

Abstract
BACKGROUIND
Angiotensin-(1-7) [Ang-(1-7)] is an endogenous, heptapeptide hormone with anti-proliferative and anti-angiogenic properties. The primary objective of this study was to determine whether Ang-(1-7) effectively reduces prostate cancer metastasis in mouse xenografts.
METHODS
Human PC3 prostate cancer cells were injected into the aortic arch via the carotid artery of SCID mice pretreated with Ang-(1-7) or injected into the tibia of athymic mice, administered Ang-(1-7) for 5 weeks beginning 2 weeks post-injection. Tumor growth and volume were determined by bioluminescent and magnetic resonance imaging. The presence of tumors was confirmed by hematoxylin and eosin staining; TRAP histochemistry was used to identify osteolytic lesions. The effect of Ang-(1-7) on osteoclastogenesis was assessed in differentiated bone marrow cells.
RESULTS
Pre-treatment with Ang-(1-7) prevented metastatic tumor formation following intra-aortic injection of PC3 cells, while 83% of untreated mice developed tumors in metastatic sites. Circulating VEGF was significantly higher in control mice compared to mice administered Ang-(1-7). A five-week regimen of the heptapeptide hormone attenuated intra-tibial tumor growth; Ang-(1-7) was significantly higher in the tibia of treated mice than in control animals. Osteoclastogenesis was reduced by 50% in bone marrow cells differentiated in the presence of Ang-(1-7), suggesting that the heptapeptide hormone prevents the formation of osteolytic lesions to reduce tumor survival in the bone microenvironment.
CONCLUSIONS
These findings suggest that Ang-(1-7) may serve as an anti-angiogenic and anti-metastatic agent for advanced prostate cancer. By extension, the heptapeptide hormone may provide effective therapy for bone metastasis produced from primary tumors of the lung and breast.
Keywords: angiotensin-(1-7), metastatic prostate cancer, vascular endothelial growth factor, osteoclasts
INTRODUCTION
Prostate cancer is the most common non-cutaneous malignancy in developed countries and the third most common cause of death [1]. Standard of care for prostate cancer patients with localized, low grade tumors includes radical prostatectomy and/or radiation therapy, generally resulting in low mortality [2-4]. Androgen ablation, either by castration or administration of gonadotropin-releasing hormone analogs, is the primary treatment regimen for progressive prostate cancer. Unfortunately, the malignancy often reoccurs as hormone-refractory prostate cancer, limiting life expectancy to approximately 18 months with fewer than 20% of patients surviving more than three years. The leading cause of prostate cancer mortality is not the primary tumor burden but the metastatic spread and subsequent complications [5, 6]. One third of patients treated with initial conventional therapy develop metastases, [7] leading to palliative care as no curative regimens are available [8]. This indicates a clear need for therapeutics that not only target tumor epithelial cells but also the tumor microenvironment to reduce metastasis [9].
While prostate cancer metastasizes to lymph nodes, lung and liver, the predominant site of metastatic prostate cancer is bone; approximately 80 - 90% of patients with advanced prostate cancer have bone metastases [10]. Metastatic prostate cancer causes bone lesions that result in severe bone pain, bone fractures, hypercalcemia, spinal cord compression and/or bone demineralization [11]. Bisphosphonates, such as pamidronate and zoledronic acid, as well as RANKL (receptor activator of nuclear factor kB ligand) inhibitors, such as denosumab, a humanized monoclonal antibody to RANKL, are currently used to treat bone metastasis [12]. Both bisphosphonates and RANKL inhibitors reduce bone metastatic growth by interfering with the growth and differentiation of osteoclasts and the osteoclast-mediated process of bone resorption [13]. Unfortunately, bisphosphonate and RANKL inhibitors are associated with renal toxicity and osteonecrosis of the jaw, indicating a clear need for novel therapeutics that not only treat primary prostate cancer but also inhibit or prevent metastatic disease.
Angiotensin-(1-7) [Ang-(1-7)] is an endogenous, heptapeptide hormone of the reninangiotensin system. In previous studies, we showed that Ang-(1-7) reduced the growth of human lung tumor xenografts with a concomitant decrease in vascular endothelial growth factor (VEGF) and reduced vessel density as well as orthotopic human estrogen receptor positive or HER2 over-expressing breast tumor xenografts [14-17]. The decrease in breast tumor growth by Ang-(1-7) was associated with a reduction in tumor fibrosis; the heptapeptide hormone inhibited the proliferation and production of fibrotic proteins in cancer-associated fibroblasts isolated from orthotopic breast tumors [17]. These results suggest that Ang-(1-7) reduces tumor size by attenuating proliferation and angiogenesis as well as by regulating the growth and activity of stromal cells in the tumor microenvironment.
In our Phase I clinical trial assessing the toxicity and efficacy of Ang-(1-7) in patients with solid tumors, a significant reduction in circulating concentrations of the angiogenic protein placental growth factor (PlGF) was observed in four of fifteen evaluable patients that had stabilization of disease or a reduction in tumor measures [18]. No change in serum PlGF was observed in patients that did not display clinical benefit, supporting our preclinical studies demonstrating a reduction in tumor blood vessel formation following Ang-(1-7) administration. One patient with stable disease for over three months had metastatic prostate malignancy, suggesting that the heptapeptide hormone may serve as a targeted therapy for prostate cancer. In this study, we investigated the effect of Ang-(1-7) on metastatic prostate cancer, using mouse models of both intra-aortic and intra-tibial injection of PC3 human prostate cancer cells, to determine whether the heptapeptide hormone reduces growth of prostate cancer in bone and to identify the molecular mechanism for the inhibition of metastatic prostate cancer growth.
METHODS
Cell culture
PC3 cells (CRL-1435), obtained from American Tissue Culture Collection, were derived from a bone metastasis of a 62-year-old male Caucasian with grade IV prostatic adenocarcinoma. DU145 cells (HTB-81), also obtained from American Tissue Culture Collection, were derived from a brain metastasis of a 69-year-old male Caucasian with carcinoma of the prostate. PC3 cells were maintained in HyClone RPMI1640 medium with 10% fetal bovine serum (FBS), 100 μg/mL penicillin and 100 U/mL streptomycin; DU145 cells were grown in Eagle’s Minimum Essential Media (EMEM) with 10% FBS, penicillin and streptomycin. PC3 cells, following their transduction (PC3LUC) were maintained in penicillin/streptomycin-free media with blasticidin. All cells were grown at 37°C in a humidified atmosphere of 5% CO2; media and growth reagents were purchased from Gibco BRL.
Generation of PC3LUC cells
PC3 cells were infected with the UBC-GFPLuc lentivirus construct (expressing a GFP-Firefly luciferase fusion protein behind the human ubiquitin C promoter) created in-house using the Gateway system (Invitrogen). The GFPluc fusion protein was amplified by polymerase chain reaction from the pEGFPLuc vector (R&D Systems). Forty-eight hours post-infection, the cells were selected with 10 μg/mL blasticidin for 2 weeks to establish a stable cell line. The percentage of GFPLuc-positive cells was determined by flow cytometry; 97% of the cells expressed GFP. Before injection, the expression of luciferase was confirmed using a commercial Luciferase Assay System kit (Promega).
Quantification of cell proliferation
PC3 and DU145 cells were seeded at a density of 0.25 × 106 cells and 0.5 × 105, respectively, in 24-well plates in RPMI or EMEM, respectively, with 1% FBS. Cells were treated daily with 100 nM Ang-(1-7) in PBS [phosphate-buffered saline; 50 mM NaPO4 (pH 7.2), 100 mM NaCl] or PBS alone. Cells were harvested from triplicate wells every third day, as indicated, and counted using a hemocytometer, to quantify cell proliferation.
Western blot hybridization
Cells lysates were obtained by solubilizing monolayers in Triton lysis buffer [100 mM NaCl, 50 mM NaF, 5 mM EDTA, 1% Triton X-100, and 50 mM Tris-HCl (pH 7.4) containing 0.01 mM NaVO4, 0.1 mM phenylmethylsulfonyl fluoride, and 0.6 μM leupeptin], protein concentration in cell lysates was quantified, [19] and Western blot hybridization was preformed as previously described [17].
In vitro migration assay
Confluent monolayers of PC3LUC and DU145 cells were scraped down the center of a 35 or 100 mm tissue culture dish with a sterile micropipet tip to create a denuded zone of constant width. Cellular debris was removed with PBS and the adherent cells were incubated in media with PBS, 100 nM Ang-(1-7), or 100 nM Ang-(1-7) and 1 μM D-alanine7-Ang-(1-7) [D-Ala7-Ang-(1-7)], an Ang-(1-7) receptor antagonist, for 24 h. The tissue culture dishes were photographed prior to and 24 h after the initiation of treatment. The number of PC3 cells that migrated into the denuded area was counted and expressed as the number of cells/50 mm2. The number of pixels in a designated field of the denuded area covered by migrating DU145 cells was measured in Adobe Photoshop and expressed as the percentage of the total number of pixels in the designated field.
In vivo model of prostate cancer metastasis
Subcutaneous osmotic minipumps infusing 24 μg/kg/h of Ang-(1-7) were implanted on the backs of six-week-old male SCID mice or sham surgery was performed for control mice 2 days prior to injection of PC3LUC cells. Animals were anesthetized with 1% isoflurane; the carotid artery was approached through a midline incision over the trachea and throat of the mouse. The carotid artery was isolated and a catheter 14 mm long was introduced into the artery, insuring that the tip of the catheter extended down the carotid artery to the aortic arch. When the animal was in the prone position with its head extended, the catheter length was empirically determined to achieve placement of the catheter tip at the aortic arch. 1 × 106 PC3LUC cells in 100 μL of Hank’s buffered saline solution (HBBS) were injected into the aortic arch catheter using a 28 gauge needle. All procedures with mice were approved by the Institutional Animal Care and Use Committee.
Orthotopic intra-tibial model
PC3LUC cells were harvested from subconfluent cultures and resuspended in Ca+2- and Mg+2-free HBSS. Five to six week old male athymic mice maintained under pathogen-free conditions were anesthetized using 1% isoflurane. A 2-3 mm longitudinal incision was made over the mid-patellar region of the right hind limb. A percutaneous interosseal bore was made in the tibia using a 27 gauge needle. A fresh needle was inserted into the bone and a 20 μL suspension of 2 × 104 PC3LUC cells was injected into the middle of the tibial plateau. A cotton swab was held over the injection site for a minute to prevent leakage of cells into surrounding areas. Skin closure was performed with 5-0 coated vicryl suture. Beginning two weeks post-injection, the mice were treated for 5 weeks via subcutaneous osmotic minipumps with Ang-(1-7) at a dose of 24 μg/kg/h or sham surgery was performed for control mice.
Bioluminescence Image Analysis
PC3LUC tumors in the mouse flank or the bone were analyzed using the IVIS 100 bioluminescence imaging system (Xenogen). Briefly, the mice were injected by an intraperitoneal route with D-Luciferin (Xenogen) in PBS at a dose of 150 mg/kg. After 15 min to allow for distribution in conscious mice, the animals were anesthetized with isofluorane (2.5-3.5% in oxygen). The acquired imaging time was between one to five min per side (dorsal/ventral).
Magnetic resonance image (MRI) analysis
MR images were acquired on a 7.0T Biospec spectrometer (Bruker Biospin) equipped with a BGA-6S 60 mm inside diameter gradient coil capable of producing a maximum gradient of 1000 mT/m. Signal transmission and reception was performed with a 35 mm inside diameter RF volume coil. Before each imaging session, the mice were anesthetized with isoflurane (3% isoflurane, 3 L/min O2); body temperature and respiration were monitored. The presence and location of the intra-tibial tumors were confirmed by oblique sagittal T2 weighted spin echo Rapid Acquisition with Relaxation Enhancement (RARE) sequence with 8 echoes in the echo train and the following parameters: TR (Repetition time) = 3000 ms, TE (Echo time) = 56 ms, FOV (field of view) = 2.5 cm, Matrix = 256 × 256 (giving 98 μm in-plane resolution), slice thickness = 0.4 mm, NEX (number of excitation) = 20, for a total acquisition time of 24 min. Tumor volume was measured as the sum of the area of each slice with tumor times the slice thickness. Tumors in the mandible were confirmed by oblique sagittal T2 weighted spin echo Rapid Acquisition with Relaxation Enhancement (RARE) sequence with 8 echoes in the echo train and the following parameters: TR = 3600 ms, TE = 45 ms, FOV = 2.0 cm, Matrix = 256 × 256 (giving 98 μm in-plane resolution), slice thickness = 0.5 mm, NEX = 24, for a total acquisition time of 34 min. Tumor volume was measured by MRI (sum of the area of each slice with tumor) X (the slice thickness); no tumors were present in mice treated with Ang-(1-7). * denotes p<0.05, n=4.
Immunohistochemistry
Mouse legs were fixed in cold 4% paraformaldehyde and after 24 h, the muscle layers were excised from the bone. Bone specimens were decalcified in PBS with 10% EDTA for 14 d. The decalcified tibias, embedded in paraffin blocks, were cut into five micron sections and every fifth section was stained with hematoxylin and eosin (H & E) to detect PC3LUC tumors. Sections were also stained with affinity-purified Ang-(1-7) antibody as previously described [20] to confirm the presence of the heptapeptide hormone in the tibia. Tartrate-resistant alkaline phosphatase (TRAP) staining was performed to identify osteoclasts as described [21].
Quantification of VEGF by ELISA
Sub-confluent PC3 or DU145 cells were serum starved overnight and stimulated with 1% FBS with or without 100 nM Ang-(1-7). Media collected from PC3 or DU145 cells after 24 h or serum from treated mice (diluted 1:5) was used to determine VEGF and/or PlGF concentrations using Human VEGF or PlGF ELISA kits according to the manufacturer’s instructions (Peprotech).
In vitro osteoclast assay
Bone marrow cells extruded from the tibias of B6 mice were cultured overnight in αMEM with 10% FBS and 10 ng/mL macrophage-colony stimulating factor (MCSF; R&D Systems). M-CSF (20 ng/mL) and RANKL (100 ng/mL; Preprotech) were added to non-adherent cells to induce osteoclast formation [22]; 100 nM Ang-(1-7) or PBS was added daily. On day 6, cells were stained for TRAP [23]; TRAP-positive multinuclear cells (containing three or more nuclei) were quantified and expressed as the average of 4 fields (0.3 mm2) per well of an 8-well chamber slide. Bone marrow cells for each well were obtained from an individual animal.
Statistic
All data are presented as the mean ± SE. Statistical differences were evaluated by Student’s t test or by one-way ANOVA followed by Dunnett’s post hoc test. The criterion for statistical significance was set at p < 0.05.
RESULTS
Ang-(1-7) reduces prostate cancer cell growth and migration
The effect of Ang-(1-7) on serum-stimulated prostate cell growth was determined in PC3 and DU145 cells plated at low density ) in the presence of 1% FBS, as shown in Figures 1A and B. Ang-(1-7) was added to a final concentration of 100 nM and replaced daily due to degradation [24]. Serum caused a time-dependent increase in the total number of cells, while Ang-(1-7) reduced PC3 cell growth by 42.8% on day 7 and 31.1% on day 10. Serum also increased the total number of DU145 cells, which exhibit aggressive growth; however, Ang-(1-7) reduced DU145 cell growth by 25.5% on day 7.
Figure 1. Ang-(1-7) inhibits prostate cancer cell growth.
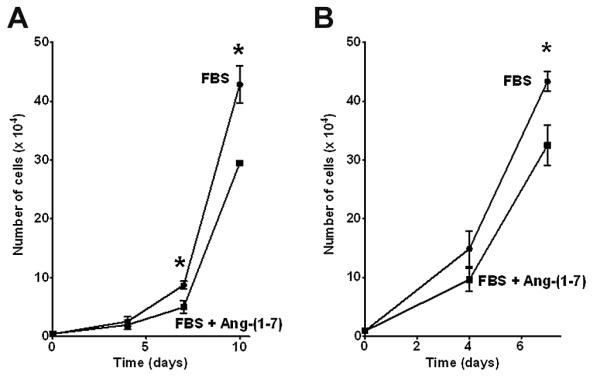
PC3 cells (Panel A) or DU145 cells (Panel B) were grown in the presence of 1% FBS or 1% FBS and 100 nM Ang-(1-7) [FBS + Ang-(1-7)]. Cell number was determined on the days indicated by counting the total number of cells per well. The data represent triplicate samples of cells from different passages. * denotes p<0.05, n=3.
Confluent monolayers of PC3LUC and DU145 cells were scraped with a micropipette tip, to evaluate the effect of Ang-(1-7) on cell migration. Cells in media alone (Control group) migrated into the “wounded” area during the subsequent 24 h incubation (Figure 2A-D). In contrast, treatment of cell monolayers with 100 nM Ang-(1-7) resulted in a significant reduction in cell migration—a 56.4% reduction in PC3 cell migration and a 43.4% reduction in DU145 cell migration. In both prostate cancer cell lines, the Ang-(1-7)-mediated decrease in cell migration was attenuated by the Ang-(1-7) receptor antagonist D-Ala7-Ang-(1-7), indicating that the reduction in cell migration was mediated by the AT(1-7) mas.
Figure 2. Ang-(1-7) inhibits prostate cancer cell migration.

Panels A and B. PC3 cells that migrated into the denuded zone were assessed after a 24 h incubation in media alone (Control, Panel A, upper panel), with 100 nM Ang-(1-7) (Panel A, middle panel) or with 100 nM Ang-(1-7) and 1 μM D-Ala7-Ang-(1-7), the AT(1-7) receptor antagonist [Ang-(1-7) + D-Ala, Panel A, lower panel]. The data were quantified as the number of cells/50 mm2 (Panel B). Ang-(1-7), A7; n=4, *** denotes p<0.001, comparing treatment with Ang-(1-7) to Control or treatment with Ang-(1-7) + D-Ala. Panels C and D. DU145 cells migrated into the denuded zone as sheets of cells in either media alone (Control, Panel C, upper panel), with 100 nM Ang-(1-7) (Panel C, middle panel) or with 100 nM Ang-(1-7) and 1 μM D-Ala7-Ang-(1-7) [Ang-(1-7) + DAla, Panel C, lower panel]. The data was calculated as the area covered by migrating cells as a percentage of the total denuded area (Panel D). n=3, ** denotes p<0.01.
Ang-(1-7) inhibits metastatic tumor growth
Injection of tumor cells into the left ventricle is a commonly used method to mimic extravasation of tumor cells from the circulation into metastatic sites. While this procedure results in tumor formed in the bone, tumors also are found in the thoracic space resulting from injury to the myocardium during the intraventricular injection and the release of cells into the thoracic space. In this study, cells were injected directly into the aortic arch through a catheter inserted into the carotid artery to prevent injury to the myocardium and deliver cells into the post-ventricular circulation. This method of delivery resulted in localized metastatic tumors in receptive skeletal sites.
Prior to the injection of tumor cells, four-week-old male SCID mice were randomly divided into 2 groups; one group was administered 24 μg/kg/h of Ang-(1-7) by subcutaneous infusion via osmotic minipumps and the other group received sham surgery. Two days after initiation of treatment, PC3LUC cells were injected into the aortic arch and imaged weekly to visualize tumor development (Figure 3A). The presence of cells in the circulation was confirmed by bioluminescent image analysis of the mice immediately post-surgery (Figure 3B). Six weeks after treatment, no detectable tumors were observed in mice treated with the Ang-(1-7) as determined by bioluminescent imaging (n = 6), whereas 83% of the control mice developed metastatic lesions in either the tibia, mandible or spine; one animal developed a tumor in the adrenal gland (Figure 3B, n = 6). The presence of tumors in the tibia, the mandible, the spine and the adrenal gland of untreated mice was confirmed by MRI, as shown in Figure 3C; no tumors were visible by MRI in similar areas of mice administered Ang-(1-7).
Figure 3. Ang-(1-7) inhibits metastatic tumor formation.

(Panel A) The diagram indicates the time line for studies to determine whether Ang-(1-7) reduces the formation of prostate tumors at metastatic target sites. (Panel B) A representative bioluminescence image of a mouse 30 min post-injection (left) demonstrates the distribution of PC3LUC cells. Representative bioluminescence images show tumors in control mice 6 weeks post-injection (upper right, 3 mice from a total of 6 animals); no bioluminescent signal was detected in mice treated with Ang-(1-7) 6 weeks post-injection (lower right, 3 mice from a total of 6 animals). (Panel C) Representative T2 MRI images demonstrate tumors at metastatic sites of control mice including the tibia, submandible, vertebral body and adrenal gland (circled in white).
Circulating VEGF was measured in control animals as compared to mice infused with Ang-(1-7), to determine whether the heptapeptide hormone reduced this pro-angiogenic factor. Circulating VEGF in untreated mice with prostate metastases averaged 261.8 ± 45.5 pg/mL (n = 6). In contrast, VEGF in mice administered Ang-(1-7) was below the detectable limit of the ELISA (n = 6), as shown in Figure 4A. In vitro incubation of PC3LUC cells with 100 nM Ang-(1-7) caused a significant decrease in secreted VEGF which was blocked by the Ang-(1-7) receptor antagonist D-Ala7-Ang-(1-7), as shown in Figure 4B. The proangiogenic factor PlGF was also significantly reduced in conditioned media from PC3LUC cells incubated overnight with Ang-(1-7), as shown in Figure 4C. DU145 prostate cancer cells derived from a brain metastasis were also incubated for 24 h with PBS or 100 nM Ang-(1-7); VEGF secreted into the media in cells treated with the heptapeptide hormone was significantly reduced compared to cells incubated with PBS, as shown in Figure 4D; PlGF secretion from DU145 cells was below the level of detection of the assay.
Figure 4. Ang-(1-7) reduces VEGF secretion in vivo and in vitro.

(Panel A) Circulating VEGF in the serum of control and Ang-(1-7)-treated mice was determined by ELISA. VEGF in the serum of mice treated with Ang-(1-7) [A7] was below the detectable limit of the assay. * denotes p<0.05, n=6. (Panel B) Sub-confluent PC3 cells were grown in media containing 1% FBS (Control), with 100 nM Ang-(1-7) [A7] or 100 nM Ang-(1-7) and 1 μM D-Ala7-Ang-(1-7), the AT(1-7) receptor antagonist [A7 + D-Ala] for 24 h. Secreted VEGF in the media were determined by ELISA. * denotes p<0.05, n=4. (Panel C) Sub-confluent PC3 cells were grown in media containing 1% FBS (Control) or with 100 nM Ang-(1-7). Secreted PlGF in the media were determined by ELISA. * denotes p<0.05, n=4. (Panel D) Sub-confluent DU145 cells were grown in media containing 1% FBS (Control) or with 100 nM Ang-(1-7). Secreted VEGF in the media was determined by ELISA. *** denotes p<0.001, n=6-7.
Ang-(1-7) reduces prostate tumor growth in the bone microenvironment
Since no tumors were detected in target tissues from mice administered Ang-(1-7) prior to injection of PC3LUC into the circulation, the cells were injected directly into the tibia to assess whether the heptapeptide hormone inhibits prostate cancer growth in the bone microenvironment. A percutaneous interosseal bore was made in the tibia and PC3LUC cells were injected into the middle of the tibial plateau. Two weeks post injection, mice were randomly divided into two groups and treated for 5 weeks with Ang-(1-7) at a dose of 24 μg/kg/h via subcutaneous osmotic minipumps; control mice were subjected to sham surgery. Tumor growth was measured weekly by bioluminescence image analysis (Figure 5A). PC3LUC cells were present in the tibias at the time of injection (Figure 5B). Control animals (n = 5) showed a time-dependent increase in bioluminescence throughout the 7-week treatment period, indicative of an increase in tumor size (Figure 5C). In contrast, the bioluminescent signal in the tibia of mice treated with Ang-(1-7) did not change (n = 5), suggesting a lack of tumor growth in the presence of the heptapeptide hormone.
Figure 5. Ang-(1-7) reduces intra-tibial prostate tumor growth.

(Panel A) The diagram shows the experimental design to determine if treatment with Ang-(1-7) regulates the growth of prostate tumors in the bone microenvironment. PC3LUC cells (2 × 105) were injected into the tibia of 4 to 5-week-old male athymic mice. Two weeks post-injection, mice were divided at random and treated by subcutaneous infusion of 24 μg/kg/h Ang-(1-7) via osmotic minipumps or subjected to sham surgery. Animals were imaged as indicated and euthanized after 7 weeks. (Panel B) Representative bioluminescence images of mice were taken on day 1 post injection (left image) or 7 weeks post injection (upper right, control mice, 2 mice from a total of 5; lower right, Ang-(1-7)-treated, 2 mice from a total of 5). (Panel C) Tumor size was measured by bioluminescence in control mice or mice treated with Ang-(1-7) and is presented in photons/sec/cm2/sterdian. * denotes p<0.05, n=5.
The tibias were imaged by MRI to determine tumor volumes in the bone. Tumors were visualized in the tibias of all control mice (Figure 6) and had an average volume of 1.29 mm3 ± 0.39 (n = 4). No tumors were detected in the tibia of mice administered Ang-(1-7). H&E staining of serial sections throughout the tibia of control mice confirmed the presence of the intra-tibial tumors, as shown in Figure 7A; abundant blood vessels were visible around the periphery of the tumors. Tibias from mice treated with Ang-(1-7) were cut into 5 micron longitudinal sections and every fifth section was stained with H & E in an attempt to detect even small clusters of PC3LUC cells. No PC3LUC cells were observed in any of the stained tibial sections from mice administered Ang-(1-7). These results suggest that Ang-(1-7) inhibited the growth of metastatic prostate tumors growing in the bone.
Figure 6. Ang-(1-7) reduces intra-tibial prostate tumor volume.

Representative MRI images show intra-tibial tumors (circled in white) at week 7 in control mice (upper image); no intratibial tumors were detected in mice treated with Ang-(1-7) (lower image).
Figure 7. Ang-(1-7) reduces intra-tibial prostate tumor growth and osteoclastogenesis in the bone microenvironment.

(Panel A) Mouse tibias were stained with H & E to visualize tumor morphology. Representative images show a tibia from a control mouse (top left) with tumor (T) surrounded by bone marrow (BM) and bone (B); blood vessels surrounding the tumor, labeled by arrows, are shown below (400X magnification). No tumors were observed in the tibias from mice treated with Ang-(1-7) (top right). (Panel B) Tibial sections from control mice (top left) and mice treated with Ang-(1-7) [top right] were stained with TRAP. Left inset – osteoclasts with multiple nuclei per cell and ruffled cell membranes (400X magnification); right inset - a few TRAP-positive cells in tibial sections from mice treated with Ang-(1-7) (400X magnification). (Panel C) Ang-(1-7) immunoreactivity in tibial sections from mice treated with the heptapeptide hormone (right) was compared to sections from control mice (left). (Panel D) Mouse bone marrow cells were differentiated with M-CSF and RANKL in the absence (Control) or presence of 100 nM Ang-(1-7); the peptide was added daily due to its degradation. The number of TRAP+, multinucleated osteoclasts was counted per field. * denotes p<0.05, n=5.
Sections of tibias from control mice were stained with tartrate resistant acid phosphatase (TRAP) to identify the presence of osteoclasts. Immunostained osteoclasts, with ruffled edges and multiple nuclei, were visible around the periphery of tumors from control mice, as shown in Figure 7B, characteristic of tumors observed in prostate cancer patients. A few TRAP-positive osteoclasts in concentrations expected for normal bone remodeling were observed throughout the tibia from the mice administered Ang-(1-7) (Figure 7B). Sections of tumors from control mice and mice treated with Ang-(1-7) were stained with an antibody to heptapeptide hormone. Positive immunoreactivity, due to endogenous Ang-(1-7), was visible in the tibia of control mice. However, a significant increase in Ang-(1-7) immunoreactivity was observed throughout the tibia of mice administered the heptapeptide hormone, as shown in Figure 7C, confirming the presence of exogenous Ang-(1-7) in the bone.
Ang-(1-7) reduces osteoclastogenesis of bone marrow cells
Bone marrow cells were isolated from mouse tibias and differentiated into osteoclasts in the presence Ang-(1-7), to determine the effect of the heptapeptide hormone on osteoclastogenesis. Bone marrow cells isolated from 4-week-old male B6 mice were treated overnight with M-CSF; subsequent non-adherent cells were incubated with M-CSF and RANKL, in the presence and absence of 100 nM Ang-(1-7), added daily due to degradation. On day 6, cells were stained for TRAP and TRAP-positive multinuclear cells (containing three or more nuclei) were quantified. Treatment with M-CSF and RANKL generated 78.6 ± 8.0 TRAP+, multi-nucleated cells/0.3 mm3. In contrast, the addition of Ang-(1-7) to the differentiation media of bone marrow cells significantly reduced the number of TRAP+, multi-nucleated cells greater than 50% (33.6 ± 4.5/0.3 mm3; p<0.01, n=4, as shown in Figure 7D). This suggests that the Ang-(1-7)-mediated inhibition of metastatic tumors following intra-tibial injection as well as the prevention of tumor formation following intra-aortic injection may be due, at least in part, to effects of the heptapeptide hormone on the bone microenvironment, to inhibit osteoclastogenesis.
DISCUSSION
The biochemical and physical properties of the bone provide a microenvironment that is ideal for tumor cell growth and colonization [25, 26]. Bone metastases occur in about 80 - 90% of prostate cancer patients with advanced disease, leading to a multitude of symptoms that adversely affect patient quality of life and survival [10]. The present study is the first demonstration of the inhibition of metastatic prostate cancer growth by the heptapeptide hormone, Ang-(1-7). Ang-(1-7) reduced the growth of PC3 prostate cancer cells derived from a bone metastasis and DU145 prostate cancer cells derived from a brain metastasis of prostate cancer patients; the heptapeptide hormone also reduced the growth of LNCaP prostate cancer cells (Krishnan, Gallagher and Tallant, unpublished results), demonstrating the anti-proliferative effect of Ang-(1-7) in three distinct prostate cancer cell lines. In addition, the heptapeptide hormone reduced migration in both PC3 and DU145 cells, an effect blocked by a specific Ang-(1-7) receptor antagonist. These results demonstrate that Ang-(1-7) inhibits prostate cancer cell growth and migration, through activation of the unique Ang-(1-7) receptor mas, suggesting that the heptapeptide hormone will reduce both the growth of the primary tumor as well as its migration to distant sites.
Tumors were formed in target metastatic sites of mice following injection of human prostate cancer cells into the aortic arch or by direct injection into the tibia, to determine the effect of the heptapeptide hormone on metastatic prostate cancer growth. However, no detectable tumors were observed in mice administered Ang-(1-7) following injection of tumor cells by either route. Circulating VEGF was significantly higher in untreated mice with tumors compared to mice treated with the heptapeptide hormone. A marked decrease in osteoclast activation was observed in differentiated bone marrow cells following incubation with Ang-(1-7), indicating that the heptapeptide hormone may attenuate angiogenesis as well as osteoclastogenesis to reduce bone metastasis. Taken together, these studies suggest that Ang-(1-7) may perturb the tumor microenvironment by multiple mechanisms to reduce or prevent prostate metastasis.
A solid tumor metastasis requires adequate vascularization during colonization to survive and grow in the bone metastatic site [27]. We and others previously showed that Ang-(1-7) has anti-angiogenic properties [16, 18, 28]. Ang-(1-7) reduced lung cancer xenograft volume with an associated decrease in vessel density and VEGF,[16] suggesting that the heptapeptide hormone attenuated tumor growth by preventing angiogenesis. In agreement, circulating PlGF was lower in patients with solid tumors displaying clinical benefit following Ang-(1-7) administration [18]. The heptapeptide hormone also attenuated human endothelial cell tubule formation, neovascularization in the chick chorioallantoic membrane,[16] and blood vessel density in a wound healing model,[28] further supporting the anti-angiogenic properties of Ang-(1-7). Ang-(1-7) reduced circulating VEGF in untreated mice with PC3 bone metastasis compared to mice treated with Ang-(1-7). In addition, VEGF and PlGF secretion from PC3 cells and VEGF production by DU145 cells was reduced by treatment with the heptapeptide hormone. In addition, we showed that Ang-(1-7) reduced angiogenesis in human LNCaP prostate cancer xenografts by decreasing the production of the angiogenic factors VEGF and PlGF as well as increasing sFlt-1, a decoy receptor that sequesters VEGF family ligands to prevent molecular signaling; both VEGF and PlGF secretion from LNCaP cells were also significantly reduced (Krishnan, Gallagher and Tallant, unpublished results). Since sFlt-1 sequesters both VEGF and PlGF, the increase in sFlt-1 may contribute to the reduction in VEGF and PlGF. In addition, Ang-(1-7) reduced HIF-1α in cobalt chloride-treated LNCaP cells (Krishnan, Gallagher and Tallant, unpublished studies), suggesting that the heptapeptide hormone may also reduce VEGF and PlGF at transcription, through effects on HIF-1α. These results demonstrate that Ang-(1-7) reduced the production of two different pro-angiogenic factors in three distinct prostate cancer cell lines (PC3, Du145 and LNCaP), in LNCaP orthotopic tumors and in the circulation of mice injected with PC3 cells and treated with Ang-(1-7). Taken together, our studies suggest that the heptapeptide hormone may prevent the formation of metastatic prostate tumors in part by reducing angiogenesis in the bone microenvironment.
Within the bone microenvironment, cytokines and growth factors secreted by prostate cancer cells cause dysregulation of normal bone homeostasis [29-31]. An overall increase in bone mass due to osteoblastogenesis generally is observed with prostate cancer metastases as well as an activation of osteoclasts resulting in pathological bone resorption. Angiogenesis plays a critical role in osteogenesis, such that the two processes are tightly regulated during bone growth, development, remodeling, and repair. Ang-(1-7) through a decrease in VEGF may reduce abnormal bone remodeling. VEGF is a potent regulator of osteoblast life-span in vitro, attenuating both spontaneous and pathological programmed cell death [32]. Mayer et al. demonstrated that increased expression of VEGF accelerated, while over-expression of sFlt-1 decreased, later stages of osteoblast differentiation and mineralization, suggesting that VEGF acts as an autocrine factor for osteoblast differentiation [33]. Osteoclast precursors are recruited to the site of bone resorption by VEGF and osteoclastogenesis is stimulated by VEGFR1 activation [34-36]. Conversely, VEGFR1-deficient mice have reduced numbers of osteoclasts and osteoblasts [36] and inhibition of VEGFR1 tyrosine kinase activity significantly reduced osteoblastic activity induced by prostate cancer cells injected into the bone [37]. VEGF121/rGel, a VEGF fusion construct composed of human VEGF121 and the highly cytotoxic plant toxin gelonin (rGel), reduced the number of mature osteoclasts at the tumor–bone interface in vivo [38]. VEGF in bone lesions is synthesized by both the metastatic prostate tumor and the surrounding stromal components [39]. VEGF produced by prostate cancer cells promotes osteoblastic activity [34] as well as facilitates tumor growth and osteolytic disease by enhancing osteoclast survival [40]. Osteoblasts release VEGF in response to a number of stimuli, including bone-derived cytokines and hypoxia, simulating bone injury [32] which enhances osteoclast differentiation and bone resorption [41]. Taken together, these studies provide strong support for a role of VEGF in the altered bone homeostasis found in metastatic lesions.
The observed decrease in circulating VEGF following Ang-(1-7) administration in part may reduce the abnormal bone remodeling that occurs following prostate cancer metastases. In support, we demonstrated a significant attenuation in osteoclastogenesis following incubation of bone marrow cells differentiated in the presence of Ang-(1-7). Studies are currently underway to determine whether the heptapeptide hormone inhibits VEGF synthesis by osteoblasts as well as reduces osteoblastogenesis. We also observe a significant reduction in both Flk-1 and Flt-1 with a concomitant 20-fold increase in sFlt-1 in the tumors of mice treated with Ang-(1-7) (Krishnan, Gallagher and Tallant, unpublished results). The inhibition of osteoclast differentiation by Ang-(1-7) could be due to the combined reduction of the anti-angiogenic factor VEGF as well as a decrease in membrane-bound VEGF receptors and an increase in sFlt-1.
No adverse reactions or gross pathological abnormalities were observed in the mice treated with Ang-(1-7), in agreement with previous studies in mice with lung or breast tumors [15, 16]. Based on favorable results and a low toxicity profile in our Phase I clinical trial,[18] a Phase II clinical trial in patients with metastatic or unresectable sarcoma is ongoing. Since Ang-(1-7) inhibited the growth of PC3 human prostate tumors in the bone of mice, the heptapeptide hormone may represent a novel treatment for metastatic prostate cancer. Pre-treatment of Ang-(1-7) before the addition of the tumor cells also blocked bone tumor formation in mice, suggesting that the heptapeptide hormone may also be an effective adjuvant therapy in patients with primary prostate cancer to prevent metastases. Other types of malignancies, particularly breast and lung cancer, also metastasize to bone. Since we previously showed that the Ang-(1-7) inhibits human lung tumor xenografts [15] and orthotopic human breast tumors [17] and has direct effects on the tumor microenvironment of the bone, to reduce angiogenesis and osteoclastogenesis, Ang-(1-7) may also be useful as adjuvant therapy for metastatic breast and lung cancer.
Conclusion
The leading cause of morbidity and mortality in patients with prostate cancer is metastatic bone disease and subsequent skeletal-related events that occur with disease progression. Over 80% of patients with advanced prostate cancer develop bone metastases, leading to palliative care, as no curative regimens are available. The results described in this paper provide pre-clinical data demonstrating the effectiveness of the heptapeptide hormone Ang-(1-7) for the treatment of metastatic prostate cancer. Since the actions of the heptapeptide hormone include a reduction in cell proliferation, angiogenesis and osteoclastogenesis, which are basic properties of many tumor types, it is expected that Ang-(1-7) would also decrease bone metastasis in lung and breast cancers. The reduction in activated osteoclasts by Ang-(1-7) also suggests that the heptapeptide hormone may provide effective therapy for bone diseases such as osteoporosis, in which osteoclastogenesis is a characteristic of the pathology.
ACKNOWLEDGMENTS
We thank J. Olsen, J. Cohen, T. Shields, R. Lanning, M. Landrum and H. Borgerink for technical assistance.
GRANT SUPPORT Funding was provided by the GT Cancer Fund, Wake Forest University Comprehensive Cancer Center, Golfers Against Cancer (Greensboro, NC), and the Farley-Hudson Foundation (Jacksonville, NC).
Footnotes
DISCLOSURE OF POTENTIAL CONFLICT OF INTEREST EA Tallant and PE Gallagher hold a patent for the treatment of cancer with Ang-(1-7). The other authors disclosed no potential conflict of interest.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Rove KO, Flaig TW. A renaissance in the medical treatment of advanced prostate cancer. Oncology (Williston Park) 2010;24:1308–1318. [PubMed] [Google Scholar]
- 3.Foley R, Marignol L, Keane JP, Lynch TH, Hollywood D. Androgen hypersensitivity in prostate cancer: molecular perspectives on androgen deprivation therapy strategies. Prostate. 2011;71:550–557. doi: 10.1002/pros.21266. [DOI] [PubMed] [Google Scholar]
- 4.Molina A, Belldegrun A. Novel therapeutic strategies for castration resistant prostate cancer: inhibition of persistent androgen production and androgen receptor mediated signaling. J Urol. 2011;185:787–794. doi: 10.1016/j.juro.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 5.Macfarlane RJ, Chi KN. Research in castration-resistant prostate cancer: what does the future hold? Curr Oncol. 2010;17(Suppl 2):S80–S86. doi: 10.3747/co.v17i0.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrissey C, Vessella RL. The role of tumor microenvironment in prostate cancer bone metastasis. J Cell Biochem. 2007;101:873–886. doi: 10.1002/jcb.21214. [DOI] [PubMed] [Google Scholar]
- 7.Fitzpatrick JM. The application of nanotechnology for the treatment of metastatic prostate cancer. BJU Int. 2009;104:i. doi: 10.1111/j.1464-410X.2009.08825.x. [DOI] [PubMed] [Google Scholar]
- 8.Msaouel P, Pissimissis N, Halapas A, Koutsilieris M. Mechanisms of bone metastasis in prostate cancer: clinical implications. Best Pract Res Clin Endocrinol Metab. 2008;22:341–355. doi: 10.1016/j.beem.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Storey JA, Torti FM. Bone metastases in prostate cancer: a targeted approach. Curr Opin Oncol. 2007;19:254–258. doi: 10.1097/CCO.0b013e32805e8787. [DOI] [PubMed] [Google Scholar]
- 10.American Cancer Society . Facts and Figures 2010. American Cancer Society; Atlanta, GA: 2010. pp. 22–37. [Google Scholar]
- 11.Macfarlane RJ, Chi KN. Novel targeted therapies for prostate cancer. Urol Clin North Am. 2010;37:105–19. doi: 10.1016/j.ucl.2009.11.011. Table. [DOI] [PubMed] [Google Scholar]
- 12.Body JJ. New developments for treatment and prevention of bone metastases. Curr Opin Oncol. 2011;23:338–342. doi: 10.1097/CCO.0b013e328347918b. [DOI] [PubMed] [Google Scholar]
- 13.Neville-Webbe HL, Holen I, Coleman RE. The anti-tumour activity of bisphosphonates. Cancer Treat Rev. 2002;28:305–319. doi: 10.1016/s0305-7372(02)00095-6. [DOI] [PubMed] [Google Scholar]
- 14.Gallagher PE, Tallant EA. Inhibition of human lung cancer cell growth by angiotensin-(1-7) Carcinogenesis. 2004;25:2045–2052. doi: 10.1093/carcin/bgh236. [DOI] [PubMed] [Google Scholar]
- 15.Menon J, Soto-Pantoja DR, Callahan MF, Cline JM, Ferrario CM, Tallant EA, Gallagher PE. Angiotensin-(1-7) inhibits growth of human lung adenocarcinoma xenografts in nude mice through a reduction in cyclooxygenase-2. Cancer Res. 2007;67:2809–2815. doi: 10.1158/0008-5472.CAN-06-3614. [DOI] [PubMed] [Google Scholar]
- 16.Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther. 2009;8:1676–1683. doi: 10.1158/1535-7163.MCT-09-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cook KL, Metheny-Barlow LJ, Tallant EA, Gallagher PE. Angiotensin-(1-7) reduces fibrosis in orthotopic breast tumors. Cancer Res. 2010;70:8319–8328. doi: 10.1158/0008-5472.CAN-10-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petty WJ, Miller AA, McCoy TP, Gallagher PE, Tallant EA, Torti FM. Phase I and pharmacokinetic study of angiotensin-(1-7), an endogenous antiangiogenic hormone. Clin Cancer Res. 2009;15:7398–7404. doi: 10.1158/1078-0432.CCR-09-1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lowry OH, Rosebrough MJ, Farr AL, Randall RJ. Protein measurement with the Folin-Phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 20.Brosnihan KB, Neves LA, Joyner J, Averill DB, Chappell MC, Sarao R, Penninger J, Ferrario CM. Enhanced renal immunocytochemical expression of ANG-(1-7) and ACE2 during pregnancy. Hypertension. 2003;42:749–753. doi: 10.1161/01.HYP.0000085220.53285.11. [DOI] [PubMed] [Google Scholar]
- 21.Erlebacher A, Derynck R. Increased expression of TGF-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol. 1996;132:195–210. doi: 10.1083/jcb.132.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coenegrachts L, Maes C, Torrekens S, Van LR, Mazzone M, Guise TA, Bouillon R, Stassen JM, Carmeliet P, Carmeliet G. Anti-placental growth factor reduces bone metastasis by blocking tumor cell engraftment and osteoclast differentiation. Cancer Res. 2010;70:6537–6547. doi: 10.1158/0008-5472.CAN-09-4092. [DOI] [PubMed] [Google Scholar]
- 23.Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, Yu-Lee LY, Zhang S, Yeh ET, Hu MC, Logothetis CJ, Lin SH. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res. 2008;6:1259–1267. doi: 10.1158/1541-7786.MCR-08-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension. 1998;31:362–367. doi: 10.1161/01.hyp.31.1.362. [DOI] [PubMed] [Google Scholar]
- 25.Guise T. Examining the metastatic niche: targeting the microenvironment. Semin Oncol. 2010;37(Suppl 2):S2–14. doi: 10.1053/j.seminoncol.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Zhang XH, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, Foekens JA, Massague J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- 28.Machado RD, Santos RA, Andrade SP. Opposing actions of angiotensins on angiogenesis. Life Sci. 2000;66:67–76. doi: 10.1016/s0024-3205(99)00562-7. [DOI] [PubMed] [Google Scholar]
- 29.Casimiro S, Guise TA, Chirgwin J. The critical role of the bone microenvironment in cancer metastases. Mol Cell Endocrinol. 2009;310:71–81. doi: 10.1016/j.mce.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Ibrahim T, Flamini E, Mercatali L, Sacanna E, Serra P, Amadori D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer. 2010;116:1406–1418. doi: 10.1002/cncr.24896. [DOI] [PubMed] [Google Scholar]
- 31.Sterling JA, Edwards JR, Martin TJ, Mundy GR. Advances in the biology of bone metastasis: how the skeleton affects tumor behavior. Bone. 2011;48:6–15. doi: 10.1016/j.bone.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Street J, Lenehan B. Vascular endothelial growth factor regulates osteoblast survival - evidence for an autocrine feedback mechanism. J Orthop Surg Res. 2009;4:19. doi: 10.1186/1749-799X-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayer H, Bertram H, Lindenmaier W, Korff T, Weber H, Weich H. Vascular endothelial growth factor (VEGF-A) expression in human mesenchymal stem cells: autocrine and paracrine role on osteoblastic and endothelial differentiation. J Cell Biochem. 2005;95:827–839. doi: 10.1002/jcb.20462. [DOI] [PubMed] [Google Scholar]
- 34.Dai J, Kitagawa Y, Zhang J, Yao Z, Mizokami A, Cheng S, Nor J, McCauley LK, Taichman RS, Keller ET. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004;64:994–999. doi: 10.1158/0008-5472.can-03-1382. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto Y, Okada Y, Fukushi J, Kamura S, Fujiwara T, Iida K, Koga M, Matsuda S, Harimaya K, Sakamoto A, Iwamoto Y. Role of the VEGF-Flt-1-FAK pathway in the pathogenesis of osteoclastic bone destruction of giant cell tumors of bone. J Orthop Surg Res. 2010;5:85. doi: 10.1186/1749-799X-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, Ikeda K, Shibuya M. VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci U S A. 2005;102:14016–14021. doi: 10.1073/pnas.0503544102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitagawa Y, Dai J, Zhang J, Keller JM, Nor J, Yao Z, Keller ET. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005;65:10921–10929. doi: 10.1158/0008-5472.CAN-05-1809. [DOI] [PubMed] [Google Scholar]
- 38.Mohamedali KA, Li ZG, Starbuck MW, Wan X, Yang J, Kim S, Zhang W, Rosenblum MG, Navone N. Inhibition of prostate cancer osteoblastic progression with VEGF121/rGel, a single agent targeting osteoblasts, osteoclasts, and tumor neovasculature. Clin Cancer Res. 2011;17:2328–2338. doi: 10.1158/1078-0432.CCR-10-2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haggstrom S, Bergh A, Damber JE. Vascular endothelial growth factor content in metastasizing and nonmetastasizing Dunning prostatic adenocarcinoma. Prostate. 2000;45:42–50. doi: 10.1002/1097-0045(20000915)45:1<42::aid-pros5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 40.Yang Q, McHugh KP, Patntirapong S, Gu X, Wunderlich L, Hauschka PV. VEGF enhancement of osteoclast survival and bone resorption involves VEGF receptor-2 signaling and beta3-integrin. Matrix Biol. 2008;27:589–599. doi: 10.1016/j.matbio.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Niida S, Kaku M, Amano H, Yoshida H, Kataoka H, Nishikawa S, Tanne K, Maeda N, Nishikawa S, Kodama H. Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J Exp Med. 1999;190:293–298. doi: 10.1084/jem.190.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]