

Abstract
Mitochondria are cellular organelles that regulate commitment and execution of apoptosis. The intrinsic apoptotic pathway culminates in the permeabilization of the mitochondrial outer membrane and dismantling of the cell. Apoptosis of cancer cells is a favorable outcome when administering chemotherapeutic treatment yet the basis for why some cancers are sensitive to chemotherapy while others are not has historically been poorly understood. In this review, we present recent work that has demonstrated the importance of mitochondrial apoptotic priming, or how close a cell is to the threshold of apoptosis, in determining whether a cell will undergo apoptosis after chemotherapy treatment. Differential levels of apoptotic priming in tumors create bona fide opportunities and challenges for effective use of targeted and cytotoxic chemotherapies.
Keywords: apoptosis, chemotherapy, mitochondrial apoptotic priming, chemotherapeutic window
Why does chemotherapy work?
A longstanding question among not only patients but also the oncologists that are treating them is “why does chemotherapy work?” This important question is asked because chemotherapy does, at times, work impressively well, leading to long-term cures of otherwise fatal neoplasms. However, despite a growing understanding of how cancers arise, grow, metastasize and eventually overcome the host, the mechanisms behind successful treatment of cancers are poorly understood[1,2]. The key determinants of response to chemotherapy will be explored in this review, with a focus on how the pathway of mitochondrial apoptosis affects treatment outcome.
Chemotherapy and apoptosis
Treatment of human malignancies with chemotherapy with curative intent has been successfully carried out for over 50 years with millions of cancer survivors enjoying long lives after treatment[3]. However, millions more have succumbed to their disease. Regardless of whether they are considered “cytotoxic” or “targeted,” most chemotherapies function by inducing a form of irreversible programmed cell death called apoptosis[4–6]. Apoptosis can proceed via two distinct pathways: intrinsic and extrinsic[7]. The extrinsic apoptotic pathway is engaged upon activation of cell surface death receptors and has a limited, controversial role in chemotherapy-induced apoptosis[2,4,6]. We will therefore focus solely on the intrinsic, or mitochondrial apoptotic pathway in this review.
Mitochondrial apoptosis is controlled by the pro- and anti-apoptotic proteins of the BCL-2 family which can be divided into three categories based on their intracellular function and sequence homology (reviewed in [8]). One category includes the anti-apoptotic proteins BCL-2, BCL-w, MCL-1, BFL-1, and BCL-XL which contain all four BCL-2 homology domains (BH1-4). These proteins prevent apoptosis by binding and sequestering their pro-apoptotic counterparts. The second category, the BH3-only proteins, includes the pro-apoptotic proteins PUMA, BIM, BID, BAD, BIK, NOXA, and BMF, which contain only the BH3 domain. The final category, the effectors, contains BAX and BAK which contain domains BH1-3 and can be activated by a subset of BH3-only proteins which include BIM, BID and, potentially, PUMA[9–15].
Activation of BAX or BAK initiates a series of steps that results in commitment to apoptotic cell death. It had been suggested that BAX and BAK are activated not by BIM and BID but instead via the inactivation of anti-apoptotic BCL-2 family members[16]. However, recent studies have confirmed the direct binding and activation of BAX by BIM and BAK by BID[9,13,17]. Upon activation, BAK and BAX oligomerize and directly cause mitochondrial outer membrane permeabilization (MOMP), a critical event during apoptosis. Cytochrome c and other factors are released after MOMP and associate with several cytosolic proteins including APAF-1 to activate caspases for dismantling of the cell[7]. Even in the absence of caspase activation, post-MOMP mitochondria are progressively impaired in their ability to generate ATP and cannot maintain cellular survival except in specific, non-physiological circumstances[7,18,19]. MOMP can thus be considered the “point of no return” in mitochondrial apoptosis.
MOMP, and consequent apoptosis, is triggered clinically when a chemotherapeutic agent induces a sufficient amount of stress within a cancer cell by damaging critical cellular components such as microtubules, DNA, or key signaling pathways[2,4,6]. The cell responds by unleashing pro-apoptotic proteins or downregulating anti-apoptotic members of the BCL-2 family, either of which can lead to a shift in the life/death balance in the cell irreversibly toward destruction[4,6]. Based on this understanding, both the state of the BCL-2 family of apoptosis-regulating proteins in the cell prior to encountering the stress as well as the magnitude of the dynamic response to the stress can have a profound impact on the eventual fate of the cell. These concepts will be explored further below.
Measuring mitochondrial apoptotic priming
Regulation of the balance of pro- and anti-apoptotic proteins within cells, and thus how close a cell is to the threshold of apoptosis, is dependent on many factors. In order to avoid apoptosis, a cell must express a sufficient amount of anti-apoptotic proteins to bind and inactivate what pro-apoptotic counterparts are also present. Furthermore, most cells contain an additional amount of “buffering” anti-apoptotic proteins that can inactivate further pro-death signals that are encountered on a stochastic basis under normal, physiological conditions[5]. The amount of buffering that a cell may employ varies by the type of cell: many cells in the hematopoietic and immune systems maintain relatively small buffers of anti-apoptotic proteins[5], likely to enable their quick and efficient elimination as the need arises[20]. Conversely, cells with a longer lifespan such as the highly specialized, fully differentiated cells that make up vital organs are more buffered against short-term, stochastic fluctuations in cellular stress[5] that could erroneously trigger cell death in a vital cell to the detriment of the host’s survival (reviewed in [21]). The specific factors that control the level of anti-apoptotic buffering within healthy cells are unknown but may involve the epigenetic programming that maintains cell identity[22].
The varying levels of anti-apoptotic buffering present within cells can affect the outcome of any event that stresses a cell, including chemotherapy treatment. For instance, a cell may express a very large reserve of unbound anti-apoptotic proteins, which are available for binding of their pro-apoptotic counterparts, and therefore be protected against even a substantial amount of subsequent pro-death signaling. Alternatively, a cell may express only enough unbound anti-apoptotic proteins to just barely keep the pro-apoptotic proteins in check and would therefore not be protected from even a small stressor. The cell that has a small reserve of unbound anti-apoptotic proteins is “primed” for apoptosis while the cell that has a large reserve is “unprimed” (Figure 1). Another way to illustrate this dichotomy is to envision two cells, one being close to the threshold (cliff’s edge) at which apoptosis is triggered while the other is far from that same threshold. The one closer to the edge is considered primed while the other is unprimed. When these two cells are treated with equal doses of chemotherapy (pushed toward the cliff’s edge), the primed cell will be more likely to trigger apoptosis than the unprimed cell if the dynamic response to chemotherapy (strength of push toward edge) is equal (Figure 2A). While it aids understanding to define priming this way, the actual measurement of priming is done by a functional test and thus a label of “primed” and “unprimed” can only be assigned based on a given cell’s response to systematic pro-apoptotic stimuli as described below.
Figure 1. A model for how pretreatment mitochondrial priming can affect response to pro-death signal.

In unprimed cells, exogenous pro-apoptotic signals are bound and sequestered by unbound anti-apoptotic proteins. In primed cells, pro-apoptotic signals displace the BH3-only proteins bound to anti-apoptotic proteins which are then able to activate BAX/BAK.
Figure 2. Apoptotic priming can affect response to chemotherapy.

(A) A cell that is primed for apoptosis is more likely to undergo cell death in response to chemotherapy than an unprimed cell. (B) A cell that is primed for apoptosis nonetheless does not undergo cell death in response to a targeted agent if it does not exhibit a dependence on the target of the therapy. A cell that is unprimed, but exhibits a strong dependence on the target of the therapy may unleash sufficient pro-apoptotic signaling to trigger apoptosis. (C) Two cells that exhibit similar dependence on the target of the therapy may have different treatment outcomes based on levels of apoptotic priming present in cells prior to therapy. (D) A cell that is unprimed yet exhibits dependence on the target of the therapy may become primed in response to the therapy if apoptosis is not triggered directly by the single agent. An additional bolus of pro-apoptotic signaling, here provided by treatment with a cytotoxic chemotherapy which would not kill an unprimed cell, is sufficient to trigger apoptosis.
Because there may be substantial utility in knowing how close a cancer cell is to the threshold of apoptosis, an assay called “BH3 profiling” has been developed to measure priming in cancerous and normal cells. The basic principle is to expose mitochondria to peptides derived from the BH3 domains of pro-death BH3-only proteins, and measure the resulting magnitude of mitochondrial outer membrane permeabilization (for full methods see [23]). Heightened sensitivity of mitochondria to BH3 peptides, such as BAD BH3 or NOXA BH3, with selective binding to anti-apoptotic proteins might indicate selective dependence on BCL-2 or MCL-1[24–26]. Heightened sensitivity to peptides that exhibit promiscuous binding to anti-apoptotic proteins, such as BIM BH3 or PUMA BH3, indicates a highly primed state, with a low reserve of unbound anti-apoptotic proteins[5,26–28].
In practice, the assay is typically performed by gently permeabilizing the plasma membrane of cells and adding fixed doses of BH3 peptides derived from the BH3 domain of pro-apoptotic BH3-only proteins. Mitochondrial potential is then monitored for detection of MOMP which occurs when the anti-apoptotic reserve is exhausted within a cell and BAX and/or BAK are activated (Figure 1). Cells can be labeled as primed or unprimed based on the extent of mitochondrial depolarization (MOMP) that occurs in response to a fixed titration of pro-death BH3 peptides – the faster that a cell is depolarized, or the lower concentration of peptide required for MOMP, the more primed it is.
Functional assays, such as BH3 profiling, are critical for measurement of apoptotic priming within a cell. The activities of the BCL-2 family of proteins is regulated by a plethora of post-translational modifications and interactions with other proteins[29]. Measuring the expression level of each member of the BCL-2 family, their modifications and the proteins they are interacting with is not a practical task and, due to additional unknown factors that may modify their activity, may not even provide an accurate assessment of priming and thus creates a need for a function-based assay. By treating a cell with fixed doses of pro-apoptotic signaling in BH3 profiling, it is possible to measure the integrated functional output of the BCL-2 family efficiently and meaningfully[5].
Mitochondrial apoptotic priming determines tumor response to cytotoxic chemotherapy
Using BH3 profiling to measure levels of apoptotic priming across a range of cancer types, it has been shown that primed cells readily undergo apoptosis in response to cytotoxic chemotherapy treatment while unprimed cells are less likely to do so (Table 1)[5]. Notably, this holds true not only in cancer cell lines but also primary tumors and can be predictive of how patients will respond to chemotherapy in the clinic[5,28]. In addition, cells within tumors that have undergone treatment and then recurred are frequently less primed, making them less sensitive to subsequent rounds of chemotherapy[5,28]. The selective pressure of chemotherapy treatment likely culls primed cancer cells, leaving only unprimed cells to repopulate the tumor[5,28]. These unprimed relapsed tumors are frequently intractable.
Table 1.
Major determinants of chemotherapy treatment success (why chemotherapy works):
| Drug Class | Targets | Examples | Determinants of Therapy Success | ||
|---|---|---|---|---|---|
| Primary | Secondary | Cancer Type | |||
| Classical cytotoxic therapy | Ubiquitous cellular elements (microtubules, DNA, topoisomerases) | Paclitaxel (microtubules), Topotecan (topoisomerase II), Doxorubicin (DNA) | Pretreatment mitochondrial apoptotic priming | Signaling dynamics post treatment | Multiple myeloma, ALL, ovarian cancer[5]; CLL[27]; AML[28] |
| Targeted therapy | Cell-specific targets (mutated oncogenes, differentially expressed oncogenes) | Erlotinib (EGFR), Vemurafenib (V600E), Imatinib (BCR-ABL) | Presence or absence of target | Pretreatment mitochondrial apoptotic priming, signaling dynamics post treatment | Non-small-cell lung cancer[81,82]; melanoma[83]; CML[84] |
| Antibody-based therapy | Cell surface markers | Rituximab (anti-CD20 antibody), brentuximab-vedotin (anti-CD30:MMAE antibody-drug conjugate) | Presence or absence of target | Immune system competence | Lymphoma[85]; lymphoma and Hodgkin’s disease[86] |
| Immune modulation (reversal of immune checkpoint blockade) | Immune system (tumor tolerance, tumor-associated macrophages) | Ipilimumab (anti-CTLA4 antibody), | Immune system anti-tumor response | Immune system competence | Melanoma[66,87] |
Experiments measuring mitochondrial priming have also uncovered a greater understanding of apoptotic signaling in cancer cells and normal cells. A longstanding misconception is that cancers must “disrupt” or “disable” apoptosis[4,30,31], which some interpret as meaning that cancer cells are inherently less sensitive to subsequent apoptotic signaling than normal cells. A “disruption” or “disabling” of apoptosis would require a complete loss of signaling in the mitochondrial apoptotic pathway via, for example, a total loss of BAX and BAK expression, yet this is rarely observed in cancers. Cancers commonly upregulate anti-apoptotic proteins or downregulate pro-apoptotic counterparts to keep apoptosis at bay (reviewed in [29,32] but this does not equate to a disruption or disabling of the apoptotic pathway. In fact, there is little evidence that cancer cells are more refractory to apoptosis than normal cells. While most cancers have an intact apoptotic pathway, some tumors are even highly primed for apoptosis as demonstrated by their efficient mitochondrial depolarization in response to pro-apoptotic signaling in the form of BH3 peptides. This depolarization often exceeds that observed in normal somatic cells[5]. This confirms that most cancers not only express the critical effectors BAX and/or BAK and have an intact mitochondrial apoptotic pathway but are also primed for apoptosis. The cancers that are very primed are those that respond most favorably to chemotherapy (acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia) while those that are unprimed respond poorly (endometrial and renal cell carcinomas, serous borderline tumors)[5,27,28]. The therapeutic index of conventional chemotherapy, which is directed against ubiquitous targets such as DNA and microtubules, probably derives largely from the fact that cancer cells are actually more ready to engage the machinery of apoptosis than normal cells, not less[5,28]. In fact, it is unlikely that anyone would ever be cured via conventional chemotherapy, if some cancer cells were not sometimes much more primed for apoptosis than most normal cells.
Other measures of the readiness of cancer cell mitochondria to undergo cell death have been utilized to predict cellular responses to chemotherapy. For example, measuring the propensity of tumor cells to spontaneously activate caspase 3 has prognostic value for response to chemotherapy in anaplastic lymphoma[33]. In colorectal cancer, measuring and integrating the protein expression levels of several BCL-2 family members may predict how patients will respond to chemotherapy[34]. Although these other measurements are not functional tests of apoptotic priming as defined here, they nonetheless validate the importance of pretreatment apoptotic signaling in cell fate decisions.
What determines how primed a tumor will be?
Although cancers have wide ranging levels of mitochondrial priming which contribute to their responses to chemotherapeutic agents[5,27,28], it is unclear what determines the level of priming within a cell or tumor. At the cellular level, an attractive hypothesis is that the level of priming evident in a tumor cell is determined, at least in part, by the level of priming evident in the cell that gave rise to the tumor itself. Lending evidence to this concept is the observation that the most highly primed cancers are derived from the most highly primed normal tissues, and vice versa. For instance, acute lymphoblastic leukemia cells are among the most highly primed and the most chemosensitive of cancers. They derive from the lymphocytic lineage, which is among the most highly primed lineages in the body[5,25,28] and is among the most chemosensitive[35]. Conversely, chemoresistant primary tumors, including renal cell carcinomas and endometrial cancers, are unprimed, as are the normal, healthy cells they are derived from[5]. Thus, cell lineage may be a strong determinant of the overall apoptotic priming of a tumor.
At the molecular level, the level of priming is regulated by the expression and post-translational modification state of BCL-2 family proteins – but what in turn regulates this? Most likely, priming results from the combined contributions of many signaling pathways. In normal cells, various signaling pathways can modulate BCL-2 family expression for regulation of survival. For example, the survival of mature B cells requires pro-survival B cell receptor signaling[36,37] which was recently shown to be mediated by PI3 Kinase (PI3K)[38]. The PI3K prosurvival signals in these cells are carried out by Akt and the FOXO family of transcription factors that eventually modulate levels of the pro-apoptotic protein BIM[38].
The same pathways that modulate apoptosis in healthy cells are aberrantly expressed in cancer cells to maintain survival. PI3K, for example, is inappropriately activated in a wide range of malignancies and confers a strong enough survival advantage to cancerous cells that it is being targeted pharmacologically for cancer therapy (reviewed in [39,40]). Other common oncogenes and tumor suppressors that are deregulated in cancer have been shown to have pro- or anti-apoptotic effects including p53[41], Notch[42] and Wnt[43] among others including, of course, the BCL-2 family. Interestingly, activation of some oncogenes, especially c-Myc, can have a strong pro-apoptotic effect[44–46] and thus likely contribute to the final overall level of priming in cancer cells.
Not surprisingly, the tumor microenvironment is also a strong modulator of apoptotic priming. Chronic lymphocytic leukemia (CLL), for example, is commonly treated with chemotherapy yet cancerous cells exhibit differing levels of chemosensitivity, with a subpopulation of cells proving to be intractable and supporting relapse[27,47]. Cells residing within lymph nodes upregulate anti-apoptotic BCL-XL and BCL2A1 (BFL-1) to reduce apoptotic priming, making them more likely to survive therapy treatment[27,47]. The interactions of cancer cells with stroma can lead to chemotherapy resistance in both solid and liquid tumors (reviewed in [48]). In addition, stimulation with exogenous growth factors[46,49,50] can affect BCL-2 family protein expression and sensitivity to chemotherapy. In one such example, hepatocyte growth factor (HGF) was recently identified in a cancer cell and stroma co-culture screen to induce a chemoresistant phenotype in tumor cells by activating PI3K[50], whose role in modulation of apoptosis has already been discussed.
The pretreatment level of priming in a tumor is likely determined by the level of priming in the cell of origin and modulated by the microenvironment of the cell and by the pro- and anti-apoptotic effects of the various perturbations that have occurred during tumorigenesis.
The chemotherapeutic window
Understanding the mitochondrial priming of the tumor alone, however, is insufficient to understanding the source of a chemotherapeutic window. The key to achieving a desirable outcome from chemotherapy treatment is as much dependent on the lack of priming in vital organs as the priming in the tumor. When comparing apoptotic priming across healthy tissues, one can see that most healthy tissues are unprimed which allows vital organs such as the liver, heart, brain and kidneys to survive high doses of chemotherapy relatively unscathed[5]. The lack of priming in these vital tissues is demonstrated by their attenuated response to relatively large doses of pro-apoptotic BH3 peptides[5]. However, the molecular mechanisms that maintain this low level of priming in vital tissues are unknown and an active area of investigation. There are, of course, some effects of chemotherapy on these tissues including long term pathologies, such as doxorubicin-induced cardiotoxicity[51], but these organs generally continue to function during and after chemotherapy treatment. Some healthy tissues, however, exhibit a high level of apoptotic priming, especially the hematopoietic system, which renders them sensitive to chemotherapy. A loss of the bulk of the hematopoietic system seems to be entirely survivable, though, as evidenced by the frequent myelosuppression that accompanies chemotherapy treatment even in those patients that emerge with cures. In general, cells from tumors that are chemosensitive are more primed than their non-transformed counterparts, thus creating a therapeutic window for tumor response to therapy[5]. For the most broadly cytotoxic chemotherapies that target common cellular elements across all cells, successful treatment relies on a level of priming in the tumor higher than other healthy tissues (Figure 3).
Figure 3.
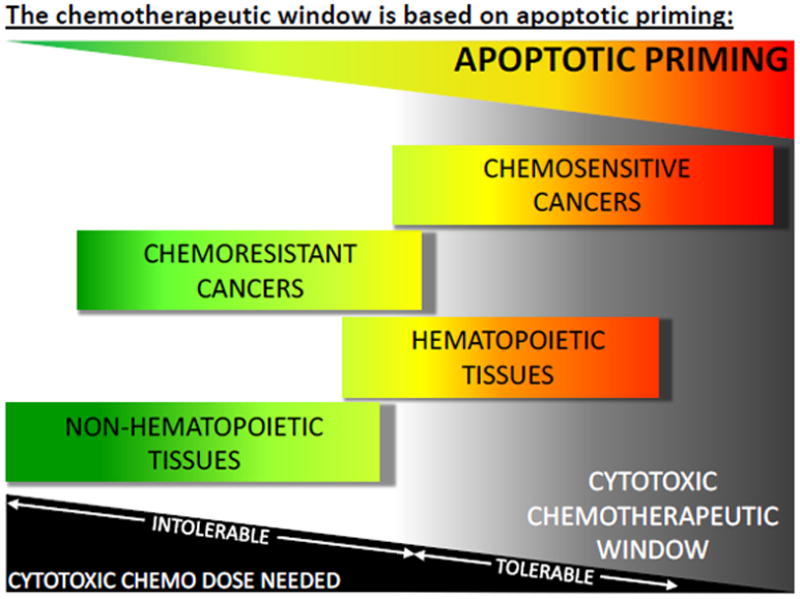
Apoptotic priming varies between tumors and healthy tissues and provides a window for chemotherapeutic efficacy.
Mitochondria in targeted chemotherapies and immunochemotherapies
Mitochondrial priming has a likely role in cellular response to “targeted” chemotherapies as well. Although “classical” cytotoxic chemotherapies can have quite specific targets, for example, paclitaxel for microtubules and topotecan for topoisomerase II, for the purpose of this review only agents that target non-ubiquitous cellular components will be considered targeted agents. Much progress has been recently made in the development of targeted agents that have the potential to be selectively toxic to cancer cells that are dependent on that target over healthy tissues that lack it (reviewed in [52]). Most of these targeted agents exert their anti-cancer effects by engaging the same mitochondrial apoptotic pathways as classical chemotherapies which proposes a potential role of apoptotic priming in the response to these agents as well[53–55]. The largest determinant of apoptotic response to targeted agents, of course, is a given cell’s dependence on the protein or signaling pathway being targeted (Figure 2B and Table 1). For example, a cell that is dependent on EGFR will upregulate pro-apoptotic BIM when EGFR signaling is inhibited, thus pushing the balance of BCL-2 family proteins toward apoptosis[55–57]. Cells lacking dependence on EGFR are unaffected by these inhibitors[55–57], and thus even a high level of mitochondrial priming in these cells is unlikely to trigger cell death (Figure 2B). Conversely, cells that are not highly primed but exhibit a strong dependence on the targeted pathway may respond to the inhibitor with enough pro-apoptotic stress signaling that apoptosis is triggered (Figure 2B). Therefore, the magnitude of the response to the inhibitor is likely the most important factor in determining whether the cell will live or die. However, in cells that exhibit a similar level of dependence on a targeted pathway and thus a similar dynamic signaling response to the inhibition of that pathway, the level of mitochondrial priming may affect whether apoptosis will occur. A highly primed cell would need a smaller dynamic pro-apoptotic response to an agent to trigger apoptosis than an unprimed cell (Figure 2C and Table 1).
It is likely that using targeted agents in cells that are dependent on the target yet are unprimed may have clinical benefits even without apoptosis being induced by the targeted therapy alone. Specifically, agents targeting cancer-specific proteins or growth signaling pathways may induce a level of apoptotic stress within a cancer cell that is perhaps not sufficient to trigger apoptosis but may upregulate pro-apoptotic proteins, thus increasing mitochondrial apoptotic priming. In this context, an additional chemotherapeutic agent, targeted or not, could provide the final bolus of pro-apoptotic signaling necessary to activate MOMP (Figure 2D). This has been convincingly demonstrated with many “priming” agents[27,54,58–60]. These priming agents, with diverse targets including histone deacetylases and PI3K, modify levels of the BCL-2 family to improve responses to cytotoxic chemotherapy[54,59]. In addition, a novel class of agents, BH3 mimetics, have been shown to potently and specifically inhibit several anti-apoptotic members of the BCL-2 family including BCL-2, BCL-XL and BCL-W. The administration of these agents can increase the priming of a cancer cell and thus make other, concurrently-administered chemotherapies, more effective[28,34,61–64].
Mitochondrial priming likely plays a smaller role in the responses of cancer cells to therapies that are based on activating an antitumor response by the immune system (Table 1). These therapies include monoclonal antibody therapies directed at cell surface markers expressed on tumor cells, immuno-modulatory agents, and allogeneic stem cell transplantation. Although monoclonal antibodies targeting tumor cells can sometimes directly induce apoptosis in target cells, they most commonly rely on antibody-dependent cellular cytotoxicity or complement-dependent cytotoxicity to activate immune-effector cells for maximal effectiveness[65]. In addition, agents that modulate the immune system directly, such as the anti-CTLA4 antibody (ipilimumab), enhance the antitumor responses of cytotoxic T cells[66]. Because the mitochondrial apoptotic pathway is involved in the direct anti-tumor cell activity of some of these therapies, priming may have a role in determining response. However, this role would likely be overshadowed by other key determinants of response and resistance including the presence or absence of the cell surface marker on the tumor cell and immune competence [65,67]. In the case of allogeneic stem cell transplant, an adaptive immune response of the donor’s T-cells against the host tumor is required. At least in acute myelogenous leukemia, success of this strategy is not affected by the apoptotic priming of tumor mitochondria[28].
Alternative determinants of chemotherapy effectiveness
Although recent results have suggested that priming is the major determinant of chemotherapy effectiveness[5,27,28], several laboratories have provided evidence of some alternatives. In mitochondria specifically, chemotherapy response has been linked to defects in mitochondrial respiratory chain complexes caused by loss of specific cytochrome c oxidase subunits[68]. The loss of these subunits was observed clinically in a subset of oesophageal adenocarcinomas and was associated with increased sensitivity to chemotherapy, suggesting that mitochondrial defects can develop during tumorigenesis and may contribute to the chemotherapeutic window.
A longstanding theory of chemotherapy effectiveness has been that the proliferation rates of cancer cells determines their sensitivity to chemotherapy. However, this explanation is inadequate since clinical observations have shown that many chemosensitive cancers proliferate very slowly while many chemoresistant cancers proliferate quickly[69–73]. In addition, some non-proliferating healthy cells (resting B cells for example) are extremely sensitive to DNA damaging agents[35], thus making it less likely that proliferation alone is the basis for chemosensitivity, despite its favored place in textbooks and general opinion in oncology.
Concluding remarks
Mitochondria have a well-established and prominent role in chemotherapy effectiveness that should be exploited for cancer therapy. Specifically targeting the BCL-2 family is a strategy that has already shown promise: the BCL-2/BCL-XL/BCL-w inhibitors ABT-737 and its derivatives have activity against multiple types of blood cancers[62] and some solid tumors[74–76]. Other strategies to inhibit these proteins are also in various stages of preclinical development (reviewed in [77]). One anti-apoptotic member of the BCL-2 family, MCL-1, is also a promising target in cancer therapy but has, so far, eluded efforts to effectively target it for cancer therapy. Other strategies to target this family, including stapled α-helices that can penetrate cells and act as inhibitors of anti-apoptotic proteins, are also being pursued[78,79]. Another interesting strategy is to overcome chemotherapy resistance is the direct pharmacological activation of the pro-apoptotic effector BAX[80].
The successful use of chemotherapy to treat malignancies for over five decades is based, at least in part, on high levels of mitochondrial priming in chemosensitive tumors, a concept only recently demonstrated[5]. As we continue to broaden our understanding of apoptosis and how it is regulated, we hope to uncover critical new vulnerabilities within this pathway in cancers.
Acknowledgments
We acknowledge the many researchers who contributed to our understanding of mitochondria, apoptosis and cancer biology, and apologize that we could not cite all of the relevant research due to space restrictions. We gratefully acknowledge funding from the American Cancer Society Postdoctoral Fellowship 121360-PF-11-256-01-TBG (K.A.S.), Women’s Cancers Program at the Dana-Farber Cancer Institute (A.L.), and NIH grants RO1CA129974 (A.L.), and P01CA139980 (A.L). A.L. is a Leukemia and Lymphoma Society Scholar. The authors have no conflict of interest to declare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rich T, et al. Defying death after DNA damage. Nature. 2000;407:777–83. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- 2.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 3.De Moor JS, et al. Cancer survivors in the United States: prevalence across the survivorship trajectory and implications for care. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive. Oncology. 2013;22:561–70. doi: 10.1158/1055-9965.EPI-12-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnstone RW, et al. Apoptosis : A Link between Cancer Genetics and Chemotherapy Defects in apoptosis underpin both tumorigenesis and. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 5.Ni Chonghaile T, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science (New York, NY ) 2011;334:1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Experimental cell research. 2000;256:42–9. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 7.Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nature reviews. Molecular cell biology. 2010;11:621–32. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 8.Chipuk JE, et al. The BCL-2 family reunion. Molecular cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Czabotar PE, et al. Bax Crystal Structures Reveal How BH3 Domains Activate Bax and Nucleate Its Oligomerization to Induce Apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Kim H, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nature cell biology. 2006;8:1348–58. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 11.Leshchiner ES, et al. Direct activation of full-length proapoptotic BAK. Proceedings of the National Academy of Sciences of the United States of America. 2013 doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Letai A, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer cell. 2002;2:183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 13.Gavathiotis E, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–81. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes & …. 2000 doi: 10.1101/gad.14.16.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Molecular cell. 2009;36:487–99. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willis SN, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science (New York, NY ) 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 17.Moldoveanu T, et al. BID-induced structural changes in BAK promote apoptosis. Nature structural & molecular biology. 2013 doi: 10.1038/nsmb.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Colell A, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–97. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- 19.Lartigue L, et al. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Molecular biology of the cell. 2009;20:4871–84. doi: 10.1091/mbc.E09-07-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nature immunology. 2003;4:410–5. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- 21.Wright KM, Deshmukh M. Restricting apoptosis for postmitotic cell survival and its relevance to cancer. Cell cycle (Georgetown, Tex ) 2006;5:1616–20. doi: 10.4161/cc.5.15.3129. [DOI] [PubMed] [Google Scholar]
- 22.Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nature reviews. Genetics. 2008;9:129–40. doi: 10.1038/nrg2295. [DOI] [PubMed] [Google Scholar]
- 23.Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods (San Diego, Calif) 2013 doi: 10.1016/j.ymeth.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunelle JK, et al. MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. The Journal of cell biology. 2009;187:429–42. doi: 10.1083/jcb.200904049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ryan Ja, et al. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12895–900. doi: 10.1073/pnas.0914878107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng J, et al. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer cell. 2007;12:171–85. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Davids MS, et al. Decreased mitochondrial apoptotic priming underlies stroma-mediated treatment resistance in chronic lymphocytic leukemia. Blood. 2012 doi: 10.1182/blood-2012-02-414060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vo T-T, et al. Relative Mitochondrial Priming of Malignant Myeloblasts and Normal HSCs Determines Chemotherapeutic Success in AML. Cell. 2012 doi: 10.1016/j.cell.2012.08.038. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yip KW, Reed JC. Bcl-2 family proteins and cancer. Oncogene. 2008;27:6398–406. doi: 10.1038/onc.2008.307. [DOI] [PubMed] [Google Scholar]
- 30.Galluzzi L, et al. Mitochondria as therapeutic targets for cancer chemotherapy. Oncogene. 2006;25:4812–30. doi: 10.1038/sj.onc.1209598. [DOI] [PubMed] [Google Scholar]
- 31.Hanahan D, Weinberg R. The hallmarks of cancer. cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 32.Juin P, et al. Decoding and unlocking the BCL-2 dependency of cancer cells. Nature Reviews Cancer. 2013:13. doi: 10.1038/nrc3538. [DOI] [PubMed] [Google Scholar]
- 33.Ten Berge RL. Expression levels of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood. 2002;99:4540–4546. doi: 10.1182/blood.v99.12.4540. [DOI] [PubMed] [Google Scholar]
- 34.Lindner AU, et al. Systems Analysis of BCL2 Protein Family Interactions Establishes a Model to Predict Responses to Chemotherapy. 2013 doi: 10.1158/0008-5472.CAN-12-2269. [DOI] [PubMed] [Google Scholar]
- 35.Stahnke K. Activation of apoptosis pathways in peripheral blood lymphocytes by in vivo chemotherapy. Blood. 2001;98:3066–3073. doi: 10.1182/blood.v98.10.3066. [DOI] [PubMed] [Google Scholar]
- 36.Kraus M, et al. Survival of resting mature B lymphocytes depends on BCR signaling via the Igalpha/beta heterodimer. Cell. 2004;117:787–800. doi: 10.1016/j.cell.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 37.Lam KP, et al. In Vivo Ablation of Surface Immunoglobulin on Mature B Cells by Inducible Gene Targeting Results in Rapid Cell Death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- 38.Srinivasan L, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–86. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochemical Journal. 2008 doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 40.Wong KK, et al. Targeting the PI3K signaling pathway in cancer. Current opinion in genetics & development. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amaral JD, et al. The role of p53 in apoptosis. Discovery medicine. 2010;9:145–52. [PubMed] [Google Scholar]
- 42.Dang TP. Notch, apoptosis and cancer. Advances in experimental medicine and biology. 2012;727:199–209. doi: 10.1007/978-1-4614-0899-4_15. [DOI] [PubMed] [Google Scholar]
- 43.Pećina-Slaus N. Wnt signal transduction pathway and apoptosis: a review. Cancer cell international. 2010;10:22. doi: 10.1186/1475-2867-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murphy DJ, et al. Distinct thresholds govern Myc’s biological output in vivo. Cancer cell. 2008;14:447–57. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evan GI, et al. Induction of Apoptosis by c-myc Protein in Fibroblasts. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 46.Sarosiek Ka, et al. Interleukin-4 distinctively modifies responses of germinal centre-like and activated B-cell-like diffuse large B-cell lymphomas to immuno-chemotherapy. British journal of haematology. 2009;147:308–18. doi: 10.1111/j.1365-2141.2009.07851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vogler M, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–13. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- 48.McMillin DW, et al. The role of tumour–stromal interactions in modifying drug response: challenges and opportunities. Nature Reviews Drug Discovery. 2013;12:217–228. doi: 10.1038/nrd3870. [DOI] [PubMed] [Google Scholar]
- 49.Roidl A, et al. Resistance to chemotherapy is associated with fibroblast growth factor receptor 4 up-regulation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:2058–66. doi: 10.1158/1078-0432.CCR-08-0890. [DOI] [PubMed] [Google Scholar]
- 50.Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang S, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nature medicine. 2012:18. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 52.Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nature reviews Cancer. 2006;6:714–27. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- 53.Gojo I, et al. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:3527–38. [PubMed] [Google Scholar]
- 54.Bender A, et al. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene. 2011;30:494–503. doi: 10.1038/onc.2010.429. [DOI] [PubMed] [Google Scholar]
- 55.Deng J, et al. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer research. 2007;67:11867–75. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 56.Costa DB, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS medicine. 2007;4:1669–79. doi: 10.1371/journal.pmed.0040315. discussion 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cragg MS, et al. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS medicine. 2007;4:1681–89. doi: 10.1371/journal.pmed.0040316. discussion 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yallapu MM, et al. Curcumin induces chemo/radio-sensitization in ovarian cancer cells and curcumin nanoparticles inhibit ovarian cancer cell growth. Journal of ovarian research. 2010;3:11. doi: 10.1186/1757-2215-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Häcker S, et al. Histone deacetylase inhibitors prime medulloblastoma cells for chemotherapy-induced apoptosis by enhancing p53-dependent Bax activation. Oncogene. 2011;30:2275–81. doi: 10.1038/onc.2010.599. [DOI] [PubMed] [Google Scholar]
- 60.Yuan L, et al. Novel chemo-sensitizing agent, ERW1227B, impairs cellular motility and enhances cell death in glioblastomas. Journal of neuro-oncology. 2011;103:207–19. doi: 10.1007/s11060-010-0379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tse C, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer research. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 62.Souers AJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature medicine. 2013;19:202–8. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 63.Muranen T, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer cell. 2012;21:227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oakes SR, et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2766–71. doi: 10.1073/pnas.1104778108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weiner LM, et al. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nature reviews. Immunology. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hodi F, O’Day S. Improved survival with ipilimumab in patients with metastatic melanoma. … England Journal of …. 2010 doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359–68. doi: 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- 68.Aichler M, et al. Clinical response to chemotherapy in oesophageal adenocarcinoma patients is linked to defects in mitochondria. The Journal of pathology. 2013 doi: 10.1002/path.4199. [DOI] [PubMed] [Google Scholar]
- 69.Hurwitz JL, et al. New advances in the second-line treatment of small cell lung cancer. The oncologist. 2009;14:986–94. doi: 10.1634/theoncologist.2009-0026. [DOI] [PubMed] [Google Scholar]
- 70.Bosch F, et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: high response rate and disease eradication. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:155–61. doi: 10.1158/1078-0432.CCR-07-1371. [DOI] [PubMed] [Google Scholar]
- 71.Marcus R, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood. 2005;105:1417–23. doi: 10.1182/blood-2004-08-3175. [DOI] [PubMed] [Google Scholar]
- 72.Pasieka JL. Anaplastic thyroid cancer. Current opinion in oncology. 2003;15:78–83. doi: 10.1097/00001622-200301000-00012. [DOI] [PubMed] [Google Scholar]
- 73.Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Molecular biology of the cell. 2012;23:1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 75.Gandhi L, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:909–16. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rudin CM, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:3163–9. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang L, et al. BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy. 2007;10:207–17. doi: 10.1016/j.drup.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cohen NA, et al. A Competitive Stapled Peptide Screen Identifies a Selective Small Molecule that Overcomes MCL-1-Dependent Leukemia Cell Survival. Chemistry & Biology. 2012;19:1175–1186. doi: 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Labelle JL, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. Journal of Clinical Investigation. 2012 doi: 10.1172/JCI46231DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gavathiotis E, et al. Direct and selective small-molecule activation of proapoptotic BAX. Nature Chemical Biology. 2012 doi: 10.1038/nchembio.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lynch T, Bell D. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. New England Journal of Medicine. 2004 doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 82.Zhu CQ, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:4268–75. doi: 10.1200/JCO.2007.14.8924. [DOI] [PubMed] [Google Scholar]
- 83.Flaherty K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. New England Journal of Medicine. 2010 doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Druker B, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996 doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 85.Kennedy G, Tey S. Incidence and nature of CD20-negative relapses following rituximab therapy in aggressive B-cell non-Hodgkin’s lymphoma: a retrospective review. British journal of …. 2002 doi: 10.1046/j.1365-2141.2002.03843.x. [DOI] [PubMed] [Google Scholar]
- 86.Francisco J, Cerveny C. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102:1458–1465. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- 87.Wolchok JD, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. The lancet oncology. 2010;11:155–64. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]