

Abstract
Purpose
The purpose of this work was to investigate the effects of triisocyanate composition on the biological and mechanical properties of biodegradable, injectable polyurethane scaffolds for bone and soft tissue engineering.
Methods
Scaffolds were synthesized using reactive liquid molding techniques, and were characterized in vivo in a rat subcutaneous model. Porosity, dynamic mechanical properties, degradation rate, and release of growth factors were also measured.
Results
Polyurethane scaffolds were elastomers with tunable damping properties and degradation rates, and they supported cellular infiltration and generation of new tissue. The scaffolds showed a two-stage release profile of platelet-derived growth factor, characterized by a 75% burst release within the first 24 h and slower release thereafter.
Conclusions
Biodegradable polyurethanes synthesized from triisocyanates exhibited tunable and superior mechanical properties compared to materials synthesized from lysine diisocyanates. Due to their injectability, biocompatibility, tunable degradation, and potential for release of growth factors, these materials are potentially promising therapies for tissue engineering.
Keywords: delivery, injectable, platelet-derived growth factor, polyurethane, scaffold
INTRODUCTION
Due to the high frequency of bone fractures, resulting in over 900,000 hospitalizations and 200,000 bone grafts each year in the United States, there is a compelling clinical need for improved fracture healing therapies (1). Fractures can result from trauma or pathologic conditions, such as osteoporotic compression fractures and osteolytic bone tumors. Autologous bone grafts are an ideal treatment due to their osteogenic, osteoinductive, and osteoconductive properties, but they are available in limited amounts and frequently result in donor site morbidity. Both synthetic and biological biomaterials have been investigated as substitutes for autogenous bone grafts, and a number of desirable properties have been identified for biomaterials designed for orthopedic applications (2). Their use can also be extended to soft tissue repair. The biomaterial and its degradation products must be biocompatible and non-cytotoxic, generating a minimal immune response. High porosity and inter-connected pores facilitate the permeation of nutrients and cells into the scaffold, as well as ingrowth of new tissue. Scaffolds should also undergo controlled degradation, preferably at a rate comparable to new tissue formation, to non-cytotoxic decomposition products. Materials that exhibit gel times of 5–10 min and low temperature exotherms are particularly suitable for clinical use as injectable therapies that can be administered percutaneously using minimally invasive surgical techniques. Additionally, scaffolds should possess sufficient biomechanical strength to withstand physiologically relevant forces. Release of growth factors with fibrogenic, angiogenic, and osteogenic properties, such as platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and bone morphogenetic protein-2 (BMP-2), may further enhance integration of the device and improved healing.
Due to their ability to meet many of the above-mentioned performance characteristics, both synthetic and biopolymers have been investigated as scaffolds for tissue engineering. The poly(α-esters), including polylactic acid (PLA), polyglycolic acid (PGA), and their copolymers (PLGA), are thermoplastic polymers incorporated in a variety of FDA-approved biomedical devices, including surgical sutures, orthopedic fixation, and drug and growth factor delivery (3). Scaffolds prepared from other thermoplastic biomaterials, such as tyrosine-derived polycarbonates and polyphosphazenes, have been shown to exhibit tunable degradation to non-cytotoxic decomposition products, high tensile strength, and bone tissue ingrowth in vivo (4–6). However, thermoplastic biomaterials cannot be injected, and must be melt- or solvent-processed ex vivo to yield solid scaffolds prior to implantation. Injectable hydrogels, such as poly(ethylene glycol) (PEG), collagen, fibrin, chitosan, alginate, and hyaluronan, have been shown to support bone ingrowth in vivo, particularly when combined with angio-osteogenic growth factors (7–13). However, hydrogels lack the robust mechanical properties of thermoplastic polymers.
Two-component reactive polymers are promising scaffolds because they can be formed in situ without the use of solvents. Poly(propylene fumarate) (PPF) can be injected as a liquid and thermally or photo cross-linked in situ with various cross-linking agents, which affect the final mechanical and degradation properties (14). Recently developed porous composite scaffolds have been formed in situ by gas foaming, with up to 61% porosity, 50–500 μm pores, and a compressive modulus of 20–40 MPa (15). PPF biomaterials have been shown to support osteoblast attachment and proliferation in vitro, and ingrowth of new bone tissue in vivo (16–19). Growth factors have been incorporated via PLGA micro-spheres into poly(propylene fumarate) materials for controlled release (20).
Two-component biodegradable polyurethane (PUR) networks have also been investigated as scaffolds for tissue engineering. Porous PUR scaffolds prepared from lysine-derived and aliphatic polyisocyanates by reactive liquid molding have been reported to degrade to non-toxic decomposition products, while supporting the migration of cells and ingrowth of new tissue in vitro and in vivo (21–24). However, many polyisocyanates are toxic by inhalation, and therefore polyisocyanates with a high vapor pressure at room temperature, such as toluene diisocyanate (TDI, 0.018 mmHg) and hexamethylene diisocyanate (HDI, 0.05 mmHg), may not be suitable for injection in a clinical environment. To overcome this limitation, we and others have formulated injectable PUR biomaterials using lysine diisocyanate (LDI, Fig. 1a), a lysine-derived polyisocyanate with a vapor pressure substantially less than that of HDI (23–25). However, two-component polyurethanes prepared from LDI exhibit microphase-mixed behavior, which inhibits the formation of hydrogen bonds between hard segments in adjacent chains and may adversely affect mechanical properties (23).
Fig. 1.
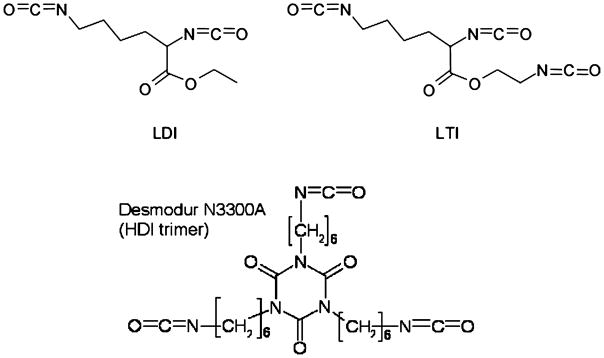
Chemical structures of lysine diisocyanate (LDI), lysine triisocyanate (LTI), and hexamethylene diisocyanate trimer (HDIt).
In this study, the biocompatibility, hydrolytic degradation, and mechanical properties of polyurethane scaffolds prepared from triisocyanates were investigated. Triisocyanates were anticipated to increase the extent of chemical crosslinking, which was conjectured to enhance the mechanical properties of microphase-mixed polyurethane networks. Lysine triisocyanate (LTI, Fig. 1b) is a lysine-derived poly-isocyanate with a vapor pressure of 7.5×10−4 mmHg at 25°C, while Desmodur N3300A is a hexamethylene diisocyanate trimer (HDIt, Fig. 1c) with a vapor pressure of 5.2×10−9 mmHg at 20°C (26). Porous scaffolds were synthesized by a one-shot foaming process, allowing for time to manipulate and inject the polymer, followed by rapid foaming and setting. In this study, the effects of the triisocyanate on biocompatibility, degradation, and mechanical properties were investigated. The potential of the biodegradable PUR scaffolds for controlled release of growth factors was also examined. As anticipated, the PUR scaffolds synthesized from triisocyanates had elastomeric mechanical properties and substantially lower permanent deformation compared to LDI scaffolds. The reaction was mildly exothermic, such that the maximum temperature attained during foaming was 40°C. In vitro, the PUR scaffolds degraded hydrolytically on the order of months at a rate controlled by triisocyanate composition, while enzymatic and locally inflammatory activity seemed to accelerate in vivo degradation. All the PUR scaffolds exhibited both in vitro and in vivo biocompatibility, with minimal immune response limited to the material surface.
MATERIALS AND METHODS
Materials
Glycolide and D,L-lactide were obtained from Polysciences (Warrington, PA), tertiary amine catalyst (TEGOAMIN33) from Goldschmidt (Hopewell, VA), polyethylene glycol (PEG, MW 600 Da) from Alfa Aesar (Ward Hill, MA), and glucose from Acros Organics (Morris Plains, NJ). Lysine triisocyanate (LTI) was purchased from Kyowa Hakko USA (New York), and hexamethylene diisocyanate trimer (HDIt, Desmodur N3300A) was received as a gift from Bayer Material Science (Pittsburgh, PA). PDGF-BB was a gift from Amgen (Thousand Oaks, CA). Sodium iodide (Na125I) for radiolabeling was purchased from New England Nuclear (part of Perkin Elmer, Waltham, MA). Reagents for cell culture were all purchased from HyClone (Logan, UT). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO). Prior to use, glycerol and PEG were dried at 10 mm Hg for 3 h at 80°C, and ε-caprolactone was dried over anhydrous magnesium sulfate, while all other materials were used as received (25).
PUR Scaffold Synthesis
Trifunctional polyester polyols of 900-Da and 1,800-Da molecular weight (abbreviated as 900 and 1,800) were prepared from a glycerol starter and 60% ε-caprolactone, 30% glycolide, and 10% D,L-lactide monomers, and stannous octoate catalyst, as published previously (23,27,28). These components were mixed in a 100-ml reaction flask with mechanical stirring under argon for 36 h at 140°C. They were then dried under vacuum at 80°C for 14 h.
PUR scaffolds were synthesized by one-shot reactive liquid molding of hexamethylene diisocyanate trimer (HDIt; Desmodur N3300A) or lysine triisocyanate (LTI) and hardener comprising either the 900-Da or 1,800-Da polyol, 1.5 parts per hundred parts polyol (pphp) water, 4.5 pphp (1.5 pphp for LTI foams) TEGOAMIN33 tertiary amine catalyst, 1.5 pphp sulfated castor oil stabilizer, and 4.0 pphp calcium stearate pore opener. The isocyanate was added to the hardener and mixed for 15 s in a Hauschild SpeedMixer™ DAC 150 FVZ-K vortex mixer (FlackTek, Inc., Landrum, SC). This reactive liquid mixture then rose freely for 10–20 min (23,27). The targeted index (the ratio of NCO to OH equivalents times 100) was 115. To examine the effects of a hydrophilic polyether segment on the material properties, some materials were synthesized with poly(ethylene glycol) (PEG, 600 Da), such that the total polyol content consisted of 30 or 50 mol% PEG and 70 or 50 mol% of the polyester polyol.
Compression set of the scaffolds was determined using a TA Instruments Q800 Dynamic Mechanical Analyzer (DMA) in static compression mode (New Castle, DE). After measuring their initial heights, triplicate 7 mm diameter cylindrical foam cores were compressed to 50% strain (i.e., 50% of their initial height) for 24 h at room temperature according to ASTM standards (29). The samples recovered for 30 min, and then their final heights were measured. Compression set was calculated as the permanent deformation after the period of compressive stress, expressed as a percentage of the original height.
Core densities were determined from mass and volume measurements of triplicate cylindrical foam cores, of 7 mm diameter×10 mm height samples (29). The core porosities (εC) were subsequently calculated from the measured density values (ρc), where ρP=1200 kg m−3 is the polyurethane specific gravity and ρA=1.29 kg m−3 is the specific gravity of air (27,30).
The pore size and distribution were also assessed by scanning electron microscopy (Hitachi S-4200 SEM, Finchampstead, UK).
Temperature profiles of the reactive mixture during foaming were assessed with a digital thermocouple at the centers of the rising foams. Scaffold degradation rates in vitro were evaluated by measuring the mass loss at various time points up to 36 weeks of incubation of triplicate 10-mg samples in 1 ml phosphate buffered saline (PBS; pH 7.4) at 37°C as described previously. At each time point, the samples were rinsed in deionized water, dried under vacuum for 48 h at room temperature, and weighed. The degradation media from 4 and 8 weeks were reserved for in vitro cell viability experiments.
Thermal Analysis
Thermal transitions of the materials were evaluated by differential scanning calorimetry (DSC) using a Thermal Analysis Q1000 Differential Scanning Calorimeter. 10-mg samples underwent two cycles of cooling (20°C/min) and heating (10°C/min), between −80°C and 100°C.
Mechanical Properties
Dynamic mechanical properties were measured using the DMA in compression and tension modes. Cylindrical 7× 6 mm samples were compressed along the axis of foam rise. The temperature-dependent storage modulus and glass transition temperature (Tg) of each material were evaluated with a temperature sweep of −80°C to 100°C, at a compression frequency of 1 Hz, 20-μm amplitude, 0.3% strain, and 0.2-N static force. The relaxation modulus was evaluated as a function of time with stress relaxation under 2% strain and 0.2-N static force. The frequency-dependent storage modulus was also evaluated with a 0.1 to 10 Hz frequency sweep at a constant temperature of 37°C, with 0.3% strain and 0.2-N static force. Stress-strain curves were generated by controlled-force compression of the cylindrical foam cores at 37°C. With an initial force of 0.1 N, each sample was deformed at 0.1 N/min until it reached 50% strain (i.e. 50% of its initial height). The Young’s (elastic) modulus was determined from the slope of the initial linear region of each stress-strain curve (31). Due to their highly elastic properties, the scaffolds could not be compressed to failure. Therefore, as a measure of compressive strength, the compressive stress of triplicate cylindrical samples after 1 min at 50% strain was measured using the DMA stress relaxation mode at 37°C (29). Calculated from the measured force and cross-sectional sample area, the compressive stress indicates material compliance such that more compliant materials require lower stress to induce a particular strain.
Tensile testing was performed on thin, rectangular scaffold samples (10 mm long×5 mm wide×1.7 mm thick). Stress-strain curves were generated by elongating the samples at 1% strain per minute at 37°C until failure. The Young’s modulus was calculated as described above, and the tensile strength was determined as the stress (kPa) at failure.
In vitro Biocompatibility
MC3T3-E1 embryonic mouse osteoblast precursor cells were statically seeded onto thin foam discs (25×1 mm) at 5×104 cells per well in 24-well tissue-culture polystyrene plates. The cells were cultured with 1 ml α-minimum essential medium (α-MEM) per well, containing 10% fetal bovine serum, 1% penicillin (100 U/ml) and streptomycin (100 μg/ml). After 5 days, the cell-seeded scaffolds were removed from culture, washed with PBS, and transferred to a new 24-well plate to verify cell adherence to the materials. 4 μM Calcein AM from the Invitrogen-Molecular Probes Live/Dead Viability/Cytotoxicity Kit for mammalian cells (Eugene, OR) was added to the samples. Calcein AM dye is retained within live cells, imparting green fluorescence (excitation/emission: 495/515 nm). Cell viability was assessed qualitatively by fluorescent images acquired with an Olympus DP71 camera attached to a fluorescent microscope (Olympus CKX41, U-RFLT50, Center Valley, PA).
In addition, PUR degradation products from 4 and 8 weeks were analyzed for cell viability and cytotoxicity. The same MC3T3-E1 cells were seeded at 5×103 cells per well in a 96-well plate with 90 μl cell culture medium (described above) and 10 μl degradation media or PBS control. After the cells were cultured for 72 h, the media was removed, the wells were rinsed with fresh PBS, and 2 μM Calcein AM was added to the wells. The percentage of viable cells was assessed by quantifying the fluorescence of the samples, in comparison to wells that were cultured with all 100 μl cell culture media, with a Biotek fluorescence microplate reader (Winooski, VT).
In vivo Biocompatibility
After polymerization, the materials were cut into 8× 2 mm discs for in vivo implantation to assess biocompatibility and degradation properties. The discs were sterilized for 5 min in ethanol prior to dorsal subcutaneous implantation in adult male Sprague-Dawley rats. Implants were retrieved from euthanized animals at 5, 14, and 21 days post-implantation, fixed in formalin for 24 h, embedded in paraffin, and processed for histological evaluation with Gomori’s trichrome as well as hematoxylin and eosin staining.
Incorporation of Radiolabeled PDGF-BB
PDGF-BB was labeled with radioactive iodine (125I) using IODO-BEADS Iodination Reagent (Pierce Biotechnology, Rockford, IL). The IODO-beads were incubated in 1 ml Reaction Buffer containing sodium iodide (approximately 1 mCi per 100 μg of protein) for 5 min at room temperature. 110 μl PDGF solution (0.43 mg/ml in PBS) was added to the IODO-BEADS reaction solution and incubated for 25 min at room temperature. The solution was then removed from the IODO-BEADS reaction tube and the 125I-labeled PDGF (125I-PDGF) was separated in a Sephadex disposable PD-10 desalting column (Sigma-Aldrich). Eluted fractions of 200 μl each were collected and analyzed by a Cobra II Autogamma counter (Packard Instrument Co, Meridien, CT) to identify the fractions containing the 125I-PDGF.
The 125I-PDGF was then co-dissolved and lyophilized with heparin and glucose in order to stabilize the protein during lyophilization and scaffold synthesis. Final dosages were 10 and 50 μg 125I-PDGF per gram of foam, each with 0.5 mg heparin and 20 mg glucose per gram of foam. The lyophilized powder was added to the polyol hardener component, which included 50 mol% PEG, before mixing with the isocyanate to prepare the PUR scaffolds.
In vitro Release of PDGF
The initial 125I-PDGF levels in triplicate 50-mg samples for each 125I-PDGF dosage were first measured by the Autogamma counter and then incubated in 1 ml MEM non-essential amino acid solution containing 1% BSA contained in glass vials while mixed end-over-end at 37°C. MEM and BSA were included to mimic the cellular growth environment and minimize adsorption of PDGF onto the scaffolds and vials. The buffer was removed and refreshed from each vial every day for the first 4 days, and then every 2 days until 28 days. The 125I-PDGF concentrations in the release samples were quantified by the Autogamma counter.
Statistical Analysis
Statistical analysis of the results was performed using single factor analysis of variance (ANOVA). In cases where statistical significance is cited, the sample size is greater than or equal to three replicates per material.
RESULTS
PUR Scaffold Characterization
The permanent deformation, or compression set, of LDI, LTI, HDIt, and HDIt + 50% PEG materials is shown in Fig. 2. The LTI (4.7±0.3%), HDIt (2.2±0.5%), and HDIt + 50% PEG (2.5±0.5%) materials exhibited minimal permanent deformation after being subjected to a 50% compressive strain for 24 h. In contrast, materials synthesized from lysine methyl diisocyanate (LDI) displayed a substantially higher compression set of 48.5±2.6%, with statistically significant differences (p< 0.005) (23). Thus the PUR scaffolds synthesized from triisocyanates were more resilient than those prepared from diisocyanates. The favorable mechanical properties of segmented polyurethane elastomers and foams have been attributed to microphase-separation of hard and soft segments and subsequent hydrogen bonding between hard segments (32). However, previous studies have shown that PUR scaffolds prepared from LDI were microphase-mixed and exhibited negligible hydrogen bonding between adjacent hard segments due to the asymmetric structure of LDI (23). Similarly, the FT-IR spectra for LTI and HDIt scaffolds also suggested minimal hydrogen-bonded urethane and urea groups (data not shown). The substantially lower compression set observed for PUR networks synthesized from triisocyanates is thus conjectured to result from the greater degree of chemical crosslinking relative to that achieved with diisocyanates.
Fig. 2.

Compression set of LTI, HDIt, HDIt + 50% PEG, and LDI scaffolds made with the 900-Da triol. LDI materials had larger permanent deformations than did materials with either of the triisocyanates.
The core densities and porosities of the scaffolds (Table I) were assessed at least 24 h after foam synthesis to ensure full curing and drying. The density of the scaffolds ranged from 86–98 kg m−3 and the porosity from 92–93 vol%. The differences between the densities and porosities measured for the materials were not statistically significant (p>0.05). SEM images (Fig. 3) illustrated that the pores were almost uniformly spherical, 200–400 μm in diameter, and interconnected by openings in the pore walls. Previous studies with LDI scaffolds have shown that MC3T3 cells penetrated up to 5 mm into the interior of the scaffolds after 21 days, suggesting that the pores were inter-connected (23). Addition of PEG had an insignificant effect on the scaffold density and porosity, but SEM showed that the pores were more irregularly shaped and variable in size, reaching 600 μm in diameter. The irregular pore shape and rough surface are thought to result from phase-separation of the PEG and polyester polyol components (32).
Table I.
PUR Scaffold Physical and Thermal Properties: Density, Porosity, and Glass Transition Temperatures as Determined by DSC and DMA
| Sample | Density (kg m-3) | Porosity (vol-%) | Tg–DSC (°C) | Tg–DMA (°C) |
|---|---|---|---|---|
| 900/LTI | 87.5±4.6 | 92.8±0.4 | 6.4 | 56.6 |
| 1800/LTI | 86.2±0.9 | 92.9±0.1 | −16.2 | 23.8 |
| 900/HDIt | 98.2±12.5 | 91.9±1.0 | 0.2 | 40.3 |
| 1800/HDIt | 92.8±7.7 | 92.4±0.6 | −20.8 | 28.2 |
| HDIt + 30% PEG | 90.2±2.6 | 92.6±0.2 | −9.8 | 24.3 |
| HDIt + 50% PEG | 93.7±11.4 | 92.3±1.0 | −30.7 | 18.5 |
Fig. 3.

SEM images of foams made with the 900-Da triol suggest interconnected pore structures with mostly uniform pore sizes of 200–500 μm. a LTI (scale bar 600 μm), b HDIt (scale bar 600 μm), c HDIt + 50% PEG (scale bar 750 μm).
A significant advantage of the PUR scaffolds is that they are injectable, as shown in Fig. 4, and therefore can potentially be administered using minimally invasive surgical techniques. Bone tissue necrosis can occur when exposed to temperatures greater than 50°C for longer than 1 min, so it is important to address the reaction temperatures of injectable systems (33). The reaction of polyester polyol and isocyanate to form urethane bonds is exothermic, although the aliphatic polyisocyanates used in this study are less reactive than aromatic polyisocyanates (32). The maximum temperature in the center of the foam was 30.5°C for HDIt materials and 40.0°C for LTI materials, both of which are significantly lower than typical exotherm temperatures of up to 110°C for poly (methyl methacrylate) (PMMA) (34). The gel times of the mixtures, estimated by observing the change in viscosity from a viscous liquid to a non-flowable gel, were approximately 3 min (LTI) and 5 min (HDIt). Despite the higher catalyst concentration used in the HDIt formulations, these polymers exhibited lower reaction exotherms and longer gel times, suggesting that HDIt is less reactive than LTI.
Fig. 4.

Injectability of PUR scaffolds: time-lapse photographs showing injection of the reactive liquid system.
The degradation rates are shown in Fig. 5. All of the materials retained 85–90% of their original mass after 8 weeks. The LTI scaffolds degraded rather quickly thereafter, with only 22% (900/LTI) and 48% (1800/LTI) mass remaining after 14 and 18 weeks, respectively, and no intact mass remaining by 36 weeks. On the other hand, the HDIt materials degraded steadily, with 52–81% mass remaining at 36 weeks. Although PEG initially accelerated degradation within the first 4 weeks, it slowed the long-term degradation rates.
Fig. 5.

In vitro degradation of PUR scaffolds. At 36 weeks, both LTI materials had completely degraded, while the HDIt materials remained at 52–81% of their original masses. Although PEG initially accelerated degradation within the first 4 weeks, it slowed the long-term degradation rates.
Thermal Analysis
DSC thermal profiles of the materials demonstrated single second-order glass transitions. The glass transition temperatures (Table I), extrapolated from the steepest point of the heat flow (mW/mg) vs. temperature (°C) curve during the second heating cycle, ranged from −30.7°C (HDIt + 50% PEG) to 6.4°C (900/LTI). The glass transition temperature of the pure polyols, −41.7°C (900 Da) and −44.7°C (1800 Da), were significantly lower than those of the PUR networks. The substantial increase in the glass transition temperatures of the PUR networks relative to those of the pure polyols suggests that microphase-mixing of hard (isocyanate) and soft (polyol) segments has occurred. Addition of PEG proportionally depressed the glass transition temperatures. Use of the 1800-Da polyol also decreased the transition temperatures, perhaps due to enhanced microphase-separation of the larger soft segments. Glass transition temperatures were also measured by DMA using temperature sweeps (Table I). Surprisingly, the values of Tg as measured by DMA were about 34–50°C higher than those measured by DSC.
Mechanical Properties
Fig. 6 shows the materials analyzed using stress relaxation and frequency sweep tests to evaluate their viscoelastic properties, which were shown to depend on the glass transition temperature. The six materials are organized into three groups in order of increasing temperature. The 900/HDIt + PEG materials (Fig. 6a and d), which had DMA glass transition temperatures of 18.5°C (50% PEG) and 24.3°C (30% PEG), exhibited dynamic mechanical behavior similar to that of an ideal elastomer in the rubbery plateau zone (35). The storage modulus E′, which represents the energy stored elastically, was nearly constant over the entire frequency range (0.1–10 Hz), while the loss modulus E″, which represents the energy lost due to viscous dissipation, was very low at low frequencies and approaches E′ at higher frequencies (e.g., >5 Hz). Similarly, the stress relaxation data showed an initial increase in the relaxation modulus when the strain was applied, followed by a negligible (50% PEG) or slight (30% PEG) decrease in relaxation modulus over 20 min due to relaxation of the polymer network. Taken together, the frequency sweep and stress relaxation data suggest that the PUR scaffolds incorporating PEG are rubbery elastomers.
Fig. 6.

Storage and loss moduli as a function of strain rate (compression) during DMA frequency sweeps from 0.1 to 10 Hz, and stress relaxation response to 2% strain over 20 min. Panels are shown in order of increasing Tg (left to right): materials with PEG (a and d), with 1800-Da polyol (b and e), and 900-Da polyol (c and f).
Frequency sweep and stress relaxation data are presented in Fig. 6b and e for the 1800/LTI and 1800/HDIt materials, which have mechanical glass transition temperatures of 23.8°C and 28.2°C, respectively. The 1800/HDIt material has a glass transition temperature closer to the experimental temperature (37°C), and therefore exhibited viscoelastic properties representative of a material approaching the transition zone, where (a) the values of E′ and E″ increase with increasing frequency, and (b) the value of E″ approaches E′ (36). As E″ approaches E′, an increasing fraction of the energy of deformation is dissipated as heat due to increased friction between polymer chains (36). The vibration damping properties of the material increase with increasing loss modulus E″. The frequency sweep data for the 1800/HDIt material show that E′ increased with increasing frequency and the value of E″ was close to that of E′, thereby suggesting that a substantial portion of the energy of deformation was dissipated as heat. The stress relaxation data are in qualitative agreement with the frequency sweep data. The relaxation modulus increased to about 10 kPa when the strain was applied, and then decreased over 20 min. At short times (corresponding to high frequencies), the period is too short to enable an active segment of the network to exhibit all possible conformations. Therefore, the strain resulting from a given stress is less than that at longer times (lower frequencies); thus the relaxation modulus is expected to decrease with increasing time (decreasing frequency) (36).
In Fig. 6c and f, the frequency sweep and stress relaxation data are presented for the 900/LTI (Tg=56.6°C) and 900/HDIt (Tg=40.3°C) materials. The 900/HDIt material has a Tg slightly greater than 37°C, and therefore exhibited properties typical of the transition zone. The moduli E′ and E″ increased with increasing frequency, and the values of E″ were close to E′. In the stress relaxation experiments, the relaxation modulus initially reached a high value when the strain was applied and then decayed over 20 min by an order of magnitude. The 900/LTI material has a Tg substantially greater than 37°C, and therefore exhibited properties typical of the glassy zone, characterized by storage modulus 2–3 orders of magnitude greater than that in the rubbery plateau. Furthermore, the values of E′ and E″ did not change substantially with increasing frequency.
Stress-strain plots show elastomeric behavior of the PUR scaffolds even up to 50% compressive strain (Fig. 7). The Young’s moduli, calculated from the slope of the initial linear region of the stress-strain curves, are listed in Table II. 900/LTI scaffolds exhibited the highest modulus values, followed by the 900/HDIt materials, while the 1,800-Da polyol or additional PEG appeared to reduce the modulus of the scaffolds. The modulus differences among the materials were statistically significant (p<0.005). The compressive stress at 50% strain ranged from 4.8 to 10.5 kPa for the different scaffold formulations (Table II), and the addition of PEG reduced the compressive stress relative to the equivalent scaffold without PEG. The two materials with PEG had nearly equivalent compressive stress values, but all other differences were statistically significant (p<0.005).
Fig. 7.

Stress-strain curves measured in compression mode. Young’s Modulus values were calculated from the initial slopes.
Table II.
PUR Scaffold Mechanical Properties, as Determined by DMA in Compression and Tension Deformation Modes
| Sample | Tension
|
Compression
|
|||
|---|---|---|---|---|---|
| Young’s modulus (kPa) | Tensile strength (kPa) | Strain at failure (%) | Young’s modulus (kPa) | Compressive stress (kPa) | |
| 900/LTI | 121.8±43.3 | 266.5±33.6 | 216.3±75.2 | 176.5±26.7 | 9.2±0.8 |
| 1800/LTI | 12.1±3.2 | 19.3±7.5 | 169.6±48.5 | 41.6±13.1 | 4.8±0.9 |
| 9000/HDIt | 38.8±8.0 | 33.6±9.1 | 103.9±34.5 | 114.5±29.7 | 10.5±1.0 |
| 1800/HDIt | 8.6±1.5 | 13.3±1.8 | 156.3±38.6 | 25.5±1.5 | 5.2±0.4 |
| HDIt + 30% PEG | 11.2±5.2 | 9.2±0.2 | 104.6±50.9 | 58.2±14.9 | 6.6±0.5 |
| HDIt + 50% PEG | 43.9±17.0 | 20.1±5.0 | 59.2±23.0 | 14.6±2.7 | 6.7±0.6 |
The tensile strength and Young’s modulus of the thin scaffold samples are given in Table II. They were both determined from stress-strain curves performed until sample failure. The trend is similar to the compressive strengths, where the 900/LTI materials had the highest tensile strength (266.5±33.6 kPa), followed by the 900/HDIt materials (33.6± 9.1 kPa). Use of the 1800-Da polyol or PEG decreased the modulus and strength. The Young’s moduli of 1800/LTI, 1800/HDIt, and 900/HDIt + 30% PEG were statistically similar (p >0.05), but all other tensile strength differences were statistically significant (p<0.005).
In vitro Biocompatibility
The MC3T3 cells permeated and adhered to the scaffold interstices, as shown by fluorescent microscope images (Fig. 8). Live cells, as indicated by dye uptake, remained attached to the scaffold during transfer procedures. The cells were easily discriminated from the autofluorescent scaffold material.
Fig. 8.

Calcein AM staining of live cells (green) seeded on PUR scaffolds, which autofluorescence red (excitation/emission 495/515 nm). a LTI, b HDIt, c HDIt + 50% PEG.
The percent viability (Table III) was determined as the proportion of live cells, or fluorescence intensity, in the wells cultured with the 4-week and 8-week degradation products, in comparison to that of cells cultured in media only. Cells cultured with 10 μl PBS exhibited 94.7% viability, while the 4-and 8-week degradation samples yielded 87.5–94.7% and 88.4–89.9% viability, respectively. All differences, including the PBS control sample, were not statistically significant (p>0.05).
Table III.
Percentage of Viable Cells Cultured for 72 h with 4- and 8-week PUR Degradation Products, as well as with PBS Control
| LTI | HDIt | HDIt + 50% PEG | Control (PBS) | |
|---|---|---|---|---|
| 4 weeks | 93.7±8.2% | 94.7±8.6% | 87.5±11.6% | 94.7±10.9% |
| 8 weeks | 89.1±8.0% | 88.4±5.5% | 89.9±10.1% | 94.7±10.9% |
In vivo Biocompatibility
Tissue response was evaluated by subcutaneous implantation of 2×8 mm discs of each formulation in rats for up to 21 days (Fig. 9). During this time, initial infiltration of plasma progressed to the formation of dense granulation tissue. Hence, the implant served as a model of deep wound healing. All of the implants showed progressive invasion of granulation tissue with little evidence of an overt inflammatory response or cytotoxicity. Fibroplasia and angiogenesis appeared to be equivalent among the different formulations. Extracellular matrix with dense collagen fibers progressively replaced the characteristic, early cellular response. The LTI scaffolds exhibited a greater extent of degradation at 21 days, although the incorporation of PEG into the HDIt scaffold accelerated its degradation significantly. Degradation rates were much higher in vivo. With time, each of the materials showed signs of fragmentation and engulfment by a transient, giant cell, foreign body response. After the remnant material was resorbed, giant cells were no longer evident.
Fig. 9.

Trichrome stain of subcutaneous in vivo implants after 5, 14, and 21 days. All scaffolds shown were made with the 900-Da triol. Material remnants are shown as white segments. Granulation tissue, collagen deposition, and giant cell response are visible.
In vitro Release of PDGF
The in vitro release of 125I-PDGF from the polyurethane scaffolds, for both 10 and 50 μg 125I-PDGF per gram of foam, is shown in Fig. 10. The release profiles are essentially identical for each of the two dosages. The cumulative % release is defined as the cumulative elution of 125I-PDGF at each time point divided by the total 125I-PDGF in each sample. The scaffolds showed a two-stage release profile, characterized by a 75% burst release within the first 24 h, and slower release thereafter. By 21 days, 89% of the 125I-PDGF had eluted from the scaffolds.
Fig. 10.

In vitro release of lyophilized 125I-PDGF (10 and 50 μg per gram of scaffold) from T6C3G1L900/HDIt + 50% PEG scaffold. Cumulative release expressed as percentage of total 125I-PDGF initially contained in sample.
DISCUSSION
Polyurethane scaffolds synthesized from aliphatic and lysine-derived polyisocyanates have been reported to support cell attachment and proliferation in vitro, as well as ingrowth of new tissue and degradation to non-cytotoxic decomposition products in vivo (23,37–39). While the low vapor pressure of LDI renders it potentially useful for injectable biomaterials, LDI-based PUR scaffolds synthesized by the gas foaming process displayed poor resiliency, with up to 50% permanent deformation when subjected to compressive loads. The high compression set of LDI-based PUR scaffolds is conjectured to result from the absence of physical crosslinks in the polymer network, as evidenced by the lack of hydrogen-bonded urethane and urea groups in the hard segment (32). For segmented PUR elastomers synthesized from LDI, the microphase morphology depends on the molecular weight of the soft segment (40,41). For LDI elastomers incorporating a 2000 g mol−1 poly(ε-caprolactone) (PCL) diol soft segment, the value of Tg was −52°C, which is close to that of pure PCL diol. However, for soft segments with molecular weights of 1250 or 530 g mol−1, the value of Tg increased 20–45°C, suggesting the presence of significant microphase-mixing that has been attributed to the asymmetric ethyl branch in LDI. Considering that microphase-mixing of LDI segmented elastomers becomes significant at soft segment equivalent weights of 625 g eq−1, it is not surprising that LDI-based PUR networks exhibited microphase-mixing at soft segment equivalent weights of 300 g eq−1. We reasoned that triisocyanates would yield PUR networks with higher chemical crosslink density, thus compensating for the lack of physical crosslinks and improving mechanical properties such as compression set. In this study, PUR scaffolds were prepared from LTI and Desmodur N3300A HDI trimer using the one-shot gas foaming process as described previously for LDI (23,27). Both HDIt and LTI have low vapor pressure at ambient temperature, thus minimizing the risk of exposure by inhalation when the materials are injected. Furthermore, it was of interest to compare the biocompatibility and degradation of PUR scaffolds synthesized from aliphatic and lysine-derived triisocyanates. While LTI and HDIt have been used to synthesize cast elastomers with improved properties, such as optical clarity and thermal stability (42), their use in biodegradable PUR scaffolds has not been previously reported. The effects of triisocyanate composition on biocompatibility, biodegradation, and mechanical properties were investigated, as well as the potential of the PUR scaffolds for release of growth factors.
The data in Fig. 2 demonstrate that the PUR networks synthesized from LTI and HDIt exhibited significantly lower permanent deformation than those synthesized from LDI. Materials in wound healing applications could benefit from greater resilience, which would allow them to better conform to the wound site and maintain contact with the host tissue when subjected to compressive or tensile forces.
Polyether and polyester polyols have been mixed in previous studies to produce foams via prepolymers and chain extension, but not for one-shot foams prepared directly from polyisocyanates without the prepolymer step (37,38,43–46). Polyethers are generally immiscible with polyesters and are typically stabilized with water-soluble polyethersiloxanes (32). However, foams with polyethersiloxane stabilizers have been reported not to support cell attachment or proliferation (27). Instead, we have shown that stable scaffolds can be synthesized with polyether-polyester mixtures using turkey red oil as a stabilizer and surfactant as previously used to stabilize polyester foams (32). These materials were stable with up to 70% PEG.
As shown in Table I, the composition of the polyol component had a substantial effect on the glass transition temperatures of the PUR scaffolds. PUR scaffolds prepared from the 1800 g mol−1 (600 g eq−1) polyol had Tg values ~20°C higher than those prepared from 900 g mol−1 (300 g eq−1) polyol, which is consistent with the effects of soft segment equivalent weight on Tg observed previously for segmented PUR elastomers prepared from LDI (40,41). The addition of PEG also reduced the Tg of the PUR networks, which is attributed to the lower Tg of PEG relative to the polyester polyols. As anticipated, the PUR networks did not display any melting transitions because amorphous polyols were used. In a previous study, PUR scaffolds synthesized from HDIt with PEG and poly(ε-caprolactone) polyols exhibited melting transitions (associated with the semi-crystalline soft segments) ranging from 39–58°C (44). However, no glass transitions were reported within the range of −20–200°C, so the extent of microphase separation of the materials is not known.
While previous studies showed that in vitro degradation is controlled by the polyol composition (23), the data in Fig. 5 demonstrate that the polyisocyanate composition also has a dramatic effect on the degradation of the PUR scaffolds. LTI scaffolds degraded faster than the HDIt materials, which has been attributed to the degradable ester linkage present in the backbone of lysine derived polyisocyanates (see Fig. 1a and b). Hydrolysis of this ester group yields a carboxylic acid group in the polymer, which has been suggested to catalyze further degradation (47). For lysine-derived polyisocyanates, hydrolysis of urethane linkages to lysine has been reported, while others have reported that urethane and urea linkages are only enzymatically degraded (24,39,48). Higher soft segment content may also explain the faster degradation of the LTI materials, due to the higher %NCO (lower equivalent weight) of LTI relative to that of HDIt. In vivo, the materials degraded significantly faster than in vitro, an observation that has been documented previously for porous poly(D-lactic-co-glycolic acid) scaffolds and most likely due to an enzymatic mechanism (49). Furthermore, enzymatic cleavage of the lysine residues likely contributes to accelerated degradation of the LTI scaffolds in vivo (48). Previous studies have shown that the addition of PEG increases the hydrolytic degradation rate, presumably due to the increased hydrophilicity with PEG (38,44–46). The addition of PEG 600 to HDIt foams increased the initial degradation rate (1–8 weeks), which is attributed to increased bulk hydrophilicity resulting from higher PEG content. This increases water absorption into the material, which results in enhanced diffusion of water to hydrolyze the ester linkages, and faster diffusion of degradation products out of the scaffold (32,46). However, at later time points (10–36 weeks), the degradation rate decreased, which is inconsistent with previous studies. Furthermore, addition of PEG was observed to increase the polymer degradation in vivo in the subcutaneous implant model. The cause of the discrepancy between the in vitro and in vivo degradation data is not known.
The PUR scaffolds exhibited elastomeric dynamic mechanical properties, as evidenced by their high elongation at break and low compression set; they ranged from ideal elastomers, where the deformation energy is primarily stored elastically, to high-damping elastomers, where the energy is both stored elastically and thermally dissipated. By varying the composition of the triisocyanate and polyol components, it is possible to prepare elastomeric PUR scaffolds with tunable damping properties. Application of rubbery elastomers (i.e., low-damping) as scaffolds for bone defects has been suggested to promote intimate contact between the implant and the host bone (37). The elastomer can be compressed prior to implantation, where it then expands in the wound to maintain intimate contact with the local tissue. Maintaining good contact between the bone and implant may promote the migration of local osteoprogenitor cells from the bone into the implant, thereby enhancing bone regeneration. It has also been suggested that elastomeric properties can protect the implant from shear forces at the bone-implant interface. However, the effects of the damping properties of the scaffold on tissue regeneration are not known. If the damping is excessive, then, upon exposure to physiological strains, the relaxation modulus may drop to values too low to provide significant support.
Materials prepared from triisocyanates in the present study displayed slightly higher densities but comparable porosities to one-shot polyurethanes made from LDI in a previous study (27). However, the compressive strength (i.e., the compressive stress measured at 50% strain) of the HDIt and LTI materials (5–15 kPa) was higher than that of the LDI materials (2–4 kPa). HDI-prepolymer foams of comparable density (80–107 kg m−3) from a previous study were generally stronger than the one-shot HDIt and LTI foams of the present study, with compressive strengths of 30–85 kPa (at 40% strain) versus 5–15 kPa (at 50% strain) for the HDIt and LTI foams (44,45). However, the Young’s moduli of the HDI-prepolymer foams are lower, at 9–21 kPa, compared to 26–202 kPa for the one-shot foams. While the HDI-prepolymer foams exhibit elastomeric mechanical properties and good biocompatibility in vivo, they are not injectable due to the high temperature (60°C) cure step.
In a previous study, endothelial cell adhesion in vitro to a poly(ether ester urethane)urea scaffold was inversely proportional to the hydrophilicity, although smooth muscle cells grew faster in the more hydrophilic scaffold (38,46). Bone regeneration occurred in polyurethane scaffolds implanted into defects of sheep iliac crests, with more calcium phosphate salts mineralized in defects with hydrophilic scaffolds, which also had the highest porosities (43). Original ilium thickness was reestablished only in defects with the most hydrophobic scaffolds, perhaps because their slow degradation rates allowed more time for bone ingrowth (37). In the PUR scaffolds of the present study, greater collagen accumulation appeared in the implants with PEG scaffolds. However, it cannot be determined conclusively whether this is a direct result of the increased hydrophilicity, the faster degradation rate, or lower damping properties of the PEG scaffolds.
A biodegradable, elastomeric polyurethane scaffold that released basic fibroblast growth factor (bFGF) has been reported for soft tissue engineering applications (50). Segmented PUR elastomers were synthesized from butane diisocyanate (BDI), putrescine, and poly(ε-caprolactone) diol. Scaffolds incorporating bFGF were processed using a thermally induced phase separation method. The scaffolds showed a two-stage release behavior characterized by an initial period of fast release (19–37% on day 1) followed by a second period of slow release over 4 weeks. The released bFGF was shown to induce proliferation of rat smooth muscle cells. However, in this study, the bFGF was released from a pre-formed polymer scaffold, not from a reactive polymer. PUR scaffolds prepared by reactive liquid molding of LDI, glycerol, water, and ascorbic acid (AA) have been shown to support controlled release of AA over 60 days (51). By dissolving the AA in the glycerol prior to adding the LDI, the AA was covalently bound to the polymer through reaction of the primary hydroxyl group in the AA with LDI to form urethane linkages. In the HDIt material of the present study, PDGF-BB was added as a lyophilized powder to minimize its reaction with the PUR. While the covalent binding approach was successful with a small molecule such as ascorbic acid, proteins were expected to lose their three-dimensional structure and denature upon reaction with the polymer. The faster release of PDGF-BB from the HDIt scaffolds compared to release of ascorbic acid from the LDI scaffolds is attributed to the absence of covalent bonds. As the scaffold swells with water, the PDGF dissolves and diffuses out of the scaffold, and the release is not dependent on the hydrolysis of covalent bonds.
CONCLUSIONS
Biodegradable PUR scaffolds prepared from triisocyanates using a one-shot process exhibited elastomeric mechanical properties and substantially lower compression set relative to scaffolds prepared from LDI. Their elastic behavior is thought to promote intimate contact between the material and surrounding tissue, which may facilitate ingrowth of new tissue and help keep the material in place when subjected to physiologically relevant strains. Both low-and high-damping elastomers can be synthesized by varying the glass transition temperature of the materials. Processing by two-component reactive liquid molding allows them to be injected and conform to the wound boundaries. The gel time of 3–5 min and moderate exotherm (e.g., < 15°C increase) suggests their potential utility for injectable wound healing applications. The materials supported cellular infiltration and generation of new tissue and facilitate neodermis formation with minimal inflammation. Signaling molecules were incorporated as labile powders upon synthesis, further enhancing their regenerative capabilities.
Acknowledgments
This work was funded by the United States Army Institute for Surgical Research (DOD-W81XWH-06–1–0654), the Orthopaedic Trauma Research Program (DOD-W81XWH-07–1–0211), Vanderbilt Skin Diseases Research Core Center (NIH-AR41943), and the Department of Veterans Affairs. The authors acknowledge Jayasri DasGupta and R. Michael Slowey for their assistance with the in vivo studies.
ABBREVIATIONS
- AA
Ascorbic acid
- HDIt
Hexamethylene diisocyanate trimer
- LTI
Lysine triisocyanate
- PDGF
Platelet-derived growth factor
- PEG
Poly(ethylene glycol)
- PUR
Polyurethane
- 900
900-MW trifunctional polyol of 60/30/10 ε-caprolactone/glycolide/lactide
- 1800
1800-MW trifunctional polyol of 60/30/10 ε-caprolactone/glycolide/lactide
References
- 1.American Academy of Orthopaedic Surgeons. website ( www.aaos.org)
- 2.Temenoff JS, Mikos AG. Injectable biodegradable materials for orthopedic tissue engineering. Biomaterials. 2000;21:2405–2412. doi: 10.1016/s0142-9612(00)00108-3. [DOI] [PubMed] [Google Scholar]
- 3.Gunatillake P, Mayadunne R, Adhikari R, El-Gewely MR. Biotechnol Annu Rev. Vol. 12. Elsevier; 2006. Recent developments in biodegradable synthetic polymers; pp. 301–347. [DOI] [PubMed] [Google Scholar]
- 4.Bourke SL, Kohn J. Polymers derived from the amino acid —tyrosine: polycarbonates, polyarylates and copolymers with poly(ethylene glycol) Adv Drug Deliv Rev. 2003;55:447–466. doi: 10.1016/s0169-409x(03)00038-3. [DOI] [PubMed] [Google Scholar]
- 5.Ertel SI, Kohn J. Evaluation of a series of tyrosine-derived polycarbonates as degradable biomaterials. J Biomed Materi Res. 1994;28:919–930. doi: 10.1002/jbm.820280811. [DOI] [PubMed] [Google Scholar]
- 6.Yu C, Kohn J. Tyrosine-PEG-derived poly(ether carbonate)s as new biomaterials: part I: synthesis and evaluation. Biomaterials. 1999;20:253–264. doi: 10.1016/s0142-9612(98)00169-0. [DOI] [PubMed] [Google Scholar]
- 7.Augst AD, Kong HJ, Mooney DJ. Alginate hydrogels as biomaterials. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 8.Chen RR, Mooney DJ. Polymeric growth factor delivery strategies for tissue engineering. Pharm Res. 2003;20:1103–1112. doi: 10.1023/a:1025034925152. [DOI] [PubMed] [Google Scholar]
- 9.Kipshidze N, Chawla P, Keelan MH. Fibrin Meshwork as a Carrier for Delivery of Angiogenic Growth Factors in Patients With Ischemic Limb. Mayo Clin Proc. 1999;74:47–848. doi: 10.4065/74.8.847-a. [DOI] [PubMed] [Google Scholar]
- 10.Labhasetwar V, Bonadio J, Goldstein S, Chen W, Levy RJ. A DNA controlled-release coating for gene transfer: transfection in skeletal and cardiac muscle. J Pharm Sci. 1998;87:1347–1350. doi: 10.1021/js980077+. [DOI] [PubMed] [Google Scholar]
- 11.Mizuno K, Yamamura K, Yano K, Osada T, Saeki S, Takimoto N, Sakurai T, Nimura Y. Effect of chitosan film containing basic fibroblast growth factor on wound healing in genetically diabetic mice. J Biomed Materi Res A. 2003;64A:177–181. doi: 10.1002/jbm.a.10396. [DOI] [PubMed] [Google Scholar]
- 12.Simmons CA, Alsberg E, Hsiong S, Kim WJ, Mooney DJ. Dual growth factor delivery and controlled scaffold degradation enhance in vivo bone formation by transplanted bone marrow stromal cells. Bone. 2004;35:562–569. doi: 10.1016/j.bone.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 13.Zawko SA, Truong Q, Schmidt CE. Drug-binding hydrogels of hyaluronic acid functionalized with β-cyclodextrin. J Biomed Materi Res A. 2008 doi: 10.1002/jbm.a.31845. 9999:NA. [DOI] [PubMed] [Google Scholar]
- 14.Timmer MD, Ambrose CG, Mikos AG. Evaluation of thermal- and photo-crosslinked biodegradable poly(propylene fumarate)-based networks. J Biomed Materi Res A. 2003;66A:811–818. doi: 10.1002/jbm.a.10011. [DOI] [PubMed] [Google Scholar]
- 15.Kim CW, Talac R, Lu L, Moore MJ, Currier BL, Yaszemski MJ. Characterization of porous injectable poly-(propylene fumarate)-based bone graft substitute. J Biomed Materi Res A. 2007 doi: 10.1002/jbm.a.31633. 9999:NA. [DOI] [PubMed] [Google Scholar]
- 16.Fisher JP, Vehof JWM, Dean D, van der Waerden JP, Holland TA, Mikos AG, Jansen JA. Soft and hard tissue response to photocrosslinked poly(propylene fumarate) scaffolds in a rabbit model. J Biomed Materi Res. 2002;59:547–556. doi: 10.1002/jbm.1268. [DOI] [PubMed] [Google Scholar]
- 17.Peter SJ, Lu L, Kim DJ, Mikos AG. Marrow stromal osteoblast function on a poly(propylene fumarate)/[beta]-tricalcium phosphate biodegradable orthopaedic composite. Biomaterials. 2000;21:1207–1213. doi: 10.1016/s0142-9612(99)00254-9. [DOI] [PubMed] [Google Scholar]
- 18.Peter SJ, Miller ST, Zhu G, Yasko AW, Mikos AG. In vivo degradation of a poly(propylene fumarate)/β-tricalcium phosphate injectable composite scaffold. J Biomed Materi Res. 1998;41:1–7. doi: 10.1002/(sici)1097-4636(199807)41:1<1::aid-jbm1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 19.Yaszemski MJ, Payne RG, Hayes WC, Langer RS, Aufdemorte TB, Mikos AG. The ingrowth of new bone tissue and initial mechanical properties of a degrading polymeric composite scaffold. Tissue Eng. 1995;1:41–52. doi: 10.1089/ten.1995.1.41. [DOI] [PubMed] [Google Scholar]
- 20.Kempen DHR, Lu L, Kim C, Zhu X, Dhert WJA, Curreir BL, Yaszemski MJ. Controlled drug release from a novel injectable biodegradable microsphere/scaffold composite based on poly(propylene fumarate) J Biomed Materi Res A. 2006;77A:103–111. doi: 10.1002/jbm.a.30336. [DOI] [PubMed] [Google Scholar]
- 21.Bonzani IC, Adhikari R, Houshyar S, Mayadunne R, Gunatillake P, Stevens MM. Synthesis of two-component injectable polyurethanes for bone tissue engineering. Biomaterials. 2007;28:423–433. doi: 10.1016/j.biomaterials.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 22.Gorna K, Gogolewski S. Preparation, degradation, and calcification of biodegradable polyurethane foams for bone graft substitutes. J Biomed Materi Res A. 2003;67A:813–827. doi: 10.1002/jbm.a.10148. [DOI] [PubMed] [Google Scholar]
- 23.Guelcher S, Srinivasan A, Hafeman A, Gallagher K, Doctor J, Khetan S, McBride S, Hollinger J. Synthesis, in vitro degradation, and mechanical properties of two-component poly (ester urethane)urea scaffolds: effects of water and polyol composition. Tissue Eng. 2007;13:2321–2333. doi: 10.1089/ten.2006.0395. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JY, Beckman EJ, Hu J, Yang GG, Agarwal S, Hollinger JO. Synthesis, biodegradability, and biocompatibility of lysine diisocyanate-glucose polymers. Tissue Eng. 2002;8:771–785. doi: 10.1089/10763270260424132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adhikari R, Gunatillake PA. Biodegradable polyurethane/urea compositions. 2004 [Google Scholar]
- 26.Kitai M, Ryuutou H, Yahata T, Hara Y, Iwane H, Soejima Y. U. PTO, editor. Aliphatic triisocyanate compound, process for producing the same, and polyurethane resin made from the compound. 2002. Vol. 20020123644 A1 (U. PTO, ed) [Google Scholar]
- 27.Guelcher SA, Patel V, Gallagher KM, Connolly S, Didier JE, Doctor JS, Hollinger JO. Synthesis and in vitro biocompatibility of injectable polyurethane foam scaffolds. Tissue Eng. 2006;12:1247–1259. doi: 10.1089/ten.2006.12.1247. [DOI] [PubMed] [Google Scholar]
- 28.Sawhney AS, Hubbell JA. Rapidly degraded terpolymers of D,L-lactide, glycolide, and ε-caprolactone with increased hydrophilicity by copolymerization with polyethers. J Biomed Mater Res. 1990;24:1397–1411. doi: 10.1002/jbm.820241011. [DOI] [PubMed] [Google Scholar]
- 29.ASTM-International. D3574–05. Standard test methods for flexible cellular materials-slab, bonded, and molded urethane foams. 08.02:360–368. 2007. [Google Scholar]
- 30.Evans G, Atkinson B, Jameson G. The Jameson cell. In: Matis K, editor. Flotation Science and Engineering. Marcel Dekker; New York: 1995. p. 353. [Google Scholar]
- 31.ASTM-International. D695–02a. Standard test method for compressive properties of rigid plastics. 14.02. 2007. [Google Scholar]
- 32.Oertel G. Polyurethane Handbook. Hanser Gardner Publications; Berlin: 1994. [Google Scholar]
- 33.Eriksson R, Albrektsson T, Magnusson B. Assessment of bone viability after heat trauma. A histological, histochemical and vital microscopic study in the rabbit. Scand J Plast Reconstr Surg. 1984;18:261–268. doi: 10.3109/02844318409052849. [DOI] [PubMed] [Google Scholar]
- 34.Meyer PJ, Lautenschlager E, Moore B. On the setting properties of acrylic bone cement. J Bone Jt Surg. 1973;55:149–156. [PubMed] [Google Scholar]
- 35.Mark JE, Erman E, Eirich FR. Science and Technology of Rubber. Academic Press, Inc; San Diego, CA: 1994. [Google Scholar]
- 36.Kramer O, Hvidt S, Ferry JD. Dynamic mechanical properties. In: Mark JE, Erman E, Eirich FR, editors. Science and Technology of Rubber. Academic Press, Inc; San Diego, CA: 1994. [Google Scholar]
- 37.Gogolewski S, Gorna K, Turner AS. Regeneration of bicortical defects in the iliac crest of estrogen-deficient sheep, using new biodegradable polyurethane bone graft substitutes. J Biomed Mater Res A. 2006;77A:802–810. doi: 10.1002/jbm.a.30669. [DOI] [PubMed] [Google Scholar]
- 38.Guan J, Fujimoto KL, Sacks MS, Wagner WR. Preparation and characterization of highly porous, biodegradable polyurethane scaffolds for soft tissue applications. Biomaterials. 2005;26:3961–3971. doi: 10.1016/j.biomaterials.2004.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang JY, Beckman EJ, Piesco NP, Agarwal S. A new peptide-based urethane polymer: synthesis, biodegradation, and potential to support cell growth in vitro. Biomaterials. 2000;21:1247–1258. doi: 10.1016/s0142-9612(00)00005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Groot JH, de Vrijer R, Wildeboer BS, Spaans CS, Pennings AJ. New biomedical polyurethane ureas with high tear strengths. Polym Bull. 1997;38:211–218. [Google Scholar]
- 41.Skarja GA, Woodhouse KA. Structure-property relationships of degradable polyurethane elastomers containing an amino acid-based chain extender. J Appl Polym Sci. 2000;75:1522–1534. [Google Scholar]
- 42.Tsutsumi N, Yoshizaki S, Sakai W, Kiyotsukuri T. Nonlinear optical polymers. 1. Novel network polyurethane with azobenzene dye in the main frame. Macromolecules. 1995;28:6437–6442. [Google Scholar]
- 43.Gogolewski S, Gorna K. Biodegradable polyurethane cancellous bone graft substitutes in the treatment of iliac crest defects. J Biomed Mater Res A. 2007;80A:94–101. doi: 10.1002/jbm.a.30834. [DOI] [PubMed] [Google Scholar]
- 44.Gorna K, Gogolewski S. Molecular stability, mechanical properties, surface characteristics and sterility of biodegradable polyurethanes treated with low-temperature plasma. Polym Degrad Stab. 2003;79:475–485. [Google Scholar]
- 45.Gorna K, Polowinski S, Gogolewski S. Synthesis and characterization of biodegradable poly(ε-caprolactone urethane) s. I. Effect of the polyol molecular weight, catalyst, and chain extender on the molecular and physical characteristics. J Polym Sci A Polym Chem. 2002;40:156–170. [Google Scholar]
- 46.Guan J, Sacks MS, Beckman EJ, Wagner WR. Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: synthesis, characterization and cytocompatibility. Biomaterials. 2004;25:85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- 47.Bruin P, Veenstra GJ, Nijenhuis AJ, Pennings AJ. Design and synthesis of biodegradable poly(ester-urethane) elastomer networks composed of non-toxic building blocks. Die Makromolekulare Chemie, Rapid Commun. 1988;9:589–594. [Google Scholar]
- 48.Elliott SL, Fromstein JD, Santerre JP, Woodhouse KA. Identification of biodegradation products formed by L-phenylalanine based segmented polyurethaneureas. J Biomater Sci Polym Ed. 2002;13:691–711. doi: 10.1163/156856202320269166. [DOI] [PubMed] [Google Scholar]
- 49.Lu L, Peter SJ, Lyman MD, Lai HL, Leite SM, Tamada JA, Uyama S, Vacanti JP, Langer R, Mikos AG. In vitro and in vivo degradation of porous poly(D-lactic-co-glycolic acid) foams. Biomaterials. 2000;21:1837–1845. doi: 10.1016/s0142-9612(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 50.Guan J, Stankus JJ, Wagner WR. Biodegradable elastomeric scaffolds with basic fibroblast growth factor release. J Control Release. 2007;120:70–78. doi: 10.1016/j.jconrel.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang JY, Doll BA, Beckman EJ, Hollinger JO. A biodegradable polyurethane-ascorbic acid scaffold for bone tissue engineering. J Biomed Materi Res A. 2003;67:389–400. doi: 10.1002/jbm.a.10015. [DOI] [PubMed] [Google Scholar]