8 min, washed in PBS, blocked with 2% donkey serum/PBS and incubated with primary antibodies against either matrilin-3 or Armet for 1 h. Cells were then washed with PBS, incubated with the secondary antibody (Alexa Fluor 555 or Fluor 488; Invitrogen) and washed in PBS before mounting in Vectashield medium with DAPI (Vector) and imaged.
Co-immunoprecipitation of interacting proteins
Co-immunoprecipitation was carried out on HEK-293 cell lysates transfected with matrilin-3 constructs using ANTI-FLAG® M2 affinity gel (Sigma Aldrich, Dorset UK) or on HT1080 cells transfected with COMP constructs using ANTI-GFP sepharose beads (Abcam). Prior to immunoprecipitation, the cells were treated with dithiobis[succinimidyl propionate] to stabilize protein complexes as previously described (30). Cell lysates were prepared by incubation in lysis buffer (50 mm Tris–HCl, pH 7.4, 1% Triton X-100, 150 mm NaCl, 1 mm EDTA). Following removal of the cell monolayer and centrifugation at 13 600g, 500 µg of cell lysate was incubated with ANTI-FLAG® M2 affinity gel or ANTI-GFP sepharose beads as per manufacturer's instructions. The resin was collected and washed three times in tris-buffered saline (50 mm Tris–HCl, pH 7.4, 150 mm NaCl), before proteins were eluted with 0.1 m glycine HCl, pH 3.5 for SDS–PAGE and western blotting.
Mass spectrometry (MS) analysis of protein precipitates
Total protein pools were excised from each lane of an SDS–PAGE gel (run as described above) before being dehydrated, reduced, alkylated and washed. Samples were then digested with trypsin overnight at 37°C and analysed by LC-MS/MS using a NanoAcquity LC (Waters, Manchester, UK) coupled to a LTQ Velos (Thermo Fisher Scientific, Waltham, MA) mass spectrometer. Peptides were concentrated on a pre-column (20 mm × 180 μm i.d, Waters) and were then separated using a gradient from 99% A (0.1% FA in water) and 1% B (0.1% FA in acetonitrile) to 25% B, in 45 min at 200 nl min-1, using a 75 mm × 250 μm i.d. 1.7 μm BEH C18, analytical column (Waters). Peptides were selected for fragmentation automatically by data-dependent analysis.
Data were interrogated using Mascot version 2.2 (Matrix Science, UK) against the UniProt database (version 2011-05) with taxonomy of Homo sapiens selected. Mascot search results were validated using Scaffold version 3.3.1 (Proteome Software, Portland, USA) to assign confidence values to peptide/protein matches, where the peptide/protein confidence values of 0.95 and 0.99 were used. Identified proteins were defined as having a number of matched peptide spectra greater than two, and the unweighted spectral count was used as a measure of quantification. Two biological replicates were used in all experiments.
Generation of Armet and Creld2 substrate-trapping mutants
cDNAs containing the entire coding sequence for Armet and Creld2 were amplified using the PCR. The forward primer included a KpnI site at the 5′ end of the sequence, while the reverse primer included a V5 tag followed by a KDEL sequence, stop codon and a XhoI restriction site to aid sub-cloning and identification. The engineered PCR products were ligated into KpnI–XhoI-digested pcDNA3.1(+) (Invitrogen). In vitro mutagenesis of the N- and C-terminal cysteines in both putative active CXXA sites of Creld2 and Armet was performed by a PCR. For Creld2, these were Cys32 and Cys264 and for Armet Cys33 and Cys154 (Supplementary Material, Figs S5 and S6). Plasmids were linearized with SspI before transfecting into subconfluent HT1080 human fibroblasts with LipofectAMINE 2000 reagent as described previously. Stable cell lines were selected with G418 for 14 days before colonies were isolated and expanded.
Identification of putative mixed disulphides using substrate-trapping mutants
HT1080 cells expressing the various forms of Armet-V5, Creld2-V5 (WT, N/C-CXXA, N-CXXA and C-CXXA) and ERp72-V5 (C-CXXA) were grown to confluence under standard conditions and then treated with NEM (25 mm) to preserve mixed disulphides. Cells were lysed in 1% (v/v) Triton X-100, 50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 2 mm ethylenediaminetetraacetic acid and 0.5 mm phenylmethylsulphonyl fluoride and 20 µg cell lysate was separated by SDS–PAGE on 4–12% Bis-Tris gels under either reducing (DTT) or non-reducing conditions. Western blotting was subsequently performed with antibodies specific to the V5 epitope (Invitrogen: 1:5000 dilution).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Wellcome Trust (M.D.B. is the recipient of a Wellcome Trust Senior Research Fellowship in Basic Biomedical Science; Grant 084353/Z/07/Z) and was undertaken in the Wellcome Trust Centre for Cell-Matrix Research (Grant 088785/Z/09/Z) and the Biomolecular Analysis Facility of the Faculty of Life Sciences at the University of Manchester. We thank Dr Katarzyna Pirog for sharing her microarray data from the T585M Comp mouse. Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Supplementary Material
Acknowledgements
The ERp72 substrate trapping mutant cell line was kindly provided by Professor Neil Bulleid, Glasgow University.
REFERENCES
- 1.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L., et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson G.C., Mittaz-Crettol L., Taylor J.A., Mortier G.R., Spranger J., Zabel B., Le Merrer M., Cormier-Daire V., Hall C.M., Offiah A., et al. Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution. Hum. Mutat. 2012;33:144–157. doi: 10.1002/humu.21611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McIntosh I., Abbott M.H., Warman M.L., Olsen B.R., Francomano C.A. Additional mutations of type X collagen confirm COL10A1 as the Schmid metaphyseal chondrodysplasia locus. Hum. Mol. Genet. 1994;3:303–307. doi: 10.1093/hmg/3.2.303. [DOI] [PubMed] [Google Scholar]
- 4.Bateman J.F., Boot-Handford R.P., Lamande S.R. Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 2009;10:173–183. doi: 10.1038/nrg2520. [DOI] [PubMed] [Google Scholar]
- 5.Boot-Handford R.P., Briggs M.D. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2009;339:197–211. doi: 10.1007/s00441-009-0877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufman R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rutkowski D.T., Kaufman R.J. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Kim P.S., Kwon O.Y., Arvan P. An endoplasmic reticulum storage disease causing congenital goiter with hypothyroidism. J. Cell Biol. 1996;133:517–527. doi: 10.1083/jcb.133.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 10.Leighton M.P., Nundlall S., Starborg T., Meadows R.S., Suleman F., Knowles L., Wagener R., Thornton D.J., Kadler K.E., Boot-Handford R.P., et al. Decreased chondrocyte proliferation and dysregulated apoptosis in the cartilage growth plate are key features of a murine model of epiphyseal dysplasia caused by a matn3 mutation. Hum. Mol. Genet. 2007;16:1728–1741. doi: 10.1093/hmg/ddm121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirog-Garcia K.A., Meadows R.S., Knowles L., Heinegard D., Thornton D.J., Kadler K.E., Boot-Handford R.P., Briggs M.D. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum. Mol. Genet. 2007;16:2072–2088. doi: 10.1093/hmg/ddm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nundlall S., Rajpar M.H., Bell P.A., Clowes C., Zeeff L.A., Gardner B., Thornton D.J., Boot-Handford R.P., Briggs M.D. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress Chaperones. 2010;15:835–849. doi: 10.1007/s12192-010-0193-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suleman F., Gualeni B., Gregson H.J., Leighton M.P., Pirog K.A., Edwards S., Holden P., Boot-Handford R.P., Briggs M.D. A novel form of chondrocyte stress is triggered by a COMP mutation causing pseudoachondroplasia. Hum. Mutat. 2012;33:218–231. doi: 10.1002/humu.21631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajpar M.H., McDermott B., Kung L., Eardley R., Knowles L., Heeran M., Thornton D.J., Wilson R., Bateman J.F., Poulsom R., et al. Targeted induction of endoplasmic reticulum stress induces cartilage pathology. PLoS Genet. 2009;5:e1000691. doi: 10.1371/journal.pgen.1000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsang K.Y., Chan D., Cheslett D., Chan W.C., So C.L., Melhado I.G., Chan T.W., Kwan K.M., Hunziker E.B., Yamada Y., et al. Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 2007;5:e44. doi: 10.1371/journal.pbio.0050044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho M.S., Tsang K.Y., Lo R.L., Susic M., Makitie O., Chan T.W., Ng V.C., Sillence D.O., Boot-Handford R.P., Gibson G., et al. COL10A1 nonsense and frame-shift mutations have a gain-of-function effect on the growth plate in human and mouse metaphyseal chondrodysplasia type Schmid. Hum. Mol. Genet. 2007;16:1201–1215. doi: 10.1093/hmg/ddm067. [DOI] [PubMed] [Google Scholar]
- 17.Klatt A.R., Becker A.K., Neacsu C.D., Paulsson M., Wagener R. The matrilins: modulators of extracellular matrix assembly. Int. J. Biochem. Cell Biol. 2011;43:320–330. doi: 10.1016/j.biocel.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 18.Chapman K.L., Mortier G.R., Chapman K., Loughlin J., Grant M.E., Briggs M.D. Mutations in the region encoding the von Willebrand factor A domain of matrilin-3 are associated with multiple epiphyseal dysplasia. Nat. Genet. 2001;28:393–396. doi: 10.1038/ng573. [DOI] [PubMed] [Google Scholar]
- 19.Jackson G.C., Barker F.S., Jakkula E., Czarny-Ratajczak M., Makitie O., Cole W.G., Wright M.J., Smithson S.F., Suri M., Rogala P., et al. Missense mutations in the beta strands of the single A-domain of matrilin-3 result in multiple epiphyseal dysplasia. J. Med. Genet. 2004;41:52–59. doi: 10.1136/jmg.2003.011429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mabuchi A., Haga N., Maeda K., Nakashima E., Manabe N., Hiraoka H., Kitoh H., Kosaki R., Nishimura G., Ohashi H., et al. Novel and recurrent mutations clustered in the von Willebrand factor A domain of MATN3 in multiple epiphyseal dysplasia. Hum. Mutat. 2004;24:439–440. doi: 10.1002/humu.9286. [DOI] [PubMed] [Google Scholar]
- 21.Fresquet M., Jackson G.C., Loughlin J., Briggs M.D. Novel mutations in exon 2 of MATN3 affect residues within the alpha-helices of the A-domain and can result in the intracellular retention of mutant matrilin-3. Hum. Mutat. 2008;29:330. doi: 10.1002/humu.9518. [DOI] [PubMed] [Google Scholar]
- 22.Ellgaard L., Ruddock L.W. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6:28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatahet F., Ruddock L.W. Substrate recognition by the protein disulfide isomerases. FEBS J. 2007;274:5223–5234. doi: 10.1111/j.1742-4658.2007.06058.x. [DOI] [PubMed] [Google Scholar]
- 24.Ferrari D.M., Soling H.D. The protein disulphide-isomerase family: unravelling a string of folds. Biochem. J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- 25.Oh-hashi K., Koga H., Ikeda S., Shimada K., Hirata Y., Kiuchi K. Creld2 is a novel endoplasmic reticulum stress-inducible gene. Biochem. Biophys. Res. Commun. 2009;387:504–510. doi: 10.1016/j.bbrc.2009.07.047. [DOI] [PubMed] [Google Scholar]
- 26.Apostolou A., Shen Y., Liang Y., Luo J., Fang S. Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp. Cell Res. 2008;314:2454–2467. doi: 10.1016/j.yexcr.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizobuchi N., Hoseki J., Kubota H., Toyokuni S., Nozaki J., Naitoh M., Koizumi A., Nagata K. Armet is a soluble ER protein induced by the unfolded protein response via ERSE-II element. Cell Struct. Funct. 2007;32:41–50. doi: 10.1247/csf.07001. [DOI] [PubMed] [Google Scholar]
- 28.Airavaara M., Shen H., Kuo C.C., Peranen J., Saarma M., Hoffer B., Wang Y. Mesencephalic astrocyte-derived neurotrophic factor reduces ischemic brain injury and promotes behavioral recovery in rats. J. Comp. Neurol. 2009;515:116–124. doi: 10.1002/cne.22039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ortiz J.A., Castillo M., del Toro E.D., Mulet J., Gerber S., Valor L.M., Sala S., Sala F., Gutierrez L.M., Criado M. The cysteine-rich with EGF-like domains 2 (Creld2) protein interacts with the large cytoplasmic domain of human neuronal nicotinic acetylcholine receptor alpha4 and beta2 subunits. J Neurochem. 2005;95:1585–1596. doi: 10.1111/j.1471-4159.2005.03473.x. [DOI] [PubMed] [Google Scholar]
- 30.Cotterill S.L., Jackson G.C., Leighton M.P., Wagener R., Makitie O., Cole W.G., Briggs M.D. Multiple epiphyseal dysplasia mutations in MATN3 cause misfolding of the A-domain and prevent secretion of mutant matrilin-3. Hum. Mutat. 2005;26:557–565. doi: 10.1002/humu.20263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cameron T.L., Bell K.M., Tatarczuch L., Mackie E.J., Rajpar M.H., McDermott B.T., Boot-Handford R.P., Bateman J.F. Transcriptional profiling of chondrodysplasia growth plate cartilage reveals adaptive ER-stress networks that allow survival but disrupt hypertrophy. PLoS One. 2011;6:e24600. doi: 10.1371/journal.pone.0024600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao L., Rosales C., Seburn K., Ron D., Ackerman S.L. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco–Sjogren syndrome. Hum. Mol. Genet. 2010;19:25–35. doi: 10.1093/hmg/ddp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindholm P., Saarma M. Novel CDNF/MANF family of neurotrophic factors. Dev. Neurobiol. 2010;70:360–371. doi: 10.1002/dneu.20760. [DOI] [PubMed] [Google Scholar]
- 34.Jessop C.E., Chakravarthi S., Garbi N., Hammerling G.J., Lovell S., Bulleid N.J. ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 2007;26:28–40. doi: 10.1038/sj.emboj.7601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bell P.A., Pirog K.A., Fresquet M., Thornton D.J., Boot-Handford R.P., Briggs M.D. Loss of matrilin 1 does not exacerbate the skeletal phenotype in a mouse model of multiple epiphyseal dysplasia caused by a Matn3 V194D mutation. Arthritis Rheum. 2012;64:1529–1539. doi: 10.1002/art.33486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansen G., Maattanen P., Denisov A.Y., Scarffe L., Schade B., Balghi H., Dejgaard K., Chen L.Y., Muller W.J., Gehring K., et al. An interaction map of endoplasmic reticulum chaperones and foldases. Mol. Cell Proteomics. 2012;11:710–723. doi: 10.1074/mcp.M111.016550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeffer S.R. Unsolved mysteries in membrane traffic. Annu. Rev. Biochem. 2007;76:629–645. doi: 10.1146/annurev.biochem.76.061705.130002. [DOI] [PubMed] [Google Scholar]
- 38.Raykhel I., Alanen H., Salo K., Jurvansuu J., Nguyen V.D., Latva-Ranta M., Ruddock L. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007;179:1193–1204. doi: 10.1083/jcb.200705180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oh-Hashi K., Tanaka K., Koga H., Hirata Y., Kiuchi K. Intracellular trafficking and secretion of mouse mesencephalic astrocyte-derived neurotrophic factor. Mol. Cell Biochem. 2012;363:35–41. doi: 10.1007/s11010-011-1155-0. [DOI] [PubMed] [Google Scholar]
- 40.Glembotski C.C. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J. Mol. Cell Cardiol. 2011;51:512–517. doi: 10.1016/j.yjmcc.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Llewellyn D.H., Roderick H.L., Rose S. KDEL receptor expression is not coordinatedly up-regulated with ER stress-induced reticuloplasmin expression in HeLa cells. Biochem. Biophys. Res. Commun. 1997;240:36–40. doi: 10.1006/bbrc.1997.7607. [DOI] [PubMed] [Google Scholar]
- 42.Lampe A.K., Bushby K.M. Collagen VI related muscle disorders. J. Med. Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mellerio J.E., Eady R.A., Atherton D.J., Lake B.D., McGrath J.A. E210 K mutation in the gene encoding the beta3 chain of laminin-5 (LAMB3) is predictive of a phenotype of generalized atrophic benign epidermolysis bullosa. Br. J. Dermatol. 1998;139:325–331. doi: 10.1046/j.1365-2133.1998.02377.x. [DOI] [PubMed] [Google Scholar]
- 44.Tooley L.D., Zamurs L.K., Beecher N., Baker N.L., Peat R.A., Adams N.E., Bateman J.F., North K.N., Baldock C., Lamande S.R. Collagen VI microfibril formation is abolished by an {alpha}2(VI) von Willebrand factor type A domain mutation in a patient with Ullrich congenital muscular dystrophy. J. Biol. Chem. 2010;285:33567–33576. doi: 10.1074/jbc.M110.152520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baker N.L., Morgelin M., Pace R.A., Peat R.A., Adams N.E., Gardner R.J., Rowland L.P., Miller G., De Jonghe P., Ceulemans B., et al. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann. Neurol. 2007;62:390–405. doi: 10.1002/ana.21213. [DOI] [PubMed] [Google Scholar]
- 46.Wang J., Takeuchi T., Tanaka S., Kubo S.K., Kayo T., Lu D., Takata K., Koizumi A., Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J. Clin. Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hartley T., Siva M., Lai E., Teodoro T., Zhang L., Volchuk A. Endoplasmic reticulum stress response in an INS-1 pancreatic beta-cell line with inducible expression of a folding-deficient proinsulin. BMC Cell Biol. 2010;11:59. doi: 10.1186/1471-2121-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson R., Freddi S., Chan D., Cheah K.S., Bateman J.F. Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J. Biol. Chem. 2005;280:15544–15552. doi: 10.1074/jbc.M410758200. [DOI] [PubMed] [Google Scholar]
- 49.Wilson R., Freddi S., Bateman J.F. Collagen X chains harboring Schmid metaphyseal chondrodysplasia NC1 domain mutations are selectively retained and degraded in stably transfected cells. J. Biol. Chem. 2002;277:12516–12524. doi: 10.1074/jbc.M112044200. [DOI] [PubMed] [Google Scholar]
- 50.Dinser R., Zaucke F., Kreppel F., Hultenby K., Kochanek S., Paulsson M., Maurer P. Pseudoachondroplasia is caused through both intra- and extracellular pathogenic pathways. J. Clin. Invest. 2002;110:505–513. doi: 10.1172/JCI14386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashimoto Y., Tomiyama T., Yamano Y., Mori H. Mutation (D472Y) in the type 3 repeat domain of cartilage oligomeric matrix protein affects its early vesicle trafficking in endoplasmic reticulum and induces apoptosis. Am. J. Pathol. 2003;163:101–110. doi: 10.1016/S0002-9440(10)63634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmitz M., Becker A., Schmitz A., Weirich C., Paulsson M., Zaucke F., Dinser R. Disruption of extracellular matrix structure may cause pseudoachondroplasia phenotypes in the absence of impaired cartilage oligomeric matrix protein secretion. J. Biol. Chem. 2006;281:32587–32595. doi: 10.1074/jbc.M601976200. [DOI] [PubMed] [Google Scholar]
- 53.Whittaker C.A., Hynes R.O. Distribution and evolution of von willebrand/integrin a domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanrahan J.W., Sampson H.M., Thomas D.Y. Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol. Sci. 2013;34:119–125. doi: 10.1016/j.tips.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 55.Sampson H.M., Lam H., Chen P.C., Zhang D., Mottillo C., Mirza M., Qasim K., Shrier A., Shyng S.L., Hanrahan J.W., et al. Compounds that correct F508del-CFTR trafficking can also correct other protein trafficking diseases: an in vitro study using cell lines. Orphanet J. Rare Dis. 2013;8:11. doi: 10.1186/1750-1172-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu S., Butkevich A.N., Yamada R., Zhou Y., Debnath B., Duncan R., Zandi E., Petasis N.A., Neamati N. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc. Natl Acad. Sci. USA. 2012;109:16348–16353. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.