

Abstract
Surfactant protein D (SP-D) is part of the innate immune system involved in lung homeostasis. SP-D knockout mice show accumulations of foamy alveolar macrophages, alveolar lipoproteinosis and pulmonary emphysema. Three single nucleotide polymorphisms (SNPs) have been described in the coding sequence of the human SP-D gene SFTPD. Clinical studies showed that the SNP SFTPD with a nucleotide change from A to C resulting in a Met to Thr substitution at position 11 in the protein (Met(11)Thr), is relevant. This study set out to create a humanised mouse model of the Met(11)Thr SNP. Transgenic mice lines expressing either Met(11) or Thr(11) SP-D under the control of the ubiquitously expressed pROSA26 promoter in C57Bl/6 SP-D deficient mice (DKO) was created. Both Met(11) (142 ± 52 ng mL−1) and Thr(11) (228 ± 76 ng mL−1) mice lines expressed human SP-D at almost similar levels. According to the literature this was within the range of SP-D levels found in wildtype (WT) mice (253 ± 22 ng mL−1). Met(11) or Thr(11) SP-D in serum from transgenic mice bound maltose in a calcium-dependent manner, and binding was inhibited in the presence of EDTA or maltose. Bronchoalveolar lavage showed for both transgenic mice lines complementation of the DKO phenotype by restoring cell counts, phospholipid levels and protein content back to WT levels. Cytospins of BAL pellet cells showed a resemblance to WT but both mice lines showed some foamy alveolar macrophages. The stereological analysis showed for none of the mice lines a complete abrogation of emphysematous alterations. However, both Met(11) and Thr(11) mice lines were partially reverted back to a WT phenotype when compared with DKO mice, indicating important effects on surfactant metabolism in vivo.
Keywords: oligomerisation, short nucleotide polymorphism, SP-D, stereology, transgenic mice
Introduction
Surfactant protein A (SP-A), SP-D and mannose binding lectin are proteins with collagenous and lectin domains belonging to the group of proteins called collectins. These are oligomerised molecules with four domains: (i) an N-terminal region with two cysteine residues involved in the final quaternary oligomerisation of the molecule, (ii) a collagenous region, (iii) a neck carbohydrate recognition domain (CRD) region where the initial oligomerisation between three polypeptide chains to form the trimeric unit takes place and (iv) the C-terminal CRD containing the C-type lectin domain responsible for molecule-binding to carbohydrates. SP-D from several species, including rat and humans, has been shown as four trimeric units oligomerised into a cross-like structure (Crouch et al. 1993a, 1994). SP-D has a molecular weight of 43 kDa under reduced conditions (Crouch et al. 1993a, 1994). Numerous in vitro and in vivo data show that these molecules are an important part of the innate immune system against potential pathogenic microorganisms, and SP-A and SP-D are also involved in normal lung homeostasis and anti-inflammatory properties, as reviewed recently (Pastva et al. 2007).
The gene for human SP-D SFTPD is encoded by 11 exons and located on the long arm of chromosome 10 (Crouch et al. 1993b). Short nucleotide polymorphisms (SNPs) have been found in three places on the translated exons, giving rise to altered amino acid residues in the mature protein (DiAngelo et al. 1999; Lahti et al. 2002). The Met11Thr SNP, with an AGT → ACT change in the N-terminal region, changes a methionine to a threonine residue at position 11 in the mature protein and the Ala160Thr (GCA → ACA) in the collagenous region, with a change of an alanine to a threonine in position 160 of the mature protein (DiAngelo et al. 1999; Lahti et al. 2002). Furthermore, at position 270, a TCT → ACT changes a serine residue to a threonine residue (Ser270Thr) in the CRD region (DiAngelo et al. 1999; Lahti et al. 2002). Several studies have shown that the short nucleotide polymorphism SFTPD aa11A→C in the gene for human SP-D that results in a change from methionine to a threonine residue in the human SP-D protein molecule has clinical implications, for example, being overrepresented in preterm infants with severe respiratory syncytial virus infections who are homozygous for Met/Met11allele. Also, there is an overrepresentation of individuals positive for a tuberculosis skin prick test and allergic rhinitis individuals that are homozygous for Thr/Thr11 allele compared with control groups (Floros et al. 2000; Lahti et al. 2002; Deng et al. 2009; Karjalainen et al. 2012). The Thr/Thr11 allele has also been associated with the development of multi-organ dysfunction syndrome and acute respiratory distress syndrome in a prospective and observational genetic study in patients with community-acquired pneumonia (García-Laorden et al. 2011). Another study investigated both exonic and intronic SNPs in the SP-D encoding gene region in four different studies of patients with chronic obstructive pulmonary disease (COPD; Foreman et al. 2011). The results showed an association with the SFTPD aa11A→C SNP (rs721917) in one of the four COPD studies investigated, but no statistical significance was found in the other three groups for this SNP (Foreman et al. 2011). Another report has studied bronchial asthma in a group of children but did not find any association between the disease and any of the three SNPs (Krueger et al. 2006).
Recently, it has become clear that the Met(11)Thr SNP of human SP-D influences the higher oligomeric state of SP-D. Gel exclusion chromatography analysis of amniotic fluid, bronchoalveolar lavage or human serum followed by fractional analysis using a sandwich human SP-D detecting ELISA showed that SP-D eluted as two peaks. Atomic force microscopy of SP-D purified from amniotic fluid from the two peaks showed that SP-D in the peak close to the void volume of the gel chromatography column was in a dodecameric or higher oligomerised state and hence was named ‘high form’ SP-D, whereas SP-D from the second peak showed only trimeric units of SP-D and therefore was named ‘low form’ SP-D (Leth-Larsen et al. 2005). When the gel exclusion chromatography analysis followed by the SP-D detecting ELISA was applied to human serum samples, SP-D from people homozygous for Met/Met11 SNP had a higher ratio of high : low peaks when compared with people homozygous for Thr/Thr11 SNP, which had a lower high : low ratio of the two SP-D forms (Leth-Larsen et al. 2005). Individuals with a heterozygous SNP (Met/Thr11) have a mix of the two forms (Leth-Larsen et al. 2005). Recombinant Met(11) or Thr(11) SP-D expressed in human HEK cells showed that recombinant Met(11) had a higher ratio of high : low forms of SP-D, whereas recombinant Thr(11)SP-D had a lower ration of high : low forms of SP-D, confirming that this single nucleotide substitution is directly involved in the oligomeric state of the human SP-D molecule (Leth-Larsen et al. 2005). However, the oligomeric state of the human SP-D molecule is not a static configuration but rather an equilibrium between the high and low forms, dependent on the local concentration of salt and pH (Sorensen et al. 2009).
We have generated a humanised mouse model of the Met(11)Thr SNP found in human SP-D to provide the platform for further study of how this SNP affects the observational and clinical findings mentioned above. The expression of SP-D is not restricted to the lung and can be found in numerous tissues with mucosal surfaces (Madsen et al. 2000; Akiyama et al. 2002). Other SP-D transgenic mouse lines have previously used the promoters for the human surfactant protein C gene or the rat Clara cell secretory protein gene, both of which restrict expression to lung epithelial cells (Fisher et al. 2000; Zhang et al. 2002; Kingma et al. 2006). To provide a mouse model that mimics the SP-D expression, the ubiquitous expressed promoter pROSA26 was used (Zambrowicz et al. 1997). Here we show data of the basic characterisation of the mouse model by bronchoalveolar lavages and design-based stereology of lungs of 24-week-old mice.
Materials and methods
Generation of the humanised Met(11)Thr mouse model
Surfactant protein D-deficient mice (Botas et al. 1998) were generated and housed in specific pathogen-free housing at the University of California San Francisco (UCSF) Laboratory Animal Resource Center and then transferred to the Biomedical Research Facility at University of Southampton (UoS), United Kingdom. Mice received sterile rodent chow and water ad libitum with a 12-h light and dark cycle. All animal procedures were approved by the Institutional Animal Care and Use Committee (UCSF) and the Home Office (UoS).
Plasmids containing either the Met(11) or the Thr(11) form of human SP-D were constructed previously for expression of the recombinant human Met(11) or Thr(11) SP-D proteins in HEK293 cells, respectively (Leth-Larsen et al. 2005). These plasmids were used as templates for PCR generation of either the Met(11) or the Thr(11) form of human SP-D with a 5′-BamH1 endonuclease restriction site and a 3′-Xba1 site. The ubiquitously expressed promoter ROSA26 was cloned into the pEGFP-N1 plasmid vector using Sal1 and BamH1 sites (a gift kindly provided by Dr Philippe Soriano, Mount Sinai School of Medicine, NY, USA). The GFP encoding segment was cut out of the vector using BamH1 and Xba1. The BamH1- and Xba1-digested SP-D fragments were cloned into the vector, transfected into Escherichia coli and the new vector containing either the Met(11) or the Thr(11) form of human SP-D was purified using the Maxiprep endotoxin free kit (Qiagen). The constructs were sequenced to verify the reading frame and the presence of the specific SNPs, respectively. Plasmids were cut with Sal1 and AfiII, respectively, to generate a fragment consisting of the ROSA26 promoter, the cDNA for either the Met(11) or the Thr(11) form of human SP-D, respectively, and the SV40 polyadenylation site. These fragments were injected directly into fertilised pro-nuclei eggs from the SP-D knock-out mice on the C57BL/6 background backcrossed at least 10 generations (Botas et al. 1998). Founders expressing either the Met(11) or the Thr(11) form of human SP-D, respectively, were identified by reduced SDS-PAGE and Western blotting of serum samples by cross-reacting to human SP-D monospecific polyclonal antibody raised against recombinant mouse SP-D as described previously (Botas et al. 1998). Founders were mated with SP-D knock-out mice and littermates carrying the transgene were identified by PCR genotyping amplifying a part of the neck and CRD of human SP-D using the following primers: HuTransSP-D F2: GTGTCGGGGAGAAGATTTTCAAG and HuTransSP-D R2: AGAACTCGCAGACCACAAGACG. The PCR programme was: one cycle of 94 °C for 1 min, followed by 35 cycles of: 94 °C for 30 s, 62 °C for 30 s and 72 °C for 30 s, followed by one cycle of 72 °C for 7 min. This gives rise to an amplicon of 250 bp.
Met(11) and Thr(11) serum SP-D-binding to maltose-agarose beads
A 1-μL aliquot of serum from either Met or Thr mice was incubated with 5 μL of maltose-agarose beads (Sigma-Aldrich) in a volume of 250 μL tris buffered saline (TBS) in the presence of 5 mm calcium chloride per tube. Increasing concentrations of maltose (5, 10, 20 and 40 mm, Sigma-Aldrich) were added to the tubes and one tube was incubated with 20 mm EDTA. All tubes were incubated on a rotator for 1 h at room temperature before being centrifuged at 15 g for 1 min to pellet beads. The beads were washed three times in TBS with 5 mm calcium chloride and after the last re-suspension, beads were transferred to a new tube. After pelleting beads by centrifugation these were re-suspended in SDS-PAGE loading buffer and reducing agent and analysed by SDS-PAGE and Western blotting as described above.
Gel permeation chromatography of mouse serum with human Met or Thr SP-D
Gel permeation chromatography was performed on 200-μL samples of mouse serum from either Met or Thr mice, respectively. The samples were applied to an analytical Superose six-column (10/30 column, GE Healthcare, Piscataway, NJ, USA) connected to a fast-performance liquid chromatography system (GE Healthcare) using TBS (pH 7.4) containing 10 mm EDTA and 0.05% emulphogene as eluent at a flow rate of 24 mL h−1 as described previously (Leth-Larsen et al. 2005). Fractions of 0.8 mL were collected, diluted 8× in TBS and analysed for the SP-D content using the human detecting SP-D ELISA as described previously (Leth-Larsen et al. 2003).
Bronchoalveolar lavage, cytospins and cell counts
The lungs of 24-week-old mice of each genotype [Met, Thr, wildtype (WT) and SP-D knock-out (DKO) mice] were lavaged four times with 1 mL of 20 mm Tris, 100 mm NaCl containing 0.2 mm EDTA, and 0.2 mm EGTA, pH 7.4, for analysis of cell counts, total protein amount and total phospholipids. The bronchoalveolar lavage (BAL) fluid was centrifuged at 140 g for 5 min at 4 °C. The cell pellet was gently resuspended in 200 μL lavage buffer for macrophage counting. Live cells were distinguished from dead cells by mixing a 1 : 1 volume with Trypan blue and counted using a haemocytometer. Cytospin slides were made using all cells from a single BAL from WT, Met and Thr mice, respectively, and no more than 200 000 cells from a single BAL from DKO mice. Cells were centrifuged onto glass slides at 140 g for 1 min at room temperature. Slides were air-dried and stained with Diff-Quik (Dade International, Miami, FL, USA) for light microscopy and photography.
Total phospholipid measurements from BALs were performed as previously described by Botas et al. (1998).
Human SP-D levels in transgenic Thr(11) and Met(11) mouse bronchoalveolar lavages
The level of human SP-D in BAL and from the transgenic Met(11) and Thr(11) mice, respectively, at 24 weeks of age was analysed by dot blot analysis using a polyclonal rabbit antibody raised against a recombinant fragment of human SP-D. The antibody has been used previously to detect human SP-D in samples by SDS-PAGE and Western blotting analysis (Jäkel et al. 2010; Duvoix et al. 2011a,b). Briefly, 1 μL of BAL was diluted in 99 μL TBS and transferred onto a nitrocellulose membrane (Bio-Rad, Hemel Hempstead, UK) using the Bio-Dot Apparatus (Bio-Rad). Known amounts of native maltose affinity chromatography purified SP-D (10–10 000 pg) from lung lavage obtained from patients with pulmonary alveolar proteinosis were also loaded onto each membrane to determine the linear range of the assay and to quantify the level of human SP-D in mouse BALs. The blots were blocked in 3% skim milk in TBS with 0.05% Tween20 (TBS/Tw) for 90 min before being incubated with 800 ng mL−1 of the polyclonal antibody overnight at room temperature on a rocking table. The membranes were washed three times in TBS/Tw for 30 min each, then incubated with 200 ng mL−1 of a goat anti-rabbit IgG antibody conjugated to HRP (Molecular Probes; Invitrogen) for 90 min before being washed again three times of 30 min each in TBS/Tw. Membranes were incubated with Lumingen TMA-6 reagent (Lumigen, Southfield, MI, USA) according to the manufacturer's instructions. The chemiluminescent signal was measured using the ChemiGenius (Syngene, Cambridge, UK) and the software genesnap (version 7.12; Syngene). The levels of SP-D were quantified using the programme genetools (version 4.02; Syngene).
Design-based stereology: sampling
All methods used in this study were based on the recently published ATS/ERS recommendations for quantitative assessment of lung structure (Hsia et al. 2010). Mice 24 weeks old (Met, Thr, WT and DKO mice) were killed by intraperitoneal phenobarbital injection, the thorax was opened and the lungs were instillation-fixed via a tracheal cannula at a hydrostatic pressure of 20 cm H2O using a mixture containing 1.5% paraformaldehyde/1.5% glutaraldehyde in Hepes buffer as described previously (Knudsen et al. 2007). After storage of the lungs in fresh fixative at 4 °C, the lungs were sampled for stereological analysis as described previously (Ochs, 2006; Weibel et al. 2007). In short, the total volume of the lungs was first measured by the fluid displacement method according to Archimedes' principle (Scherle, 1970). Afterwards, the lungs were embedded in agar and cut from base to apex into tissue slices of 1 mm thickness. Starting randomly with the first or second slice, every second slice was sampled for embedding in glycol methacrylate for light microscopy (Technovit 7100; Heraeus Kulzer, Wehrheim, Germany). The remaining slices were further sampled by projecting a point grid onto the slices and taking a sample from each position hit by one of the points. These samples were further embedded in Araldite® (SERVA Electrophoresis GmbH, Heidelberg, Germany) for transmission electron microscopy.
Stereological analysis
Systematic uniform random sampling was applied at all subsequent stages of microscopic analysis. Investigations were carried out using an Axioskop light microscope (Zeiss, Oberkochen, Germany) equipped with a computer-assisted stereology system (CAST 2.0; Olympus, Ballerup, Denmark) and a CM12 electron microscope (Philips, Eindhoven, Netherlands) equipped with a digital camera (Morad; Olympus Soft Imaging Systems, Münster, Germany), respectively.
Four tissue blocks embedded in glycol methacrylate were randomly chosen. From each tissue block, the first and third section of a consecutive row of 1.5-μm-thick sections was mounted on one glass slide and stained with orcein. Using the first section, the volume density of parenchyma within the lungs or of ductal and alveolar airspaces and septal wall tissue within parenchyma was determined by point counting. The surface area of alveolar epithelium was calculated by intersection counting (Weibel, 1979). The volume and surface densities were multiplied by the reference volume to obtain absolute values per lung, e.g. total volume of parenchyma [V (par, lung)] or total alveolar epithelial surface area per lung [S (alvepi, lung)]. Using a physical disector (Sterio, 1984) with a height of 3 μm the number of alveoli was determined (Hyde et al. 2004; Ochs et al. 2004b). By estimating the connectivity of the network of alveolar openings we established the Euler number from which the numerical density [NV (alv/par)] and the number [N (alv, lung)] of alveolar openings and, hence, the number of alveoli, could be calculated (Ochs et al. 2004b).
Four Araldite-embedded tissue blocks were randomly chosen for sectioning. From each tissue block, the first and fourth section of a consecutive row of 1 μm semithin sections was mounted on one glass slide and stained with toluidine blue. These sections were used as physical disector pairs with a disector height of 3 μm. At an objective magnification of 63× oil immersion, the numerical density [NV (type II/lung)], the number [N (type II, lung)] and the mean volume [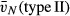 ] of alveolar type II cells based on nucleoli sampling were estimated (Ochs et al. 2004a). The number-weighted mean volume of AEII cells was estimated using the rotator for isotropic uniform random sections (Vedel-Jensen et al. 1993).
] of alveolar type II cells based on nucleoli sampling were estimated (Ochs et al. 2004a). The number-weighted mean volume of AEII cells was estimated using the rotator for isotropic uniform random sections (Vedel-Jensen et al. 1993).
After cutting of the semithin sections, ultrathin sections (40–60 nm) were prepared for transmission electron microscopy. At a primary magnification of 7100×, systematic uniform random sampling was performed to gather digital images of AEII cells which were subsequently used to estimate the volume of lamellar bodies per AEII cell [V (lb, type II)] by point counting (Weibel, 1979; Ochs, 2006).
Statistical analysis
Bronchoalveolar lavage characterisation values are presented as ‘mean’ and stereological data are given as ‘mean (standard deviation)’. Differences between groups were determined by one-way analysis of variance (anova) test using post hoc Bonferroni analysis with the predictive analytics software (pasw, version 18) for BAL data and prism graphpad for stereological data. Data were regarded as statistically significant when P < 0.05.
Results
Generation of the humanised Met(11)Thr mouse model
A humanised mouse model resembling the human SNP Met(11)Thr was created by generating mice expressing either the Met(11) or the Thr(11) form of human SP-D on the background of mice made deficient for mouse SP-D. Founders, expressing either the Met(11) or the Thr(11) form of human SP-D, respectively, were identified by SDS-PAGE and Western blotting of serum samples by cross-reacting to human SP-D monospecific polyclonal antibody raised against recombinant mouse SP-D (Fig. 1). The human SP-D in transgenic lines migrated with a molecular weight of ∼ 43 kDa in the reduced state (Fig. 1). No bands were seen in serum samples from wildtype or DKO mice (Fig. 1). Three lines for Met(11) [M(11)-line1, M(11)-line14 and M(11)-line20] and two lines for Thr(11) [T(11)-line135 and T(11)-line707] were identified in this way [M(11)-line14, M(11)-line20 and T(11)-line707 shown in Fig. 1]. The lines were mated with DKO mice and offspring carrying the construct was identified with PCR from genomic DNA combined with the SDS-PAGE and Western blotting analysis of serum (data not shown). M(11)-line1 and M(11)-line20 were not stable lines and the T(11)-line135 had lower SP-D levels compared with the T(11)-line707 (data not shown). The M(11)-line14 line (or Met mice in the remainder of the paper) and the Thr(11)-line707 (Thr mice, respectively) had the highest levels of human SP-D in BAL samples (see below) and offspring carrying either the Met(11) or the Thr(11) gene in these lines inherited in Mendelian proportions (data not shown). Real-time PCR analyses of liver, salivary gland and spleen also demonstrated expression of human SP-D in other organs than the lung.
Fig. 1.
SDS-PAGE and Western blotting of serum from transgenic founders M(11)-line 14, M(11)-line 20 and T(11)-line 707. Serum (1 μL) from each animal was analysed by reduced SDS-PAGE and Western blots by cross-reaction with human SP-D polyclonal rabbit anti-mouse SP-D antibody. WT, wild-type, DKO, SP-D deficient mice (SP-D−/−).
Met(11) and Thr(11) serum SP-D-binding to maltose-agarose beads
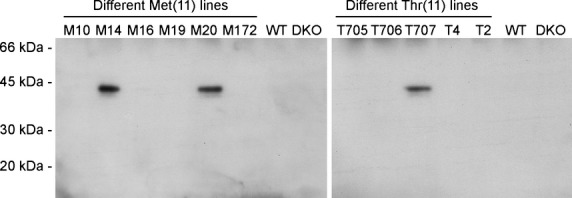
To ensure that the human SP-D expressed in the transgenic mice was folded correctly and thereby possessed normal biological activity, SP-D was tested in a binding assay to maltose-agarose beads in the presence of calcium (Fig. 2). Serum from each mouse line was incubated with maltose-agarose beads in the presence of calcium and increasing concentrations of maltose. The binding of human SP-D to maltose-agarose beads decreased in the presence of increasing concentrations of maltose and no binding was seen when 20 mm maltose or more was added (Fig. 2). Furthermore, no binding was observed between maltose-agarose beads and human SP-D in the presence of 10 mm EDTA (Fig. 2).
Fig. 2.
SDS-PAGE and Western blotting of sera from transgenic mice. Sera were mixed with increasing concentrations of maltose (mm) in the presence of calcium followed by addition of maltose beads. The beads were washed three times and analysed for human SP-D by SDS-PAGE and Western blotting by cross-reaction with human SP-D polyclonal rabbit anti-mouse SP-D antibody. (A) Serum from Met mice. (B) Serum from Thr mice. Experiments were performed three times with serum from different animals from each mouse line.
Gel permeation chromatography of serum from Met and Thr mice
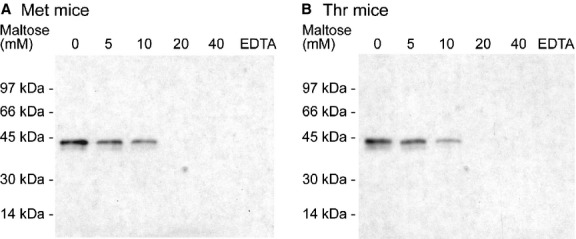
Gel permeation chromatography was performed on serum from Met or Thr mice. Fractions were collected, diluted and analysed for SP-D using a human SP-D detecting ELISA (Fig. 3). Met SP-D eluted as two peaks with a tall peak in the ‘high form’ of SP-D close to void volume of the column and followed by a smaller peak corresponding to the ‘low form’ of SP-D. Thr SP-D had a tall peak with the ‘high form’ but also a tall peak with the ‘low form’ of SP-D (Fig. 3). Quantification of the area under the curve for the two peaks revealed that the ratio of high : low forms of human SP-D was almost twice as high for the Met mice than for the Thr mice, with a ratio of 3.3 : 1 for Met mice and 1.8 : 1 for Thr mice.
Fig. 3.
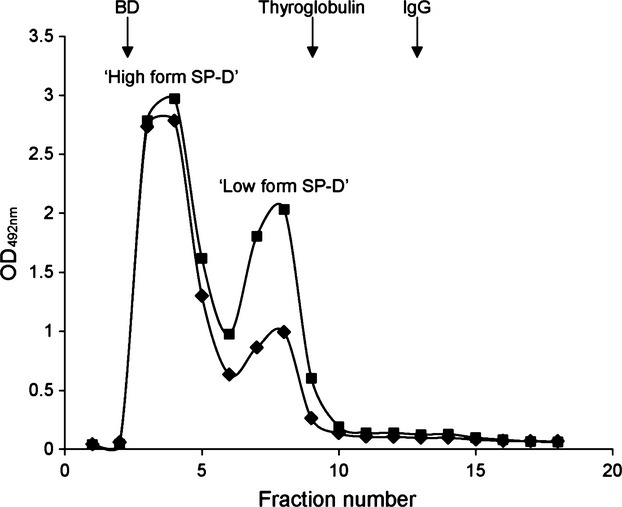
Gel permeation chromatography of mouse serum followed by the human SP-D detecting ELISA of all fractions. (♦) Met mice and (▪) Thr mice. This is a representative chromatogram of n = 3 mice per genotype. Blue dextran (BD): 2000 kDa, thyroglobulin: 670 kDa and IgG: 158 kDa.
Characterisation of bronchoalveolar lavage from 24-week-old mice
Bronchoalveolar lavages were performed on 24-week-old mice from each group and analysed by means of cell numbers, total phospholipids and total protein content using the anova post hoc Bonferroni test allowing comparison between all groups (Fig. 4A–C). Wildtype mice had a mean of 107 878 cells per BAL and similar numbers were observed in Met (111 028 cells per BAL) and Thr mice (113 073 cells per BAL), whereas DKO mice had statistically significant more cells per BAL (400 431 cells per BAL, P < 0.001) when compared with WT mice (Fig. 4A). The same pattern was observed in the total phospholipid content per BAL where the mean levels in WT, Met and Thr mice were similar (421, 420 and 382 μg per BAL, respectively), whereas DKO mice had statistically significantly higher mean levels [DKO: 1386 μg per BAL, P < 0.001 compared with WT mice (Fig. 4B)]. This pattern was also reflected in total protein levels, where WT, Met and Thr mice had similar mean levels (593, 843 and 764 μg per BAL, respectively), whereas DKO mice had statistically significantly higher mean levels [DKO: 1135 μg per BAL, P < 0.001 when compared with WT mice (Fig. 4B)].
Fig. 4.
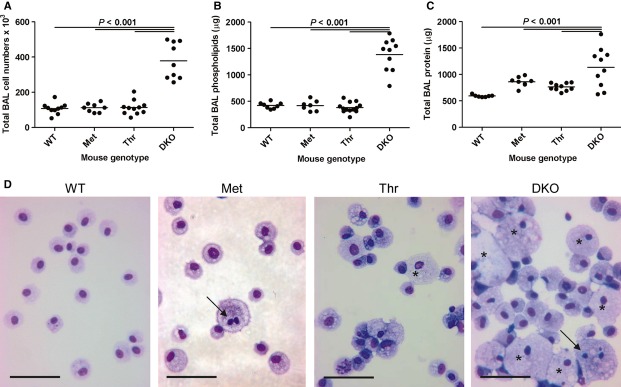
BAL analysis of mouse groups from 24-week-old mice. n = 7–12 mice per group. WT, wild-type; Met, Met(11) mice, Thr, Thr(11) mice; DKO, SP-D-deficient mice (SP-D−/−). Dot plots showing (A) number of BAL cells per mouse, (B) total BAL phospholipid per mouse, (C) total BAL protein per mouse. Line indicates mean of each mouse group. (D) Cytospins of cell pellets of BALs from 24-week-old mice. Arrows show multinucleated macrophages and asterisks show examples of heavy lipid-loaded macrophages. Scale bars: 50 mm. WT, wild-type; Met, Met(11) mice; Thr, Thr(11) mice; DKO, SP-D-deficient mice (SP-D−/−). All cells from a single BAL were used for cytospins, except for DKO mice, where 200 000 were used per slide to prevent dense crowding of cells on the glass slide.
Cytospins of cells from BALs were performed from all mouse groups (Fig. 4D). In all mouse groups, alveolar macrophages were the predominant cell type in BALs. In wildtype mice, the appearance of macrophages was normal, whereas in DKO mice there were cells were enhanced in number and size, lipid-loaded, with some multinucleated macrophages as well (Fig. 4D). Met and Thr mice resembled WT mice in terms of cell numbers. However, in Met and Thr mice some BAL macrophages were lipid-loaded and occasionally multinucleated macrophages were seen as well (Fig. 4D).
Human SP-D levels were measured in BAL from the 24-week-old mice. The transgenic mice have no endogenous mouse SP-D as they are made on the background of DKO mice. The level of SP-D in the mouse BAL was quantified by chemiluminescent dot blot analysis and quantified using a standard of native purified human SP-D. The analysis showed that Met mice had an average human SP-D level of 142.2 ± 51.6 ng mL−1 (mean ± SD) and Thr mice 228.2 ± 75.8 ng mL−1 (Fig. 5).
Fig. 5.
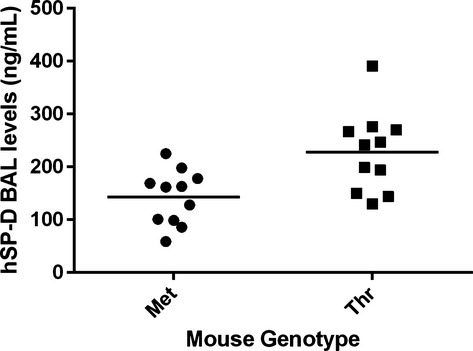
Quantification of human SP-D (hSP-D) levels in mouse BAL from 24-week-old mice. Met mice (n = 11 mice), Thr mice (n = 11). Dot plots showing the level of human SP-D in mouse BALs when analysed by chemiluminescent dot blot analysis. Vertical lines show the mean for each group.
Qualitative findings at light and electron microscopic level
Distal airspaces in WT lungs were well inflated and the septal walls were slim, indicating an appropriate fixation without any signs of cell swelling (Fig. 6A). Characteristic alterations of SP-D deficiency were observed in DKO (Fig. 6D). In some regions of lung parenchyma, the distal airspaces were enlarged and sometimes filled with foamy alveolar macrophages as well as intra-alveolar surfactant components. Occasionally, foamy alveolar macrophages were also seen in Thr and Met and, although less pronounced as in DKO, some regions of lung parenchyma demonstrated slight dilations of the distal airspaces compared with WT (Fig. 6B,C).
Fig. 6.
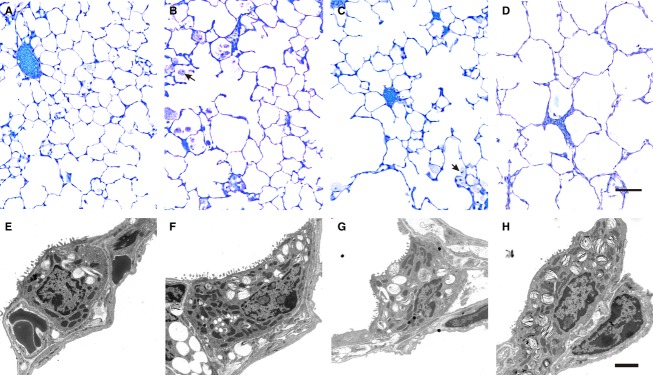
Microscopic images of lungs from 24-week-old mice. Light microscopic images of WT (A), Met (B), Thr (C) and DKO (D). Distal airspaces are enlarged in some regions in DKO. Slight rarification of septal wall tissue can also be recognised in Met and Thr, although to a much lesser degree than seen in DKO. Foamy alveolar macrophages are observed in both Met and Thr (arrows), and also in DKO. Scale bar: 100 μm. Representative electron microscopic images of AEII cells from WT (E), Met (F), Thr (G) and DKO (H). In general, the profiles of AEII cells were larger in DKO and contained more laminar bodies (LB) than AEII cells from WT lungs. The amount of LB within AEII cells from Met or Thr appeared to be reduced in both transgenic lines. Scale bar: 2 μm.
At the electron microscopic level, some AEII cells in DKO were completely filled with lamellar bodies; the amount of lamellar bodies per AEII cell appeared to be reduced as compared with DKO in the transgenic humanised groups but was still increased compared with WT (Fig. 6).
Design-based stereology
Stereological data are summarised in Table 1 and visualised in Fig. 7A–F. The current study confirms previous analyses demonstrating that DKO has an emphysema-like phenotype. The number of alveoli was slightly decreased, whereas the volume-weighted and number-weighted mean volume of alveoli increased in DKO compared with WT (Fig. 7A,B). In addition, the lung volume was higher in DKO, which was mostly a consequence of an increased volume of the distal airspaces. The surface density of the alveolar epithelium was decreased in DKO. The DKO mice had alveolar epithelial type II (AEII) cell hyperplasia and hypertrophy validated by a significantly larger number and number-weighted mean volume of AEII cells than found in the WT mice (Fig. 7C,D). The AEII cell hypertrophy resulted in part from an increased lamellar body volume per cell (Fig. 7E). Moreover, the total volume of lamellar bodies per lung was markedly increased (Fig. 7F). The transgenic mice (Met and Thr) were found to be intermediate between WT and DKO mice depending on the parameter being measured, with no clear difference between them (Fig. 7A–F and Table 1). None of the humanised transgenic mice demonstrated a complete abrogation of the emphysema-like phenotype. Alveolar numbers and sizes ranged between DKO and WT without being significantly different from either of these groups (Fig. 7A,B). The emphysema-like phenotype was thus attenuated in both humanised transgenic mice to a similar degree but not entirely corrected.
Table 1.
Summary of stereological data, grouped into parameters related to parenchymal architecture, type II cells and lamellar bodies
| Parameter | WT | DKO | Met | Thr |
|---|---|---|---|---|
| V (lung) [cm3] | 0.67 (0.04) | 1.01 (0.12)* | 0.87 (0.12) | 0.94 (0.13)‡ |
| VV (par/lung) [%] | 88.2 (3.8) | 87.8 (2.6) | 80.2 (4.1) | 87.7 (2.8) |
| N (alv, lung) 106 | 8.31 (0.8) | 6.42 (0.69)* | 7.68 (1.15) | 7.46 (0.66) |
| NV (alv/par) [1 mm−3] | 14 030.05 (732.98) | 7309.89 (732.98)* | 11 070.3 (1208.9)‡,§ | 9107.31 (625.72)‡ |
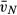 (alv) [103 μm3] (alv) [103 μm3] |
34.13 (4.02) | 63.08 (9.33)* | 54.37 (6.40)‡ | 51.23 (10.22)‡ |
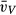 (alv) [103 μm3] (alv) [103 μm3] |
74.32 (13.88) | 152.81 (37.49) | 240.53 (93.70)‡ | 115.83 (32.93)** |
CV [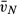 (alv)] (alv)] |
1.08 (0.23) | 1.18 (0.27) | 1.80 (0.34)‡,§ | 1.17 (0.30)** |
| S (alvepi, lung), cm2 | 469.50 (27.73) | 538.36 (44.91) | 457.31 (87.38) | 604.29 (59.13)‡,** |
| SV (alvepi/par) [1 cm−1] | 721.2 (102.87) | 614.84 (95.64)* | 652.6 (20.57)‡ | 736.55 (37.71) |
| V (alvair, lung), mL | 0.28 (0.03) | 0.40 (0.04)* | 0.40 (0.07)‡ | 0.38 (0.07) |
| V (ductair, lung), mL | 0.24 (0.05) | 0.38 (0.08)* | 0.22 (0.04)§ | 0.35 (0.08)** |
| V (sep, lung), mL | 0.06 (0.01) | 0.10 (0.01)* | 0.08 (0.02) | 0.09 (0.02) |
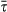 (sep) [μm] (sep) [μm] |
3.08 (0.73) | 3.42 (0.13) | 3.27 (0.19) | 2.93 (0.42) |
| N (type II, lung) [106] | 6.21 (0.53) | 12.27 (0.94)* | 9.63 (1.54)‡,§ | 10.48 (1.83)‡ |
| NV (type II/par) [103 mm−3] | 10.55 (0.91) | 14.08 (1.5)* | 13.84 (1.14)‡ | 12.72 (1.46) |
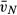 (type II) [μm3] (type II) [μm3] |
403.81 (20.0) | 598.29 (25.84)* | 516.8 (27.0)‡,§ | 524.68 (45.52)‡,¶ |
| VV (LB/type II) [%] | 22.0 (1.9) | 28.6 (2.1)* | 24.4 (3.1) | 22.7 (3.1)¶ |
| V (LB, type II) [μm3] | 89.0 (10.52) | 170.76 (7.51)* | 126.03 (16.82)‡,§ | 118.71 (17.57)‡,¶ |
| V (LB, lung) [mm3] | 0.55 (0.07) | 2.08 (0.12)* | 1.2 (0.12)‡,§ | 1.26 (0.36)‡,¶ |
WT, wild-type; Met, SP-D knock-out group expressing Met/Met11 human SP-D; Thr, SP-D knock-out group expressing Thr/Thr11 human SP-D; DKO, SP-D knock-out group; V, volume; VV, volume density; S, surface area; SV, surface area density; 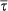 , mean thickness; N, number; NV, numerical density;
, mean thickness; N, number; NV, numerical density; 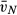 , number-weighted mean volume;
, number-weighted mean volume; 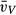 , volume-weighted mean volume; CV, coefficient of variation; par, parenchyma; alvair, alveolar airspace; ductair, ductal airspace; sep, septal tissue; alvepi, alveolar epithelium; alv, alveoli; typeII, type II cells; LB, lamellar bodies.
, volume-weighted mean volume; CV, coefficient of variation; par, parenchyma; alvair, alveolar airspace; ductair, ductal airspace; sep, septal tissue; alvepi, alveolar epithelium; alv, alveoli; typeII, type II cells; LB, lamellar bodies.
Statistically significant differences between groups are indicated as: *DKO vs. WT; †Met vs. WT; ‡Thr vs. WT; §Met vs. DKO; ¶Thr vs. DKO; **Thr vs. Met. Values are presented as mean (± SD) of n = 5 lungs.
Fig. 7.
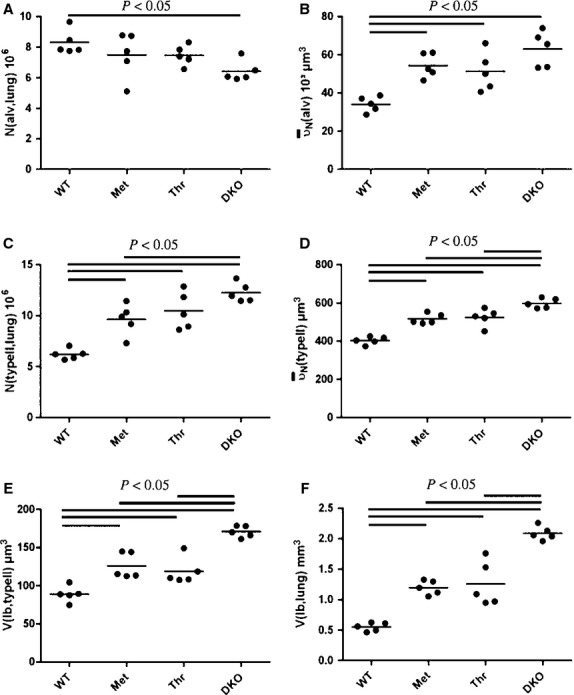
Individual stereological data presenting number of alveoli per lung (A), mean alveolar volume (B), total number of alveolar type II cells per lung (C), number-weighted mean volume of alveolar type II cells (D), mean volume of lamellar bodies per alveolar type II cell (E), and per whole lung (F).
Regarding the alterations of the AEII cells and the intracellular surfactant pool, partial reconstitution could be observed in both Met and Thr. Hypertrophy and hyperplasia of AEII cells, as characterised by the number-weighted mean volume and total number of AEII cells, were reduced to a similar degree as DKO mice (Fig. 7C,D). However, compared with WT, number and mean volume of AEII cells was still increased in Met and Thr. With respect to the intracellular surfactant pool, similar findings could be observed. The total volume of lamellar bodies per AEII cell and per lung was reduced in Met and Thr compared with DKO but was still increased compared with WT (Fig. 7E,F).
Discussion
Here we have created a humanised mouse model on the background of mice deficient in mouse SP-D, resembling the SNP Met(11)Thr found in the human population. Compared with results from the human population, Met(11) and Thr(11) SP-D in the transgenic mice demonstrated a similar behaviour regarding the degree of oligomerisation, with Met(11) SP-D having a higher high : low ratio than Thr(11) SP-D. Thus, these transgenic mice models have the potential to elucidate the pathophysiological relevance of the different gene-products using animal models of lung diseases. Expression of SP-D is not limited to the lung but can also be found in diverse other tissues/organs, as observed both in humans and mice (Madsen et al. 2000; Akiyama et al. 2002). Former studies used rat SP-D linked to a lung-specific promoter such as the surfactant protein C promoter to analyse the efficiency of different SP-D constructs in converting the phenotype related to SP-D deficiency to normal (Fisher et al. 2000; Kingma et al. 2006). To mimic the physiological expression of human SP-D in a more realistic way, we used the ubiquitously expressed promoter pROSA26. Hence, our model differs from previous models of SP-D gene-manipulated mice as it used SNP of human SP-D instead of rat SP-D and a ubiquitous promoter instead of a lung-specific promoter. The Met(11) and Thr(11) mice showed more WT characteristics compared with DKO mice but did not look fully rescued when considering the alveolar macrophages, as they exhibited signs of lipid load and occasionally multinucleated macrophages. In addition, neither the stereological parameters characterising the degree of the pulmonary emphysema nor the alterations of the AEII cells were completely normalised. We have no explanation for these results but one possibility could be the concentration of SP-D in the alveolar compartment. The level of mouse SP-D in BAL samples from C57Bl/6 wildtype mice has been quantified by ELISA with a standard of recombinant mouse SP-D, to 253 ± 22 ng mL−1 for a 3-mL BAL procedure (Gaunsbaek et al. 2013). Here we have used a 4-mL BAL procedure, which would result in a lower SP-D concentration. However, Hansen et al. (2008) have shown that there is a range in the SP-D concentration in BAL from various mouse strains (147–726 ng mL−1). Although both the dot blot analysis and the ELISA quantified the levels of SP-D using purified SP-D, these numbers should be viewed with caution as these were analysed using different antibodies and compared with standards using SP-D from different species and sources (recombinant vs. native SP-D). However, we show here that human SP-D is sufficient for a partial rescue of the phenotype of a DKO mouse and converts it to a partial WT phenotype. Transgenic mice deficient in mouse SP-D but expressing rat SP-D, which was reverted back to a wildtype phenotype, was found to have a level of rat SP-D in their BAL ∼ 20–30× higher than in wildtype mice (Fisher et al. 2000). One possible explanation for the not completely restored phenotype of the Met and Thr transgenic mice could be that human SP-D is not capable of restoring the WT phenotype. Mouse SP-D and rat SP-D show 92% identity at the overall protein level, whereas human SP-D and mouse SP-D only show an overall identity of 76% (Motwani et al. 1995). We have previously shown that intranasal treatment of DKO mice with a high dose (10 μg) of a recombinant fragment of human SP-D lead to a reduction in the degree of emphysema and a correction of type II cell hyperplasia and hypertrophy when compared with non-treated DKO mice (Knudsen et al. 2007). However, the treated DKO mice still had non-significant alterations in the stereological parameters when compared with WT mice (Knudsen et al. 2007), similar to what we observed here. This raises the questions of whether human SP-D is capable of entirely replacing mouse SP-D functions in vivo and what doses of human SP-D would be required to convert DKO mice back to a completely restored WT phenotype.
The ROSA26 promoter has previously been used for successful expression of several transgenic genes in lung tissue (Wang et al. 2008; Johnson et al. 2012). The construct, with expression of human SP-D driven by the ROSA26 promoter, was successful, as it resulted in high levels of human SP-D in serum from the mice (Fig. 1) and expression, as measured by real-time PCR, was also seen in other tissues such as liver, salivary gland and spleen (Madsen J, unpublished data). In addition, the high to low ratio of serum SP-D as found in human blood samples (Leth-Larsen et al. 2005) could be mimicked by this expression system (Fig. 3). The level of SP-D in the serum in these mice is higher than seen in humans, as 1 μL of serum gave a clear signal in Western blotting (Fig. 1), whereas this was not observed in human plasma samples when using the same sample volume (Madsen J, unpublished results). No side effects from local high levels of SP-D have been reported and another transgenic mouse line expressing rat SP-D in the lung was found to have 30–50 times higher SP-D levels compared with wildtype mice without having any effect on lung morphology (Fisher et al. 2000). The level of SP-D in the lung is critical for the phenotype of the mice. We have observed in another mouse line expressing Thr(11) SP-D, where the SP-D BAL level was found to be four times lower than seen in the Thr(11) mice in this report, that these mice had a phenotype resembling DKO mice with an increased number of macrophages in BAL, higher phospholipids and proteins levels and enlarged distal airspaces with a decreased number of alveoli (Knudsen L & Madsen J, unpublished results). This has also previously been observed in other SP-D transgenic mice where mice with low levels of rat SP-D in BAL showed a resemblance to DKO mice (Fisher et al. 2000). However, no previous reports have quantified the level of SP-D required for a mouse to revert from a DKO phenotype to a wildtype phenotype, or the levels required when substituting SP-D from one species with SP-D from another species.
Several reports have found that individuals with Thr/Thr11 have lower serum levels of SP-D than people with Met/Met11 SP-D (Leth-Larsen et al. 2005; Foreman et al. 2011). However, the strongest influence on the level of SP-D in serum has recently been found to be a non-coding SNP (rs1885551nn) located in the promoter region of human SP-D gene; however, no linkage disequilibrium map was established for the identified promoter SNP and the Thr/Thr11 SNP in the study (Foreman et al. 2011). This could mean that a subset of people with the Thr/Thr11 SNP and the non-coding SNP in the promoter region would potentially have very low serum concentrations of SP-D and be more susceptible to certain infections than would people not having this combination. Several association studies have investigated genotypes of serum mannose-binding lectin (MBL), levels of MBL and different diseases including virus infections such as HIV infection (recently reviewed by Heitzeneder et al. 2012). There is no clear picture as to whether low levels of MBL in general are disadvantageous or beneficial for an individual but the emerging picture is that this should be put into a disease-specific context for every single case. This is probably the same picture emerging for human SP-D serum levels combined with the oligomeric state of the human SP-D molecule. A specific SP-D serum level and oligomeric state could therefore be advantageous in one particular disease but disadvantageous in another disease. Gel permeation chromatography of mouse serum showed that Met mice had approximately twice as much of the ‘high form’ of SP-D compared with Thr mice. This is in accordance with the literature on what was observed in human serum for people homozygous for the Met or Thr SNP (Leth-Larsen et al. 2005). The Thr mice here had more of the high form/oligomerised form of SP-D than seen in human serum. This could be due to differences in serum between mouse and human serum, as the oligomeric structure of human SP-D has also been found to be dependent on salt and pH conditions (Sorensen et al. 2009).
In unchallenging conditions, transgenic mice expressing either the Met(11) or the Thr(11) SNP have the same ability to attenuate the phenotype of SP-D deficient mice as shown by means of biochemical and stereological data in the present study. However, clinical studies showing associations of these SNPs with several diseases suggest differential potencies of Met(11) and Thr(11) under infectious (Floros et al. 2000; Lahti et al. 2002; Karjalainen et al. 2012) or chronic inflammatory conditions (Deng et al. 2009; Foreman et al. 2011). To date there is no mechanistic explanation for why preterm infants with a Met/Met11 genotype are more susceptible to infections with respiratory syncytial virus (RSV) or why individuals with the Thr/Thr11 genotype are overrepresented in patients with allergic rhinitis compared with a control population (Lahti et al. 2002; Deng et al. 2009). SP-D has been shown to augment bacterial association but attenuate major histocompatibility complex class II presentation of bacterial antigens in alveolar macrophages and dendritic cells, and to inhibit proliferation of T lymphocytes and their production of interleukin (IL)-2 (Borron et al. 1998; Hansen et al. 2007). This highlights not only the important role of SP-D in the primary infection of an individual but also the possible effect on the immune response in the ensuing infections. How this SNP influences the adaptive immune system remains to be investigated. For clarification of the specific roles of the different gene-products of the Met11Thr SNPs of SP-D, these transgenic humanised mice (Met and Thr) represent a novel approach to be studied in animal models of different infectious or inflammatory diseases in the future.
Conclusion
We generated and thoroughly characterised a transgenetic mouse model, expressing clinical relevant SNPs of human SP-D in the lung. Both genotypes Met/Met11 and Thr/Thr11 demonstrated a similar potency to attenuate pathological alterations resulting from SP-D deficiency under unchallenging conditions. These mice models have the potential to elucidate in the future the pathophysiological relevance of the different gene products, including the observed different oligomerisation state (high : low ratio) in mice models of human lung diseases.
Acknowledgments
The authors thank Beat Haenni (University of Bern) and Marita Peter (Hannover Medical School) for excellent technical assistance. This work was supported by grants from the Wessex Medical Research (J.M.), the Alfred Benzon Foundation (J.M.) and the German Research Foundation DFG (SFB587-B18) (L.K. and M.O.).
Authors' contributions
L.K. participated in the design of the study, performed the design-based stereology, interpretation of results and drafted the manuscript. K.O. and L.A. performed the design-based stereology and interpretation of results. I.T. participated in the design of the study, performed the gel filtration and ELISA, and interpretation of results. R.M.M. participated in the design of the study, performed some of the BALs and interpretation of results. G.L.S., H.W.C. and U.H. participated in the design of the study and interpretation of results. M.O. participated in the design of the study, interpretation of results and drafted the manuscript. J.M. participated in the design of the study, performed the construction of the mice, the genotyping of the mice, the BAL analysis, the interpretation of results and drafted the manuscript. All authors read and approved the final manuscript.
Disclosures
The authors have no conflicts of interest.
References
- Akiyama J, Hoffman A, Brown C, et al. Tissue distribution of surfactant proteins A and D in the mouse. J Histochem Cytochem. 2002;50:993–996. doi: 10.1177/002215540205000713. [DOI] [PubMed] [Google Scholar]
- Borron PJ, Crouch EC, Lewis JF, et al. Recombinant rat surfactant-associated protein D inhibits human T lymphocyte proliferation and IL-2 production. J Immunol. 1998;161:4599–4603. [PubMed] [Google Scholar]
- Botas C, Poulain F, Akiyama J, et al. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc Natl Acad Sci U S A. 1998;95:11869–11874. doi: 10.1073/pnas.95.20.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch E, Persson A, Chang D. Accumulation of surfactant protein D in human pulmonary alveolar proteinosis. Am J Pathol. 1993a;142:241–248. [PMC free article] [PubMed] [Google Scholar]
- Crouch E, Rust K, Veile R, et al. Genomic organization of human surfactant protein D (SP-D). SP-D is encoded on chromosome 10q22.2-23.1. J Biol Chem. 1993b;268:2976–2983. [PubMed] [Google Scholar]
- Crouch E, Persson A, Chang D, et al. Molecular structure of pulmonary surfactant protein D (SP-D) J Biol Chem. 1994;269:17311–17319. [PubMed] [Google Scholar]
- Deng YQ, Tao ZZ, Kong YG, et al. Association between single nucleotide polymorphisms of surfactant protein D and allergic rhinitis in Chinese patients. Tissue Antigens. 2009;73:546–552. doi: 10.1111/j.1399-0039.2009.01232.x. [DOI] [PubMed] [Google Scholar]
- DiAngelo S, Lin Z, Wang G, et al. Novel, non-radioactive, simple and multiplex PCR-cRFLP methods for genotyping human SP-A and SP-D marker alleles. Dis Markers. 1999;15:269–281. doi: 10.1155/1999/961430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvoix A, Mackay RM, Henderson N, et al. Physiological concentration of calcium inhibits elastase-induced cleavage of a functional recombinant fragment of surfactant protein D. Immunobiology. 2011a;216:72–79. doi: 10.1016/j.imbio.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Duvoix A, Miranda E, Perez J, et al. Evaluation of full-length, cleaved and nitrosylated serum surfactant protein D as biomarkers for COPD. COPD. 2011b;8:79–95. doi: 10.3109/15412555.2011.558542. [DOI] [PubMed] [Google Scholar]
- Fisher JH, Sheftelyevich V, Ho YS, et al. Pulmonary-specific expression of SP-D corrects pulmonary lipid accumulation in SP-D gene-targeted mice. Am J Physiol Lung Cell Mol Physiol. 2000;278:L365–L373. doi: 10.1152/ajplung.2000.278.2.L365. [DOI] [PubMed] [Google Scholar]
- Floros J, Lin HM, Garcia A, et al. Surfactant protein genetic marker alleles identify a subgroup of tuberculosis in a Mexican population. J Infect Dis. 2000;182:1473–1478. doi: 10.1086/315866. [DOI] [PubMed] [Google Scholar]
- Foreman MG, Kong X, DeMeo DL, et al. Polymorphisms in surfactant protein-D are associated with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2011;44:316–322. doi: 10.1165/rcmb.2009-0360OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Laorden MI, Rodríguez de Castro F, Solé-Violán J, et al. Influence of genetic variability at the surfactant proteins A and D in community-acquired pneumonia: a prospective, observational, genetic study. Crit Care. 2011;15:R57. doi: 10.1186/cc10030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunsbaek MQ, Rasmussen KJ, Beers MF, et al. Lung surfactant protein D (SP-D) response and regulation during acute and chronic lung injury. Lung. 2013;191:295–303. doi: 10.1007/s00408-013-9452-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S, Lo B, Evans K, et al. Surfactant protein D augments bacterial association but attenuates major histocompatibility complex class II presentation of bacterial antigens. Am J Respir Cell Mol Biol. 2007;36:94–102. doi: 10.1165/rcmb.2006-0195OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S, Schmidt V, Steffensen MA, et al. An enzyme-linked immunosorbent assay (ELISA) for quantification of mouse surfactant protein D (SP-D) J Immunol Methods. 2008;330:75–85. doi: 10.1016/j.jim.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Heitzeneder S, Seidel M, Förster-Waldl E, et al. Mannan-binding lectin deficiency - Good news, bad news, doesn't matter? Clin Immunol. 2012;143:22–38. doi: 10.1016/j.clim.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Hsia CC, Hyde DM, Ochs M, et al. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde DM, Tyler NK, Putney LF, et al. Total number and mean size of alveoli in mammalian lung estimated using fractionator sampling and unbiased estimates of the Euler characteristic of alveolar openings. Anat Rec A Discov Mol Cell Evol Biol. 2004;277:216–226. doi: 10.1002/ar.a.20012. [DOI] [PubMed] [Google Scholar]
- Jäkel A, Clark H, Reid KB, et al. Surface-bound myeloperoxidase is a ligand for recognition of late apoptotic neutrophils by human lung surfactant proteins A and D. Protein Cell. 2010;1:563–572. doi: 10.1007/s13238-010-0076-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Hemnes AR, Perrien DS, et al. Cytoskeletal defects in Bmpr2-associated pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2012;302:L474–L484. doi: 10.1152/ajplung.00202.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karjalainen MK, Huusko JM, Tuohimaa A, et al. A study of collectin genes in spontaneous preterm birth reveals an association with a common surfactant protein D gene polymorphism. Pediatr Res. 2012;71:93–99. doi: 10.1038/pr.2011.2. [DOI] [PubMed] [Google Scholar]
- Kingma P, Zhang L, Ikegami M, et al. Correction of pulmonary abnormalities in Sftpd−/− mice requires the collagenous domain of surfactant protein D. J Biol Chem. 2006;281:24496–24505. doi: 10.1074/jbc.M600651200. [DOI] [PubMed] [Google Scholar]
- Knudsen L, Ochs M, Mackay R, et al. Truncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient mice. Respir Res. 2007;8:70. doi: 10.1186/1465-9921-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger M, Puthothu B, Gropp E, et al. Amino acid variants in Surfactant protein D are not associated with bronchial asthma. Pediatr Allergy Immunol. 2006;17:77–81. doi: 10.1111/j.1399-3038.2005.00353.x. [DOI] [PubMed] [Google Scholar]
- Lahti M, Lofgren J, Marttila R, et al. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res. 2002;51:696–699. doi: 10.1203/00006450-200206000-00006. [DOI] [PubMed] [Google Scholar]
- Leth-Larsen R, Nordenbaek C, Tornoe I, et al. Surfactant protein D (SP-D) serum levels in patients with community-acquired pneumonia. Clin Immunol. 2003;108:29–37. doi: 10.1016/s1521-6616(03)00042-1. [DOI] [PubMed] [Google Scholar]
- Leth-Larsen R, Garred P, Jensenius H, et al. A common polymorphism in the SFTPD gene influences assembly, function, and concentration of surfactant protein D. J Immunol. 2005;174:1532–1538. doi: 10.4049/jimmunol.174.3.1532. [DOI] [PubMed] [Google Scholar]
- Madsen J, Kliem A, Tornoe I, et al. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J Immunol. 2000;164:5866–5870. doi: 10.4049/jimmunol.164.11.5866. [DOI] [PubMed] [Google Scholar]
- Motwani M, White RA, Guo N, et al. Mouse surfactant protein-D. cDNA cloning, characterization, and gene localization to chromosome 14. J Immunol. 1995;155:5671–5677. [PubMed] [Google Scholar]
- Ochs M. A brief update on lung stereology. J Microsc. 2006;222:188–200. doi: 10.1111/j.1365-2818.2006.01587.x. [DOI] [PubMed] [Google Scholar]
- Ochs M, Knudsen L, Allen L, et al. GM-CSF mediates alveolar epithelial type II cell changes, but not emphysema-like pathology, in SP-D-deficient mice. Am J Physiol Lung Cell Mol Physiol. 2004a;287:L1333–L1341. doi: 10.1152/ajplung.00137.2004. [DOI] [PubMed] [Google Scholar]
- Ochs M, Nyengaard JR, Jung A, et al. The number of alveoli in the human lung. Am J Respir Crit Care Med. 2004b;169:120–124. doi: 10.1164/rccm.200308-1107OC. [DOI] [PubMed] [Google Scholar]
- Pastva AM, Wright JR, Williams KL. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc. 2007;4:252–257. doi: 10.1513/pats.200701-018AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherle W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie. 1970;26:57–60. [PubMed] [Google Scholar]
- Sorensen GL, Hoegh SV, Leth-Larsen R, et al. Multimeric and trimeric subunit SP-D are interconvertible structures with distinct ligand interaction. Mol Immunol. 2009;46:3060–3069. doi: 10.1016/j.molimm.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134:127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- Vedel-Jensen EB, Gundersen HJ. The rotator. J Microsc. 1993;170:35–44. [Google Scholar]
- Wang IC, Meliton L, Tretiakova M, et al. Transgenic expression of the forkhead box M1 transcription factor induces formation of lung tumors. Oncogene. 2008;27:4137–4149. doi: 10.1038/onc.2008.60. [DOI] [PubMed] [Google Scholar]
- Weibel ER. Stereological Methods. Vol. 1: Practical Methods for Biological Morphometry. London: Academic Press; 1979. [Google Scholar]
- Weibel ER, Hsia CC, Ochs M. How much is there really? Why stereology is essential in lung morphometry. J Appl Physiol. 2007;102:459–467. doi: 10.1152/japplphysiol.00808.2006. [DOI] [PubMed] [Google Scholar]
- Zambrowicz BP, Imamoto A, Fiering S, et al. Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci U S A. 1997;94:3789–3794. doi: 10.1073/pnas.94.8.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hartshorn K, Crouch E, et al. Complementation of pulmonary abnormalities in SP-D−/− mice with an SP-D/conglutinin fusion protein. J Biol Chem. 2002;277:22453–22459. doi: 10.1074/jbc.M201632200. [DOI] [PubMed] [Google Scholar]