

Abstract
Dyrk1A is a member of the mammalian Dyrk [dual-specificity tyrosine-(Y)-phosphorylation regulated kinase] family of protein kinases that is expressed at high levels in the brain, but its role in the development and function of this organ is not well understood. The human DYRK1A gene is located on trisomic chromosome 21 in Down syndrome (DS) patients, leading to its overexpression. Dyrk1A is also overexpressed in animal models of DS and in gene-specific transgenic mice that consistently exhibit cognitive impairment. To elucidate the cellular and molecular mechanisms that are affected by increased levels of Dyrk1A in the developing brain, we overexpressed this kinase in the embryonic mouse neocortex using the in utero electroporation technique. We found that Dyrk1A overexpression inhibits neural cell proliferation and promotes premature neuronal differentiation in the developing cerebral cortex without affecting cell fate and layer positioning. These effects are dependent on the Dyrk1A kinase activity and are mediated by the nuclear export and degradation of cyclin D1. This study identifies specific Dyrk1A-induced mechanisms that disrupt the normal process of corticogenesis and possibly contribute to cognitive impairment observed in DS patients and animal models.
Introduction
Down syndrome (DS) is caused by the triplication of genes on chromosome 21 and is characterized by several physical and cognitive abnormalities (Nadel, 2003). Mental retardation is a prominent phenotype observed in virtually all DS patients. However, despite of genomic advances that led to the complete cataloging of the genes on chromosome 21, the specific genes that alter brain development and cause mental retardation in DS have not yet been identified. The dual-specificity tyrosine (Y)-phosphorylation regulated kinase 1A (DYRK1A) gene is located within the DS critical region (Song et al., 1996), and it is overexpressed in the DS brain (Guimera et al., 1999). Several studies using transgenic mice have demonstrated that Dyrk1A overexpression leads to cognitive impairment (Smith and Rubin, 1997; Altafaj et al., 2001; Branchi et al., 2004; Ahn et al., 2006). However, the molecular and cellular bases of this behavioral phenotype are not understood.
Members of the Dyrk family of protein kinases play a role in proliferation and differentiation in a variety of cell types. Dyrk kinases, which include several vertebrate, invertebrate, and lower eukaryotic orthologs, are characterized by highly conserved Dyrk-homology (DH) and kinase domains. Dyrk1A is the best-characterized mammalian member of this family. In addition to the DH and kinase domains, Dyrk1A also contains a bipartite nuclear localization signal (NLS) and a PEST domain regulating protein stability. A unique C-terminal region characterized by a stretch of histidine residues and a serine/threonine-rich sequence distinguishes Dyrk1A from its closest homologs, the Drosophila Minibrain (mnb) and the mammalian Dyrk1B kinases (Kentrup et al., 1996; Becker and Joost, 1999; Himpel et al., 2000). mnb and Dyrk1B have both been shown to affect proliferation and/or differentiation of progenitor cells. In Drosophila, the loss-of-function mnb mutation primarily affects postembryonic neurogenesis, resulting in abnormal neuroblast proliferation. This defect leads to the generation of an insufficient number of neurons in the larval brain and correlates with an impairment in odor-discrimination learning (Tejedor et al., 1995). In mammals, Dyrk1B controls cell cycle progression, differentiation, and the survival of myoblasts through the regulation of the cell cycle proteins p27 and cyclin D1 (Mercer and Friedman, 2006).
Dyrk1A is the only member of its family that is abundantly expressed in the brain (Song et al., 1996; Rahmani et al., 1998; Hämmerle et al., 2008). Genetic disruptions in the human DYRK1A lead to microcephaly, seizures, and developmental delay (Møller et al., 2008). In the mouse, homozygous loss of Dyrk1A causes early embryonic lethality, whereas heterozygous mutant mice appear be growth retarded (Fotaki et al., 2004). Expression studies in the chick CNS (Hämmerle et al., 2002) and in the mouse forebrain (Hämmerle et al., 2008) suggested a role in the regulation of the transition from proliferation to differentiation. Based on these observations, we postulated that altered Dyrk1A expression in neural cells might adversely affect the normal process of neurogenesis. Here we demonstrate that Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells in the developing mouse cerebral cortex through a cyclin D1-mediated mechanism.
Materials and Methods
Expression vectors.
An expression plasmid encoding Dyrk1A–green fluorescent protein (GFP) fusion protein under the cytomegalovirus (CMV) promoter was constructed by subcloning the full-length mouse Dyrk1A cDNA in-frame into the phosphorylated enhanced GFP (pEGFP)–N1 vector (Clontech). To generate the kinase mutant pDyrk1AK188REGFP, the codon corresponding to lysine residue 188 was converted to arginine by site-directed mutagenesis. This mutation abolishes kinase activity by preventing ATP binding to the catalytic domain (Becker et al., 1998). To generate a construct that lacks the kinase domain (pDyrk1AΔ165-478EGFP), we digested pDyrk1A–EGFP with HindIII and SacII restriction enzymes to linearize the plasmid and remove amino acids containing the kinase domain and the C terminus of Dyrk1A. Subsequently, we amplified a C-terminus fragment of Dyrk1A by PCR using forward and reverse primers in which HindIII and SacII were added. The resulting 816 bp fragment was reintroduced into the linearized pDyrk1A–EGFP plasmid by ligation at the HindIII and SacII sites to generate the pDyrk1AΔ165-478EGFP. The pCyclin D1–hemagglutinin (HA) plasmid was a gift from Sarah J. Freemantle (Dartmouth Medical School, Hanover, NH). pmCherry encoding a red fluorescent protein was a gift from Richard Behringer (M. D. Anderson Cancer Center, Houston, TX). All expression plasmids used for in utero electroporation (IUE) were constructed by subcloning the wild-type, kinase mutant, and kinase deleted Dyrk1A, cyclin D1–HA, and mCherry cDNAs into a vector containing the CMV–actin–β-globin (CAG) promoter (a gift from Dennis O'Leary, The Salk Institute for Biological Studies, La Jolla, CA).
In utero electroporation and 5-bromo-2-deoxyuridine labeling.
In utero electroporation experiments were performed using ICR mice (Taconic Farms) housed in fully accredited Association for Assessment and Accreditation of Laboratory Animal Care facilities at Rutgers, the State University of New Jersey in accordance with an approved animal protocol. Expression plasmids under the control of the CAG promoter were used as described previously (Saito and Nakatsuji, 2001). Briefly, ∼3 μl of a plasmid mix (3 μg) containing Fast Green dye (Sigma) was injected and electroporated into the lateral ventricle of embryonic day 14.5 (E14.5) embryos using the ElectroSquarePorator ECM 830 (BTX) set at five 50 ms pulses of 40 V with 950 ms intervals. For cotransfection, plasmids were used at the 1:1 ratio. Unless otherwise stated, embryos were killed 24 h after electroporation, and the brain tissue was dissected and fixed overnight in 4% paraformaldehyde (PFA) at 4°C and then placed in 30% sucrose/1× PBS mix for cryoprotection. Brains were frozen in OCT (Tissue-Tek) and sectioned coronally at 20 μm. For 5-bromo-2-deoxyuridine (BrdU) (Sigma) labeling, electroporated pregnant dams were treated with 100 μg/g BrdU by intraperitoneal injection 20 h after IUE. All experiments were conducted at least in triplicates.
Autofluorescence and immunofluorescence of brain tissue sections.
Brain sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) and directly imaged by confocal microscopy to visualize EGFP autofluorescence. For immunofluorescence, sections were permeabilized in 0.01% Triton X-100 in PBS, blocked with 10% normal goat serum/0.01% Triton X-100/1× PBS for 1 h at room temperature, and incubated overnight at 4°C in primary antibodies diluted in blocking solution. Primary antibodies were as follows: rabbit anti-EGFP (1:1000 dilution; Abcam); rabbit anti-Tbr1 (1:1000 dilution; Millipore Bioscience Research Reagents); rabbit anti-Tbr2 (1:1000 dilution; Abcam); rabbit anti-phospho-Histone H3 (1:250 dilution; Millipore Bioscience Research Reagents); rabbit anti-Cux1 (1:100 dilution; Santa Cruz Biotechnology); mouse anti-neuronal-specific nuclear protein (NeuN) (1:500 dilution; Millipore Bioscience Research Reagents); mouse anti-Tuj1 (1:500 dilution; Covance); and mouse anti-HA (1:1000 dilution; Covance). Sections were then washed and incubated in AlexaFluor 488-, AlexaFluor 594-, or AlexaFluor 647-conjugated secondary antibodies (1:1000 dilution; Invitrogen) for 1 h at room temperature. Finally, sections were mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories). For BrdU labeling, sections were incubated in 2N hydrochloric acid for 30 min at 37°C, followed by neutralization with 100 mm sodium borate, pH 8.5, for 10 min at room temperature. Immunofluorescence staining proceeded as above, using primary rat anti-BrdU antibody (1:100 dilution; Abcam). For terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay, we used the In Situ Cell Death Detection kit (Roche) according to the protocol of the manufacturer.
Imaging and quantification of fluorescent cells.
Confocal z-stack images were acquired using a Carl Zeiss LSM 510 Meta confocal microscope. For quantification of EGFP-positive (EGFP+) cell distribution in cortical regions, sections were stained for TuJ1 and a line was drawn between the strongly labeled intermediate zone (IZ) and the poorly labeled subventricular (SVZ)/ventricular (VZ). This latter region was further divided in two: the top part was designated SVZ, and the bottom part was designated VZ. The percentage of EGFP+ cells in each region was then calculated from multiple sections and triplicate IUE experiments. For quantification of double-labeled cells, the NIH ImageJ software was used to measure cells from confocal z-stack images. All experiments were conducted in triplicates and quantified in a blind manner.
Neuro2a cell culture and transfection.
Neuro2a cells were grown and maintained in DMEM (low glucose; Invitrogen) with 10% fetal bovine serum (Invitrogen) at 37°C in 5% CO2. Transfection of expression constructs (1:1 ratio) was conducted using Fugene 6 (Roche) according to the protocol of the manufacturer. For cell counts, transfected Neuro2a cells were trypsinized to obtain single-cell suspensions. A hemocytometer was used to count the number of EGFP+ and EGFP-negative (EGFP−) cells from each well under an inverted fluorescence microscope. All experiments were conducted in triplicates and quantified in a blind manner.
Immunofluorescence of Neuro2a cells.
Neuro2a cells were fixed 48 h after transfection using ice-cold 4% PFA at room temperature for 20 min. Cells were then washed with 1× PBS, permeabilized in 0.01% Triton X-100/1× PBS, blocked with 10% normal serum/0.1% Triton X-100/1× PBS for 20 min at room temperature, and incubated in mouse anti-HA (1:1000 dilution; Covance) diluted in blocking buffer for 1 h at room temperature. After primary antibody incubation, cells were washed and incubated in AlexaFluor 594-conjugated goat anti-mouse IgG (1:1000 dilution; Invitrogen) for 30 min at room temperature. Cells were then washed with 1× PBS and mounted on cover-glass slides using Vectashield Mounting Medium with DAPI (Vector Laboratories). Cells were imaged using a Carl Zeiss LSM 510 Meta confocal microscope. The number of double-labeled and single-labeled cells was quantified from captured images using NIH ImageJ software. All experiments were conducted in triplicates and quantified in a blind manner.
Western blot analysis.
Neuro2a cells were lysed in radioimmunoprecipitation assay buffer and cleared by centrifugation at 14,000 rpm for 10 min at 4°C. Soluble extracts (30 μl) were loaded onto 4–12% Tris–glycine SDS-PAGE gels (Invitrogen), separated at 120 V for 2 h, and transferred to 0.22 μm nitrocellulose membrane at 200 mA for 2 h. The membranes were blocked with 3% milk/1× TBS-T (Tris-buffered saline with 0.1% Tween 20) for 1 h at room temperature, followed by incubation with primary antibodies diluted in 0.3% milk/1× TBS-T overnight at 4°C, and incubation with secondary antibodies (1:1000 dilution; goat anti-mouse IgG; Invitrogen) for 1 h at room temperature. Membranes were washed three to four times in 1× TBS-T for 1 h, incubated with ECL-Plus Western Blotting Detection System (GE Healthcare) for 5 min, and exposed to autoradiographic film (Denville). The following antibodies were used: mouse monoclonal anti-cyclin D1, 1:500 dilution (Santa Cruz Biotechnology); mouse anti-actin, 1:1000 dilution (Millipore Bioscience Research Reagents); and rabbit anti-EGFP, 1:1000 dilution (Abcam).
Statistical analysis.
We used the Student's t test for all statistical analysis.
Results
Expression of Dyrk1A–EGFP plasmids in the embryonic mouse neocortex by in utero electroporation
Dyrk1A is normally expressed in neurogenic regions of the embryonic and postnatal mouse brain (Song et al., 1996; Hämmerle et al., 2008). These regions include the VZ and SVZ of the mouse embryonic neocortex. To examine the effect of Dyrk1A overexpression on neurogenesis in vivo, we used the IUE technique to target neural progenitor cells that line the lateral ventricle of the neocortex (Saito and Nakatsuji, 2001). For these experiments, we generated EGFP fusion constructs expressing wild-type Dyrk1A, the catalytically inactive Dyrk1AK188R point mutant (Kentrup et al., 1996; Becker et al., 1998) or a mutant protein that lacks the kinase domain (Dyrk1AD165–478) under the under the control of the CAG hybrid promoter (Fig. 1A). As a control, we used a plasmid encoding a nuclear-targeted EGFP (pCAG–nEGFP). When these plasmids were electroporated into the mouse brain at E14.5, all proteins were readily detectable by autofluorescence as early as 24 h after transfection (Fig. 1C), and their expression persisted for several days (see Fig. 5). Confocal analysis of transfected neural cells revealed that all proteins were localized to the nucleus and that wild-type as well as mutant Dyrk1A proteins exhibited a typical speckle-like subnuclear localization described previously in clonal cell lines (Fig. 1B) (Alvarez et al., 2003). These data indicate that the subnuclear localization of Dyrk1A in neural progenitor cells is independent on its kinase activity.
Figure 1.
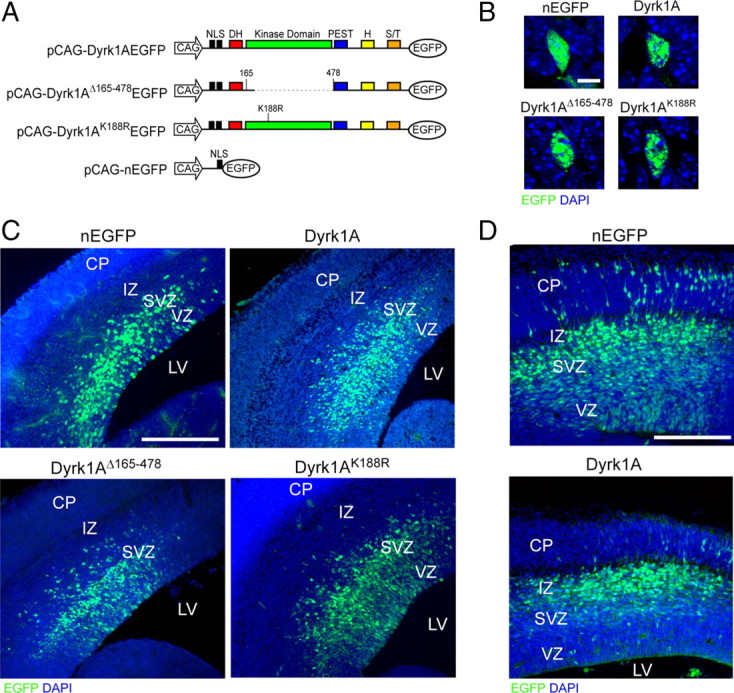
In utero electroporation of Dyrk1A–EGFP constructs in the mouse neocortex. A, Diagram of the plasmids used for IUE. All constructs contained the CAG hybrid promoter, an NLS, and the EGFP reporter fused in-frame to Dyrk1A coding sequences. Expression constructs encode wild-type Dyrk1A (pCAG–Dyrk1AEGFP), a kinase domain deletion (pCAG–Dyrk1AΔ165-478EGFP), and a kinase mutation (pCAG–Dyrk1AK188REGFP). A construct encoding nuclear EGFP alone (pCAG–nEGFP) with an NLS was used as a control. H, Histidine repeat; S/T, serine/threonine-rich regions. B, Subcellular localization of Dyrk1A proteins in transfected neural cells. Confocal images of individual neural cells in the embryonic neocortex expressing Dyrk1A proteins or control nEGFP (green). All proteins accumulated in the nucleus as indicated by colocalization with DAPI (blue), but Dyrk1A proteins exhibited a speckle-like distribution pattern. C, Expression pattern of transfected cells 24 h after IUE. Coronal sections of the lateral neocortex were obtained and imaged by direct EGFP fluorescence (green) and DAPI counterstain (blue) using confocal microscopy. Cells expressing nEGFP as well as Dyrk1AK188R and Dyrk1AΔ165-478 appeared to be widely distributed in the VZ, SVZ, and IZ. Dyrk1A+ cells were primarily excluded from the VZ. D, Expression of transfected cells 48 h after IUE. Representative images of the neocortex were obtained as above, showing that Dyrk1A+ cells were preferentially excluded from the VZ. Scale bars: B, 5 μm; C, D, 500 μm. LV, Lateral ventricle; CP, cortical plate.
Figure 5.
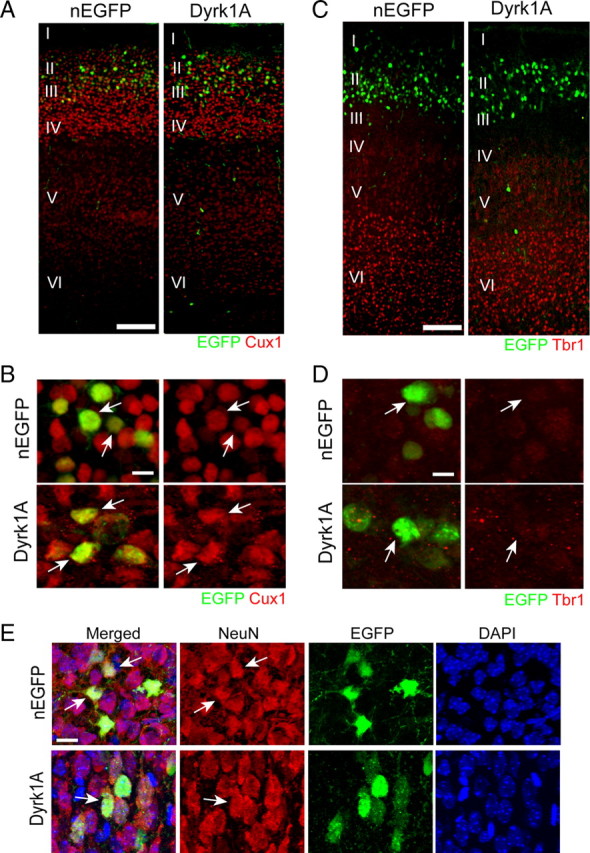
Dyrk1A-overexpressing cells migrated and differentiated normally in the postnatal neocortex. Postnatal day 6 brains were obtained from pups electroporated at E14.5 with plasmids encoding Dyrk1A–EGFP or nEGFP. A–E, Coronal sections from the lateral neocortex were subjected to immunofluorescence staining against the upper layer marker Cux1 (A, B), the deep layer marker Tbr1 (C, D), or the mature neuronal marker NeuN (E). Confocal z-stack images show that Dyrk1A+ and nEGFP+ cells (green) were positioned in the upper cortical layers, expressed Cux1 but not Tbr1, and also expressed the mature neuronal marker NeuN. Scale bars: A, C, 100 μm; B, D, E, 25 μm.
In the E14.5 embryonic cortex, most progenitor cells in the VZ, namely radial glial (RG) cells, divide asymmetrically to generate either another progenitor cell and a postmitotic neuron or another neuronal progenitor and an intermediate progenitor (IP) cell, which will then divide again symmetrically to generate two neurons in the SVZ (Miyata et al., 2004; Noctor et al., 2004). RG cells remain in the VZ, whereas IP cells leave the VZ to populate the SVZ, and postmitotic neurons migrate through the IZ toward the developing cortical plate. As expected, control EGFP-transfected cells were present in the VZ, SVZ, and lower IZ 24 h after electroporation. At 48 h, these cells localized mostly to the IZ and began to invade the cortical plate (Fig. 1C,D). At both time points, however, some EGFP+ cells remained detectable in the VZ and SVZ, likely corresponding to cells that had maintained a progenitor identity and reentered the cell cycle. Cells that expressed wild-type Dyrk1A appeared preferentially localized to the SVZ and IZ at 24 h (Fig. 1C) and mostly to the IZ at 48 h (Fig. 1D). Unlike control cells, there were essentially no detectable Dyrk1A+ cells in the VZ at both time points (Fig. 1C,D), suggesting that these cells did not maintain the progenitor identity. Mutant Dyrk1AK188R+ and Dyrk1AΔ165-478+ cells appear to behave similarly to control nEGFP (Fig. 1C), suggesting that the observed shift in cellular localization depends on the kinase activity of Dyrk1A. Similar results were obtained when plasmids were electroporated in the lateral (Fig. 1 C,D) or medial (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) neocortex.
Dyrk1A-overexpressing cells are excluded from the proliferative zone of the neocortex
To further examine the localization of cells transfected in utero in a quantitative manner, we sectioned brains 24 h after electroporation and conducted immunofluorescence staining using the marker antibody Tuj1, which strongly labels the processes of early postmitotic neurons in the IZ. We designated the heavily Tuj1-labeled region as IZ, whereas the unstained region near the lateral ventricles was designated as the SVZ (top half) or the VZ (bottom half) (Fig. 2A). The distribution of nEGFP+, Dyrk1A–EGFP+ and Dyrk1AK188REGFP+ cells within each region was then quantified from triplicate experiments (Fig. 2B). Confirming our initial observations, we found that a significantly smaller percentage of Dyrk1A–EGFP+ cells (4.7 ± 0.34%, n = 534) was present in the VZ compared with nEGFP+ (19 ± 1.6%, n = 559) or Dyrk1AK188REGFP+ (17.5 ± 3.1%, n = 530) cells. In the SVZ, a significantly lower percentage of Dyrk1A–EGFP+ cells (25 ± 1.0%) was also observed compared with nEGFP+ (35 ± 2.1%) or Dyrk1AK188REGFP+ (31 ± 1.8%) cells. Conversely, a significantly higher percentage of Dyrk1A+ cells (70 ± 1.3%) was found in the IZ compared with nEGFP+ (45 ± 0.65%) or Dyrk1AK188REGFP+ (51 ± 2.6%) cells. The shift of Dyrk1A–EGFP+ cells from the proliferative to the nonproliferative regions of the neocortex suggests that they may have exited the cell cycle and undergone differentiation. Because this shift was not observed in kinase-deficient Dyrk1AK188REGFP+ cells, our results also indicate that overexpression of Dyrk1A alters the localization of transfected neural cells in a manner that is dependent on its kinase activity.
Figure 2.

Neural cells expressing Dyrk1A–EGFP preferentially exit the ventricular zone. A, Embryos were electroporated with the indicated constructs and analyzed 24 h after IUE. Coronal sections of the lateral neocortex were imaged by direct EGFP fluorescence (green) and Tuj1 immunofluorescence (red) using confocal microscopy. LV, Lateral ventricle. Scale bar, 50 μm. B, The percentage of EGFP fluorescent cells in the VZ, SVZ, and IZ was quantified using confocal z-stack images collected as in A. The percentage of cells expressing wild-type Dyrk1A was significantly reduced in the VZ and in the SVZ, and it was increased in the IZ compared with control nEGFP or mutant Dyrk1AK188R. Error bars indicate SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
The absence of Dyrk1A–EGFP+ cells from the proliferative regions of the neocortex is not attributable to cell death or asymmetric protein distribution
To investigate the possibility that the observed loss in Dyrk1A–EGFP+ cells in the VZ and SVZ may result from cell death, we conducted a fluorescent TUNEL assay to identify apoptotic cells in embryonic brains electroporated with either pCAG–Dyrk1AEGFP or pCAG–nEGFP. As controls, we used a DNase-treated E15.5 brain section as a positive control and excluded the terminal deoxynucleotidyl transferase enzyme in another brain section as a negative control. As expected, there were virtually no apoptotic cells at this stage of corticogenesis in either nEGFP- or Dyrk1A-electroporated brains (Fig. 3A).
Figure 3.
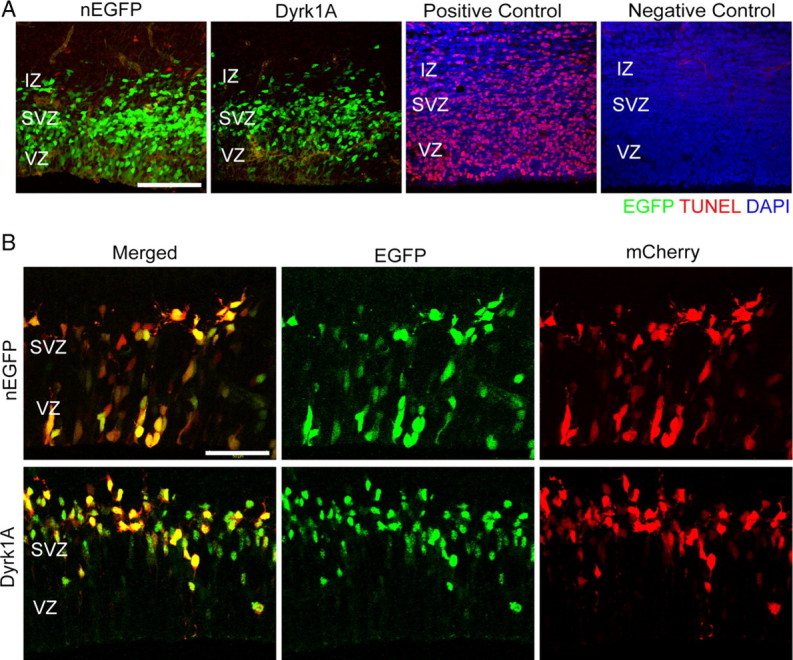
The absence of Dyrk1A–EGFP cells in the VZ is not attributable to cell death or asymmetric protein localization. A, Dyrk1A overexpression in neural progenitor cells does not induce cell death. nEGFP or Dyrk1A–EGFP constructs were electroporated at E14.5, and TUNEL assay was conducted on E15.5 sections. Virtually no TUNEL staining (red) was observed in nEGFP+ and Dyrk1A+ (green) cells. DNase-treated sections were used as a positive control. As a negative control, sections were not treated with terminal deoxyribonucleotide transferase enzyme. Control sections are shown with DAPI counterstaining (blue). Scale bar, 100 μm. B, Coexpression of mCherry and Dyrk1A–EGFP or control nEGFP. Coronal sections of the medial neocortex were imaged to visualize EGFP and mCherry. The distribution of the mCherry signal was similar to that of EGFP for all electroporated constructs. Scale bar, 50 μm.
Previous studies by Hämmerle et al. (2002, 2008) indicated that Dyrk1A mRNA is asymmetrically distributed in proliferating chick or mouse neuroblasts at early stages of corticogenesis. This raised the possibility that the Dyrk1A–EGFP protein may only label one of the two daughter cells resulting from each division event, corresponding to the postmitotic neuron, which would then preferentially leave the proliferative regions of the neocortex. To visualize all daughter cells resulting from cell division events, we coelectroporated E14.5 embryos with pDyrk1A–EGFP and a plasmid encoding the red fluorescent protein mCherry. The localization and expression of EGFP+ and mCherry+ cells were analyzed 24 h after IUE. The data reveal that the distribution of mCherry primarily overlapped and was similar to that of EGFP or Dyrk1A–EGFP (Fig. 3B). The finding that essentially no mCherry signal was present in the VZ of pDyrk1A–EGFP cotransfected embryos strongly suggest that the entire cell progeny resulting from division events of Dyrk1A-overexpressing progenitor cells shifted toward nonproliferative regions of the neocortex.
Overexpression of Dyrk1A inhibits proliferation of neural progenitor cells
The preferential localization of Dyrk1A+ cells along the SVZ/IZ boundary suggested that these cells stopped proliferating and entered the differentiated state. To verify that this was indeed the case, we labeled embryos with BrdU 20 h after electroporation. BrdU labels cells that are in S phase and thus identifies cells that are actively proliferating at this stage. Four hours after BrdU injection in the pregnant dam, embryos were dissected and subjected to immunofluorescence staining with BrdU antibodies. We observed that Dyrk1A–EGFP+ cells were rarely double labeled with BrdU antibodies, whereas approximately half of the Dyrk1AK188R EGFP+ and control nEGFP+ cells exhibited BrdU colabeling (Fig. 4A). Quantification of the data confirmed these observations, revealing that a significantly lower percentage of Dyrk1A+ cells (12.5 ± 3.6%, n = 243) was labeled with BrdU compared with nEGFP+ (43 ± 2.4%, n = 210) or Dyrk1AK188R+ (52 ± 7%, n = 214) cells (Fig. 4B). We also conducted immunofluorescence staining using phospho-Histone H3 antibodies to label cells in the M phase of the cell cycle. We observed that a subset of nEGFP+ and Dyrk1AK188R+, mostly located in close apposition to the ventricular surface, coexpressed phospho-Histone H3 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). However, we found that Dyrk1A+ cells almost never coexpressed this marker. Together with the BrdU labeling data, these results demonstrate that overexpression of Dyrk1A in vivo inhibits cell cycle progression in a kinase-dependent manner.
Figure 4.
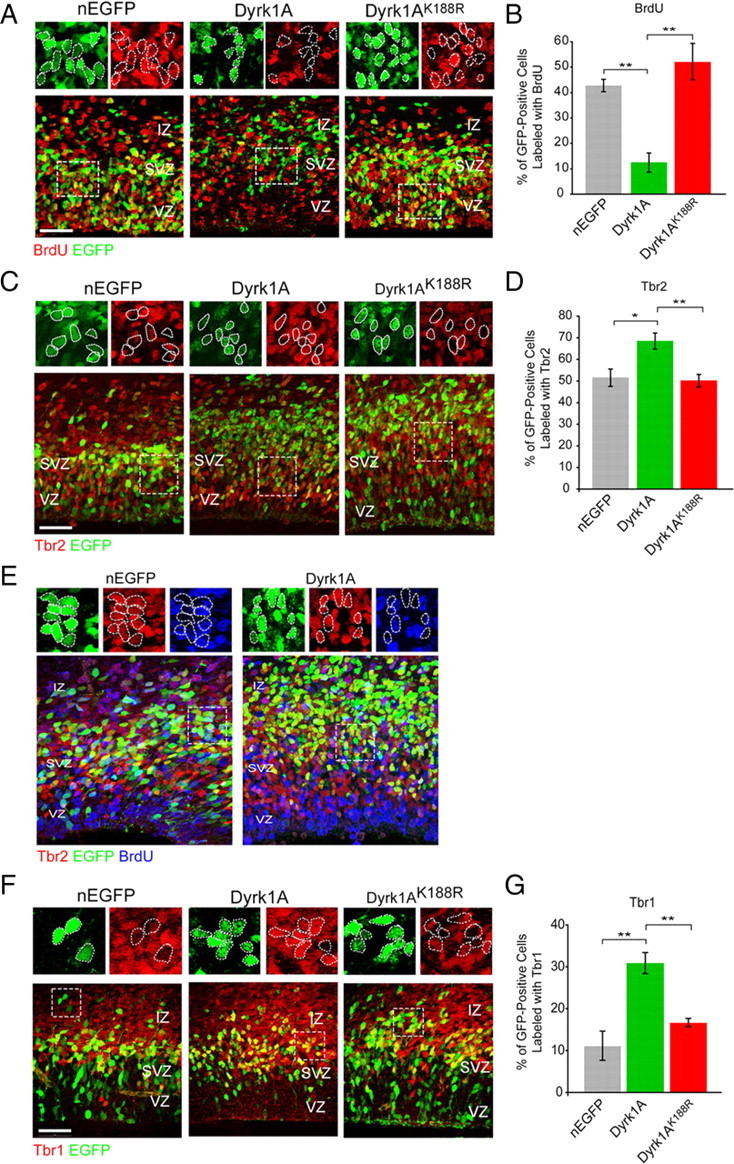
Dyrk1A overexpression inhibits cell cycle progression and induces premature neuronal differentiation in a kinase-dependent manner. E14.5 mouse embryos were electroporated in utero with the indicated plasmids. Representative confocal images were obtained from the lateral neocortex 24 h after IUE. Sections were imaged by EGFP autofluorescence (green) to visualize the transfected proteins, immunofluorescence for the indicated antibodies (red), and DAPI staining (blue). Higher magnifications of the boxed areas are shown above each overlay image to illustrate representative patterns of colocalization. A, BrdU was injected in the pregnant dam 4 h before killing the embryos to identify proliferating cells. B, Quantification of the BrdU colabeling data. The percentage of cells expressing wild-type Dyrk1A that were positive for BrdU was significantly reduced compared with cells expressing control nEGFP or mutant Dyrk1AK188R. C, Coexpression of the intermediate progenitor marker Tbr2. D, Quantification of Tbr2 coexpression data. The percentage of Dyrk1A+ cells that coexpressed Tbr2 was significantly higher compared with nEGFP+ and mutant Dyrk1AK188R+ cells. E, BrdU labeling of Tbr2+ cells. F, Coexpression of the early postmitotic neuronal marker Tbr1. G, Quantification of the Tbr1 coexpression data. The percentage of cells expressing wild-type Dyrk1A that was positive for Tbr1 was significantly increased compared with cells expressing control nEGFP+ and mutant Dyrk1AK188R. Scale bars, 50 μm. Bar graphs show SEM. *p ≤ 0.05; **p ≤ 0.01.
Dyrk1A overexpression in progenitor cells induces premature neuronal differentiation
Previous studies have suggested that Dyrk1A may act as a switch from proliferative to neurogenic divisions in the developing nervous system (Hämmerle et al., 2002, 2008). Our finding that Dyrk1A+ cells preferentially localize to the SVZ and IZ regions of the neocortex supports this hypothesis, because these regions are primarily populated by either IP cells or postmitotic neurons. To directly examine the effect of Dyrk1A overexpression on the genesis of IP cells, we conducted immunofluorescence staining against the IP cell marker Tbr2 using sections of electroporated brains 24 h after transfection (Englund et al., 2005). Confocal imaging and quantification of Tbr2 coexpression revealed that a significantly higher percentage of Dyrk1A+ cells coexpressed Tbr2 (68.6 ± 3.8%, n = 209) compared with Dyrk1AK188R+ (50.2 ± 2.9%, n = 209) or nEGFP+ (51.5 ± 4%, n = 168) cells (Fig. 4C,D). The high rate of Tbr2 coexpression suggest that most of the progeny of Dyrk1A-overexpressing radial progenitor cells preferentially acquired an intermediate progenitor identity. Because IP cells normally proliferate in the SVZ, we also determined whether Dyrk1A overexpression in this population affected their ability to incorporate BrdU. Sections from embryos electroporated with Dyrk1A–EGFP or control nEGFP were thus immunolabeled with BrdU as well as Tbr2 antibodies (Fig. 4E). The data indicate that a significantly smaller percentage of transfected IP cells overexpressing Dyrk1A (EGFP+ and Tbr2+) also incorporated BrdU (10.1 ± 1.4%, n = 282 cells) compared with nEGFP controls (44.6 ± 2.7%, n = 194 cells) (p ≤ 0.01). These findings indicate that the overexpression of Dyrk1A in Tbr2+ IP cells also inhibited their proliferation.
To determine whether Dyrk1A overexpression affected the generation of neuroblasts, we conducted similar experiments using the antibody marker Tbr1, which is expressed immediately after cells become postmitotic in the upper SVZ and IZ (Englund et al., 2005). Quantification of the Tbr1 coexpression data revealed a significant increase in the percentage of Dyrk1A+ cells (30.9 ± 2.6%; n = 196) that expressed Tbr1 (Fig. 4F,G) compared with nEGFP+ (11.1 ± 3.5%, n = 197) or Dyrk1AK188R+ (16.6 ± 1%, n = 200) cells. To determine whether Dyrk1A overexpression induced premature glial differentiation, we also conducted immunofluorescence staining using the astrocyte-specific marker glial fibrillary acidic protein (GFAP). Consistent with previous reports (Fox et al., 2004), we found that GFAP was absent in the embryonic neocortex at this age (E15.5) and was also not expressed in EGFP+ or Dyrk1A+ cells (data not shown). These experiments demonstrate that Dyrk1A overexpression in neural progenitor cells promotes premature neuronal differentiation.
Dyrk1A-expressing cells undergo normal migration and differentiation in the postnatal neocortex
The majority of neurons generated between E14.5 and E15.5 migrate to the developing neocortex and form the upper cortical layers II/III. The observed premature differentiation of Dyrk1A+ cells prompted us to determine whether these cells can migrate normally into the cortical plate and express birth date-appropriate cell markers. Therefore, we examined the localization of Dyrk1A+ and nEGFP+ cells at postnatal day 6, a time when cortical layer formation is mostly completed. We conducted immunofluorescence staining against cortical layer markers Cux1 and Tbr1. Cux1 is expressed by neurons that populate layers II–IV of the neocortex (Nieto et al., 2004), whereas Tbr1 is expressed at high levels by neurons located in the deep cortical layer VI in the caudal neocortex (Hevner et al., 2003). Our experiments indicate that the great majority of Dyrk1A+ and nEGFP+ cells localized similarly within Cux1+ upper cortical layers and were excluded from the Tbr1+ deep layer (Fig. 5A,C). Higher-magnification confocal images further reveal that Dyrk1A+ as well as nEGFP+ cells appropriately express high levels of Cux1 (Fig. 5B) but only very low levels of Tbr1 (Fig. 5D). Furthermore, we found that Dyrk1A+ cells expressed the neuronal marker NeuN, suggesting that they are able to differentiate into mature neurons (Fig. 5E). These findings demonstrate that Dyrk1A overexpression does not alter the normal program of neuronal specification and differentiation in the developing neocortex.
Expression of Dyrk1A in Neuro2a cells inhibits proliferation
To investigate the mechanisms involved in the inhibition of proliferation induced by Dyrk1A overexpression, we used the mouse neural cell line Neuro2a. For these studies, we used Dyrk1A–EGFP fusion constructs (pDyrk1AEGFP) under the control of the CMV promoter. As a control, we used a plasmid encoding EGFP alone (pEGFP). These cells did not express detectable levels of endogenous Dyrk1A protein as determined by Western blot and immunofluorescence assays (data not shown). To examine cell proliferation, we transfected Neuro2a cells with pDyrk1AEGFP or pEGFP and monitored the growth of transfected, fluorescent cells over a 4 d period using a hemocytometer. Results from triplicate experiments indicated that control EGFP+ and untransfected (EGFP−) cells grew rapidly during this time, whereas Dyrk1A–EGFP+ cells failed to do so (Fig. 6A). These experiments suggest that, similar to neural progenitor cells in vivo, the expression of Dyrk1A inhibits the proliferation of Neuro2a cells in vitro.
Figure 6.

Dyrk1A expression in Neuro2a cells causes proliferation arrest and cyclin D1 nuclear export. A, Neuro2a cells expressing Dyrk1A failed to proliferate. Cells were transfected with pDyrk1A–EGFP or pEGFP, and the number of fluorescent cells was determined in triplicate samples using a hemocytometer. Untransfected (EGFP−) cells from the same cultures were also counted. Line graphs show the SEM. B, Dyrk1A expression induces cyclin D1 degradation. Neuro2a cells were transfected with pDyrk1A–EGFP or pEGFP plasmids and lysed at 0, 6, 12, and 20 h after transfection. Western blot analysis was conducted using antibodies against cyclin D1, EGFP, and actin as a loading control. C, Neuro2a cells cotransfected with plasmids encoding HA-tagged cyclin D1 and EGFP (top row) or Dyrk1A–EGFP (bottom row) were subjected to immunofluorescence staining using anti-HA antibodies. Confocal z-stack images were obtained to analyze the subcellular localization of HA-labeled cyclin D1 (red) in EGFP+ cells (green). Cyclin D1 was predominantly nuclear in cells transfected with pEGFP but appeared to be mostly cytoplasmic in cells transfected with pDyrk1A–EGFP. Scale bar, 25 μm. D, Dyrk1A and cyclin D1 colocalized in nuclear aggregates in transfected Neuro2a cells. High-resolution confocal images of a single 1 μm optical section showing the colocalization of Dyrk1A–EGFP and cyclin D1–HA in many nuclear aggregates near the nuclear membrane. Scale bar, 10 μm.
Dyrk1A expression in Neuro2a cells causes nuclear export and degradation of cyclin D1
To understand the mechanisms involved in the observed cell cycle arrest, we examined the effect of Dyrk1A on the known cell cycle regulating protein cyclin D1. This cyclin is required for cell proliferation by allowing entry into the cell cycle. Its activation and complex formation with Cdk4 in the G1 phase of the cell cycle is necessary for cells to enter the S phase. To investigate whether Dyrk1A affects cyclin D1 protein levels, we transfected Neuro2a cells with either pDyrk1A–EGFP or pEGFP and monitored protein levels by Western blot analysis over the course of 20 h. The data indicate that cyclin D1 levels decreased concurrently with the increased expression of Dyrk1A, whereas they did not decrease in pEGFP-transfected cells over the same time period (Fig. 6B). These experiments suggest that Dyrk1A expression leads to cyclin D1 degradation.
Because cyclin D1 degradation is typically attributable to proteasome activity in the cytoplasm, we investigated whether the expression of Dyrk1A resulted in the nuclear export of cyclin D1. For these experiments, we cotransfected Neuro2a cells with pDyrk1A–EGFP or pEGFP together with a plasmid encoding cyclin D1 tagged with the HA epitope (pCyclin D1–HA). Then, we conducted double-immunofluorescence analysis using anti-HA antibodies to determine the cellular localization of cyclin D1 in transfected cells. In EGFP-expressing cells, cyclin D1 appeared to be predominantly or exclusively nuclear, whereas it appeared to be mostly localized in the cytoplasm of cells that expressed Dyrk1A–EGFP (Fig. 6C). Quantification of the cyclin D1 localization data from triplicate experiments confirmed our observations and demonstrated that the percentage of cells that expressed cyclin D1 exclusively in the nucleus was significantly reduced in the presence of Dyrk1A (7.3%, n = 133) compared with EGFP (51.8%, n = 133) (p < 0.01). Furthermore, single-plane confocal analysis of Dyrk1A–EGFP-transfected cells expressing significant levels of nuclear cyclin D1 revealed that these proteins colocalized in aggregates present mostly around the nuclear membrane (Fig. 6D). Together, these data suggest that Dyrk1A directly or indirectly interacts with cyclin D1 in protein aggregates and promotes its export and degradation in the cytoplasm.
Dyrk1A overexpression leads to the nuclear export of cyclin D1 in vivo
To determine whether Dyrk1A overexpression also affects cyclin D1 localization in vivo, we cotransfected Dyrk1A–EGFP or EGFP with cyclin D1–HA expression plasmids in the E14.5 mouse cortex by in utero electroporation. Brain sections were analyzed 24 h later by confocal microscopy using HA antibodies to determine the localization of cyclin D1 in EGFP+ cells. These experiments revealed that cyclin D1 was expressed predominantly in the nucleus of control nEGFP+ cells, although it could also be detected in the cytoplasm surrounding the nucleus in some cells that expressed high levels of the cyclin (Fig. 7A). In Dyrk1A+ cells, the expression of cyclin D1 appeared to be predominantly cytoplasmic in most cells and could also be detected in cytoplasmic regions far removed from the nucleus (Fig. 7A). In many Dyrk1A+ cells, cyclin D1 aggregates were present in the nucleus, in which they colocalized with Dyrk1A–EGFP (Fig. 7B), a finding similar to our in vitro observations. In kinase-deficient Dyrk1AK188R+ cells, cyclin D1 was observed predominantly in the nucleus and did not form aggregates, similarly to control nEGFP+ cells (Fig. 7A). These findings suggest that Dyrk1A affects the subcellular localization cyclin D1 in vivo in a kinase-dependent manner.
Figure 7.
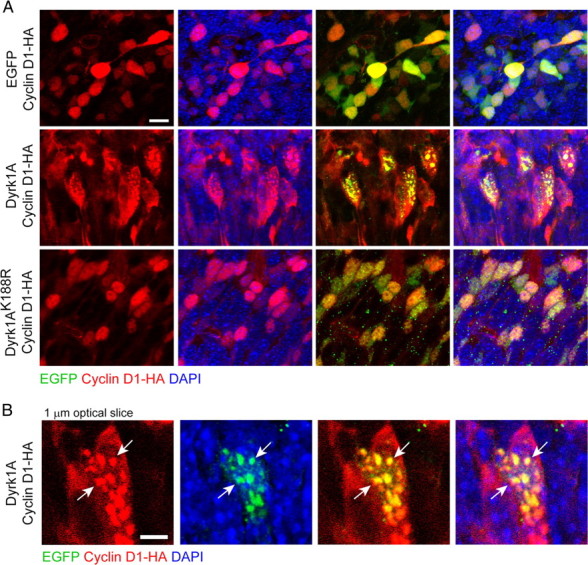
Dyrk1A overexpression in neural cells in vivo promotes cyclin D1 nuclear export. E14.5 mouse embryos were coelectroporated with pCAG–cyclin D1–HA and pCAG–nEGFP, pCAG–Dyrk1AEGFP, or pCAG–Dyrk1AK188REGFP. After 24 h, coronal sections of the lateral neocortex were obtained, and confocal z-stack images were acquired to visualize EGFP expression (green) and cyclin D1–HA localization (red). Sections were counterstained with the nuclear marker DAPI (blue). A, Cyclin D1 appeared to be mostly nuclear in nEGFP+ or Dyrk1AK188R+ cells. Cells expressing Dyrk1A exhibited either mostly cytoplasmic cyclin D1 or nuclear aggregates containing the cyclin. Scale bar, 10 μm. B, High-resolution confocal images of a single 1 μm optical section showing the colocalization of Dyrk1A–EGFP and cyclin D1 in nuclear aggregates. Scale bar, 5 μm.
Cyclin D1 rescues the proliferation inhibition induced by Dyrk1A overexpression in vivo
The data described above raised the possibility that Dyrk1A induces cell cycle arrest by depleting the nuclei of transfected cells of cyclin D1. To test this hypothesis, we set out to determine whether cyclin D1 overexpression could rescue the Dyrk1A-induced phenotype in vivo. Plasmids encoding Dyrk1A–EGFP or EGFP alone were coelectroporated with cyclin D1–HA in the E14.5 brain, and the localization of EGFP+ cells in the neocortex was determined 24 h after in utero electroporation using TuJ1 immunofluorescence to delineate the IZ. Confocal images of electroporated cortices reveal that, in the presence of exogenous cyclin D1–HA, many Dyrk1A+ cells could be detected in the proliferative VZ and SVZ regions, similarly to control nEGFP+ cells (Fig. 8A). This pattern is dramatically different from that of Dyrk1A+ cells in the absence of cotransfected cyclin D1, which shows an accumulation of transfected cells at the SVZ/IZ boundary (Figs. 1–4). When cyclin D1 was coexpressed, the distribution of Dyrk1A+ cells in each cortical zone (VZ, 30.9 ± 1.2%; SVZ, 37.5 ± 2.2%; IZ, 31.6 ± 2.2%; n = 348 cells) did not significantly differ from that of nEGFP+ cells (VZ, 29.1 ± 0.8%; SVZ, 31.7 ± 5.9%; IZ, 43.5 ± 4.0%; n = 346 cells) (Fig. 8B). To further examine cell proliferation in double-transfected embryos, we conducted BrdU labeling. The data reveal that the percentage of BrdU-labeled Dyrk1A+ cells (58.5 ± 2.7%, n = 152 cells) was not significantly different from that of nEGFP+ cells (47.6 ± 4.4%, n = 177 cells) (Fig. 8C). Thus, these results demonstrate that increased levels of cyclin D1 suppress the proliferation defects caused by Dyrk1A overexpression in vivo.
Figure 8.
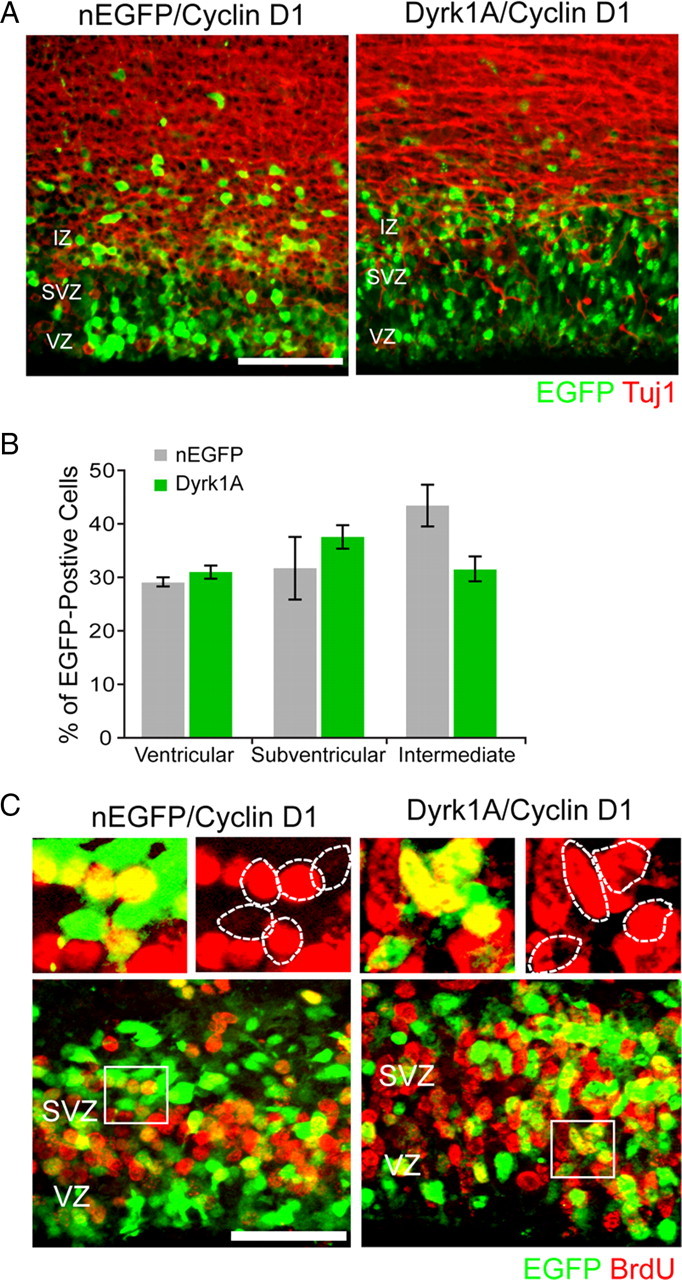
Cyclin D1 rescued the proliferation defect induced by Dyrk1A overexpression in vivo. E14.5 embryos were coelectroporated with pCAG–cyclin D1–HA and pCAG–nEGFP or pCAG–Dyrk1AEGFP and pulsed by BrdU injection. At 24 h after IUE, coronal sections of the lateral neocortex were subjected to immunofluorescence staining against Tuj1 (A) or BrdU (C). A, Representative confocal images showing the localization of transfected cells (green) and Tuj1 immunofluorescence (red). B, Quantification of the data obtained as in A. The localization of cells expressing Dyrk1A in different zones of the neocortex was comparable with that of control nEGFP-expressing cells. C, BrdU labeling of embryos coelectroporated in utero with cyclin D1 and nEGFP or Dyrk1A plasmids. Higher magnifications of the boxed areas are shown above each overlay image to illustrate representative patterns of colocalization. The percentage of cells expressing Dyrk1A that was positive for BrdU did not differ significantly when compared with control nEGFP+ cells. Scale bars: A, 50 μm; C, 100 μm. Bar graphs show SEM.
Discussion
DYRK1A is widely regarded as a candidate gene for mental retardation in DS (Park et al., 2009), but the cellular function of the encoded protein in mammalian brain development is poorly understood. Previous studies demonstrated that Dyrk1A overexpression in transgenic mice consistently results in cognitive dysfunction (Smith and Rubin, 1997; Altafaj et al., 2001; Branchi et al., 2004; Ahn et al., 2006), but the mechanisms underlying the etiology of behavioral defects remained elusive. Dyrk1A is also overexpressed, although to low levels, in DS patients as well as in animal models of DS, such as Ts65Dn mice (Guimera et al., 1999). In this study, we sought to fill a gap in our understanding of the Dyrk1A role in abnormal brain development by investigating the cellular events affected by Dyrk1A overexpression in the developing mouse brain. Our approach has been to overexpress this protein at high levels in the embryonic neocortex in vivo to reveal defective cellular and molecular mechanisms that alter the development of this structure and thereby may contribute to cognitive dysfunction. Our results demonstrate that increased levels of Dyrk1A in the developing neocortex alter the timing of neural cell proliferation and differentiation. Specifically, Dyrk1A overexpression inhibited cell cycle progression and resulted in premature neuronal differentiation. This activity correlated with a shift in the distribution of transfected cells from the proliferative zones of the neocortex toward nonproliferative zones. The observed phenotype in vivo was completely dependent on the kinase activity of Dyrk1A, suggesting that yet unidentified protein substrates mediate these events.
We also investigated the molecular mechanism underlying the proliferation defects and discovered that Dyrk1A overexpression promotes the nuclear export of cyclin D1 in vivo and in vitro. These observations are reminiscent of the activity of Dyrk1B, the closest homolog of Dyrk1A, which has been shown to cause cell cycle arrest in proliferating epithelial cells (Zou et al., 2004). Dyrk1B appears to exert this effect by directly phosphorylating cyclin D1 on threonine 228, which correlates with its cytoplasmic localization and degradation (Zou et al., 2004). Biochemical studies are currently under way to determine whether Dyrk1A can also directly phosphorylate cyclin D1. Because Dyrk1A has been shown previously to function as a priming kinase for several substrates of the guanine synthase kinase 3β (GSK3β) (Woods et al., 2001; Skurat and Dietrich, 2004), it is also possible that the phosphorylation of cyclin D1 by Dyrk1A may facilitate additional phosphorylation of the cyclin by GSK3β on the well known threonine 286 residue (Diehl et al., 1998). This scenario would be consistent with the previously established role of GSK3β as a negative regulator of cyclin D1 (Alao, 2007). However, preliminary experiments indicate that Dyrk1A overexpression in Neuro2a cells failed to increase the levels of phosphor-T286–cyclin D1 (data not shown). Another possibility is that Dyrk1A promotes the nuclear export of cyclin D1 without directly affecting its phosphorylation state. This mechanism could be similar to that used by Dyrk1A in regulating the subcellular localization of the transcription factor NFAT (Arron et al., 2006). However, it is unlikely that Dyrk1A functions as a general nuclear export chaperone because it does not appear to bind to Crm1, a nuclear export protein also involved in cyclin D1 export (Benzeno et al., 2006) in immunoprecipitation assays (data not shown). Thus, additional studies will be necessary to dissect the exact molecular mechanism of cyclin D1 regulation by Dyrk1A.
The observed shift of Dyrk1A-overexpressing cells toward nonproliferative zones of the neocortex, the reduction in BrdU incorporation, and the high rate of coexpression of the early neuronal marker Tbr1 strongly suggest that Dyrk1A overexpression prematurely drives cells into a neuronal fate. Normally, postmitotic neurons are produced by either the asymmetric division of radial glia cells or the symmetric division of intermediate progenitor cells. Both these types of division take place in the neocortex of control EGFP-electroporated embryos at E14.5, as demonstrated by the presence of BrdU+ fluorescent cells in the VZ (radial progenitors) and BrdU+;Tbr2+ fluorescent cells (intermediate progenitor cells) in the SVZ. However, when Dyrk1A expression plasmids were electroporated, no fluorescent cells were detected in the VZ, suggesting that cells overexpressing Dyrk1A can no longer reenter the cell cycle as radial progenitor cells. Furthermore, very few fluorescent cells (∼10%) could incorporate BrdU in the whole neocortex compared with controls (∼40–50%). Triple-labeling experiments suggest that these BrdU+;Dyrk1A+ cells correspond to a small subset of Tbr2+ IP cells in the SVZ (∼10%). Thus, despite the increase in the overall population of Tbr2+ cells in Dyrk1A-electroporated cortices, fewer IP cells can incorporate BrdU 24 h after electroporation. Our data indicate that Dyrk1A-overexpressing cells cannot reenter the cell cycle regardless of their radial or intermediate progenitor identity, at least within the time frame examined in these studies. These cells may prematurely differentiate contributing to the observed increase in the Tbr1+ population. Alternatively, the excess of in Tbr1+ cells in the Dyrk1A-transfected embryos may be generated directly from radial progenitor cells. These cells may have undergone asymmetric division, yielding one postmitotic neuron and one cell cycle-arrested radial progenitor cell per event, which then both shifted to the SZV/IZ. It is also conceivable that radial progenitor cells overexpressing Dyrk1A could have divided symmetrically to generate two postmitotic neurons per division event. Additional developmental studies and time-lapse imaging studies on cortical slice cultures will be required to fully resolve this issue.
Although the progeny of Dyrk1A-transfected cells was often reduced in number compared with control EGFP, we did not observe obvious migration and differentiation defects in the postnatal neocortex of electroporated embryos. Dyrk1A+ cells appeared to position themselves in the appropriate cellular layer and to express a mature neuronal marker, suggesting that the overexpression of Dyrk1A during neurogenesis does not alter their cell identity or their ability to integrate into the cortical plate. However, additional studies will be necessary to determine whether Dyrk1A+ cells exhibit defects in the rate of migration, morphological defects in dendrite or axon elongation, or perhaps synaptic connectivity defects. These future studies are necessary to conclusively determine whether or not Dyrk1A-overexpressing neurons are able to develop normally in the postnatal neocortex.
Our present findings identify for the first time a specific cellular process that is altered when Dyrk1A is overexpressed in the developing mammalian brain. The premature differentiation of progenitor cells likely affects the size of the progenitor pool, thereby resulting in a reduced number of neurons. Interestingly, recent studies have reported reduced proliferation in neurogenic forebrain regions of the DS mouse model Ts65Dn and DS human fetuses (Chakrabarti et al., 2007; Contestabile et al., 2007; Guidi et al., 2008). Because Dyrk1A is overexpressed in both Ts65Dn mice and DS patients (Guimera et al., 1999), these observations support the possibility that Dyrk1A overexpression may be responsible for the generation of an inadequate number of neurons in cortical structures of the brain. Alterations in the number of neurons as well as in the timing of differentiation could lead to the formation of abnormal cortical circuits contributing to the cognitive deficits observed in transgenic mice and DS patients.
Footnotes
Support for the initial stages of this work was provided by a Basil O'Connor Starter Scholar Research Award from The March of Dimes and National Institutes of Health (NIH) Grant R03 HD39914 from National Institute of Child Health and Human Development (G.D.). Additional support was provided by NIH Grant R01 NS042616 from National Institute of Neurological Disorders and Stroke (G.D.) and a Research Supplement to Promote Diversity in Health-Related Research (O.Y.). We thank the following: A. Renfro-Spelman for assistance with plasmid construction; S. Freemantle, R. Behringer, and D. O'Leary for plasmid constructs; B. Antalffy, J. Zhang, and K. Herrup for technical and scientific advice; C.-C. Huang, C. Ljungberg, and W. Krause for critical reading of this manuscript and helpful discussions; and N. Kane-Goldsmith and the Neuroscience Imaging Facility in the W. M. Keck Center for Collaborative Neuroscience at Rutgers for assistance with confocal microscopy.
References
- Ahn KJ, Jeong HK, Choi HS, Ryoo SR, Kim YJ, Goo JS, Choi SY, Han JS, Ha I, Song WJ. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol Dis. 2006;22:463–472. doi: 10.1016/j.nbd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer. 2007;6:24. doi: 10.1186/1476-4598-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altafaj X, Dierssen M, Baamonde C, Martí E, Visa J, Guimerà J, Oset M, González JR, Flórez J, Fillat C, Estivill X. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down's syndrome. Hum Mol Genet. 2001;10:1915–1923. doi: 10.1093/hmg/10.18.1915. [DOI] [PubMed] [Google Scholar]
- Alvarez M, Estivill X, de la Luna S. DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J Cell Sci. 2003;116:3099–3107. doi: 10.1242/jcs.00618. [DOI] [PubMed] [Google Scholar]
- Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Becker W, Joost HG. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog Nucleic Acid Res Mol Biol. 1999;62:1–17. doi: 10.1016/s0079-6603(08)60503-6. [DOI] [PubMed] [Google Scholar]
- Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost HG. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem. 1998;273:25893–25902. doi: 10.1074/jbc.273.40.25893. [DOI] [PubMed] [Google Scholar]
- Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, Klein PS, Rustgi A, Diehl JA. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene. 2006;25:6291–6303. doi: 10.1038/sj.onc.1209644. [DOI] [PubMed] [Google Scholar]
- Branchi I, Bichler Z, Minghetti L, Delabar JM, Malchiodi-Albedi F, Gonzalez MC, Chettouh Z, Nicolini A, Chabert C, Smith DJ, Rubin EM, Migliore-Samour D, Alleva E. Transgenic mouse in vivo library of human Down syndrome critical region 1: association between DYRK1A overexpression, brain development abnormalities, and cell cycle protein alteration. J Neuropathol Exp Neurol. 2004;63:429–440. doi: 10.1093/jnen/63.5.429. [DOI] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, Santini D, Bartesaghi R, Ciani E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus. 2007;17:665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotaki V, Martínez De Lagrán M, Estivill X, Arbonés M, Dierssen M. Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav Neurosci. 2004;118:815–821. doi: 10.1037/0735-7044.118.4.815. [DOI] [PubMed] [Google Scholar]
- Fox IJ, Paucar AA, Nakano I, Mottahedeh J, Dougherty JD, Kornblum HI. Developmental expression of glial fibrillary acidic protein mRNA in mouse forebrain germinal zones: implications for stem cell biology. Brain Res Dev Brain Res. 2004;153:121–125. doi: 10.1016/j.devbrainres.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Guidi S, Bonasoni P, Ceccarelli C, Santini D, Gualtieri F, Ciani E, Bartesaghi R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008;18:180–197. doi: 10.1111/j.1750-3639.2007.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimera J, Casas C, Estivill X, Pritchard M. Human minibrain homologue (MNBH/DYRK1): characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics. 1999;57:407–418. doi: 10.1006/geno.1999.5775. [DOI] [PubMed] [Google Scholar]
- Hämmerle B, Vera-Samper E, Speicher S, Arencibia R, Martínez S, Tejedor FJ. Mnb/Dyrk1A is transiently expressed and asymmetrically segregated in neural progenitor cells at the transition to neurogenic divisions. Dev Biol. 2002;246:259–273. doi: 10.1006/dbio.2002.0675. [DOI] [PubMed] [Google Scholar]
- Hämmerle B, Elizalde C, Tejedor FJ. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur J Neurosci. 2008;27:1061–1074. doi: 10.1111/j.1460-9568.2008.06092.x. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Daza RA, Rubenstein JL, Stunnenberg H, Olavarria JF, Englund C. Beyond laminar fate: toward a molecular classification of cortical projection/pyramidal neurons. Dev Neurosci. 2003;25:139–151. doi: 10.1159/000072263. [DOI] [PubMed] [Google Scholar]
- Himpel S, Tegge W, Frank R, Leder S, Joost HG, Becker W. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J Biol Chem. 2000;275:2431–2438. doi: 10.1074/jbc.275.4.2431. [DOI] [PubMed] [Google Scholar]
- Kentrup H, Becker W, Heukelbach J, Wilmes A, Schürmann A, Huppertz C, Kainulainen H, Joost HG. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J Biol Chem. 1996;271:3488–3495. doi: 10.1074/jbc.271.7.3488. [DOI] [PubMed] [Google Scholar]
- Mercer SE, Friedman E. Mirk/Dyrk1B: a multifunctional dual-specificity kinase involved in growth arrest, differentiation, and cell survival. Cell Biochem Biophys. 2006;45:303–315. doi: 10.1385/CBB:45:3:303. [DOI] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M. Asymmetric production of surface-dividing and non-surface-dividing cortical progenitor cells. Development. 2004;131:3133–3145. doi: 10.1242/dev.01173. [DOI] [PubMed] [Google Scholar]
- Møller RS, Kübart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP, Menzel C, Ullmann R, Tommerup N, Ropers HH, Tümer Z, Kalscheuer VM. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet. 2008;82:1165–1170. doi: 10.1016/j.ajhg.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel L. Down's syndrome: a genetic disorder in biobehavioral perspective. Genes Brain Behav. 2003;2:156–166. doi: 10.1034/j.1601-183x.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Park J, Song WJ, Chung KC. Function and regulation of Dyrk1A: towards understanding Down syndrome. Cell Mol Life Sci. 2009;66:3235–3240. doi: 10.1007/s00018-009-0123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani Z, Lopes C, Rachidi M, Delabar JM. Expression of the mnb (dyrk) protein in adult and embryonic mouse tissues. Biochem Biophys Res Commun. 1998;253:514–518. doi: 10.1006/bbrc.1998.9803. [DOI] [PubMed] [Google Scholar]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev Biol. 2001;240:237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Skurat AV, Dietrich AD. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J Biol Chem. 2004;279:2490–2498. doi: 10.1074/jbc.M301769200. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Rubin EM. Functional screening and complex traits: human 21q22.2 sequences affecting learning in mice. Hum Mol Genet. 1997;6:1729–1733. doi: 10.1093/hmg/6.10.1729. [DOI] [PubMed] [Google Scholar]
- Song WJ, Sternberg LR, Kasten-Sportès C, Keuren ML, Chung SH, Slack AC, Miller DE, Glover TW, Chiang PW, Lou L, Kurnit DM. Isolation of human and murine homologues of the Drosophila minibrain gene: human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics. 1996;38:331–339. doi: 10.1006/geno.1996.0636. [DOI] [PubMed] [Google Scholar]
- Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, Heisenberg M, Fischbach KF, Pongs O. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J. 2001;355:609–615. doi: 10.1042/bj3550609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Ewton DZ, Deng X, Mercer SE, Friedman E. Mirk/dyrk1B kinase destabilizes cyclin D1 by phosphorylation at threonine 288. J Biol Chem. 2004;279:27790–27798. doi: 10.1074/jbc.M403042200. [DOI] [PubMed] [Google Scholar]