

Abstract
Epidemiological studies have revealed a significantly reduced risk of Parkinson's disease (PD) among coffee and tobacco users, although it is unclear whether these correlations reflect neuroprotective/symptomatic effects of these agents or preexisting differences in the brains of tobacco and coffee users. Here, we report that coffee and tobacco, but not caffeine or nicotine, are neuroprotective in fly PD models. We further report that decaffeinated coffee and nicotine-free tobacco are as neuroprotective as their caffeine and nicotine-containing counterparts and that the neuroprotective effects of decaffeinated coffee and nicotine-free tobacco are also evident in Drosophila models of Alzheimer's disease and polyglutamine disease. Finally, we report that the neuroprotective effects of decaffeinated coffee and nicotine-free tobacco require the cytoprotective transcription factor Nrf2 and that a known Nrf2 activator in coffee, cafestol, is also able to confer neuroprotection in our fly models of PD. Our findings indicate that coffee and tobacco contain Nrf2-activating compounds that may account for the reduced risk of PD among coffee and tobacco users. These compounds represent attractive candidates for therapeutic intervention in PD and perhaps other neurodegenerative diseases.
Introduction
Parkinson's disease (PD) is a prevalent movement disorder caused by dopamine (DA) neuron death in the midbrain. The mechanisms underlying DA neuron death in PD are incompletely understood, and there are currently no preventative therapies for this disorder. Over the past decade, linkage studies have identified a number of gene defects that confer simple Mendelian forms of parkinsonism (Biskup et al., 2008; Mizuno et al., 2008). Although these mutations appear to account for only a small fraction of PD, studies of the genes associated with these rare Mendelian forms of parkinsonism have provided important insight to our understanding of the pathogenesis of both genetic and sporadic forms of PD (Biskup et al., 2008; Mizuno et al., 2008).
Epidemiological studies have also contributed important clues to the pathogenesis of PD by identifying environmental factors associated with increased or decreased risk of PD (de Lau and Breteler, 2006; Elbaz and Tranchant, 2007). Among the more intriguing findings to emerge from epidemiological studies are that coffee and tobacco users exhibit a significantly reduced risk of developing PD relative to the general population (Ross and Petrovitch, 2001; de Lau and Breteler, 2006; Elbaz and Tranchant, 2007; Ritz et al., 2007). Three hypotheses have been proposed to explain this relationship: intrinsic differences in the brains of individuals that convey habitual usage of tobacco and coffee might also confer a decreased risk of developing PD (Arias-Carrion and Poppel, 2007); coffee and tobacco may contain chemical agents, such as caffeine or nicotine, that confer symptomatic relief (Schwarzschild et al., 2006; Quik et al., 2008); and coffee and tobacco may contain neuroprotective agents (Schwarzschild et al., 2006; Quik et al., 2008).
To test the hypothesis that coffee and tobacco contain neuroprotective agents, we analyzed the neuroprotective potential of these agents using two different fly genetic models of PD: a transgenic strain of flies that overexpress the human α-synuclein protein in DA neurons (Trinh et al., 2008) and a strain of flies that bears a homozygous loss-of-function mutation in the parkin gene (Greene et al., 2003). Both of these fly models of PD bear genetic alterations akin to those responsible for familial forms of parkinsonism, and both models recapitulate multiple features of PD, most notably progressive death of DA neurons in the brain (Whitworth et al., 2005; Trinh et al., 2008). We report that coffee and tobacco confer neuroprotection in both of our fly PD models. We further find that decaffeinated coffee and nicotine-free tobacco are as effective in suppressing neurodegeneration as their caffeine and nicotine-containing counterparts, and we failed to detect a neuroprotective role of caffeine or nicotine alone. Finally, we find that the neuroprotective effects of decaffeinated coffee and nicotine-free tobacco act through an Nrf2-dependent mechanism and that these neuroprotective effects are also evident in transgenic fly models of Alzheimer's disease (AD) and polyglutamine disease. Our work suggests that coffee and tobacco contain chemical species that could be used to treat or prevent PD and perhaps other neurodegenerative disorders.
Materials and Methods
Drosophila strains and culture.
The upstream activating sequence (UAS)–α-synuclein, UAS–β-amyloid 42 (Αβ42), UAS–polyQ78, tyrosine hydroxylase (TH)–galactosidase-4 (Gal4), embryonic lethal-abnormal vision (Elav)–Gal4, and glutathione S-transferase D1 (gstD1)–green fluorescent protein (GFP) transgenic lines and park25 and parkZ472 parkin mutants used in our studies have been described previously (Brand and Perrimon, 1993; Warrick et al., 1998; Friggi-Grelin et al., 2003; Greene et al., 2003; Finelli et al., 2004; Iijima et al., 2004; Whitworth et al., 2005; Sykiotis and Bohmann, 2008; Trinh et al., 2008). The Cap-n-collar RNA interference (CncRNAi), P{TRiP.JF02006}attP2 transgenic line was obtained from the Bloomington Drosophila Stock Center (http://flystocks.bio.indiana.edu). Flies were raised on standard cornmeal–molasses food at 25°C. Exposure of Drosophila to the dietary supplements used in our analyses was performed by allowing flies to lay eggs on supplemented food or by transferring first-instar larvae to supplemented food and allowing them to develop to the adult stage. Adult flies were then collected during eclosion and transferred to fresh supplemented food.
Preparation of supplemented food.
Coffee extracts were prepared using Starbucks House blend (Starbucks Corporation) or Tully House blend (Tully Corporation) for regular coffee and Starbucks decaffeinated House blend for decaffeinated coffee (Starbucks Corporation). Tobacco extracts were made using Eve light (Liggett) or Skoal Smokeless tobacco (Skoal) for regular tobacco and Quest2 (Vector Tobacco) for nicotine-free tobacco. Extracts were prepared by adding 18.4 g of ground coffee and 50 mg of dried tobacco separately to 100 ml of water and boiling for 30 min. The extracts were then filtered through Whatman filter paper to remove particulate matter and added to standard cornmeal–molasses fly food at varying concentrations. Pure caffeine (Sigma) or nicotine (Sigma) were added to standard cornmeal–molasses fly food at concentrations that approximate their concentrations in our coffee and tobacco extracts, respectively. Pure cafestol (Sigma) was added to standard cornmeal–molasses fly food at a concentration of 0.2 μg/ml.
Dopamine neuron immunostaining.
Dopamine neuron integrity was analyzed as described previously (Whitworth et al., 2005). Briefly, adult fly brains were dissected in 1× PBS and fixed for 30 min in 4% paraformaldehyde. Fixed brains were blocked with 10% serum and stained with a rabbit antibody against tyrosine hydroxylase (TH) overnight. Dopamine neurons were visualized with an Alexa anti-rabbit secondary antibody using confocal microscopy. The experimenter was blinded to the treatment conditions, and a Student's t test was used for statistical analysis. A minimum of 20 brains were analyzed for each treatment condition. Positive and/or negative control genotypes were included in each experiment to control for subtle variation in technique between investigators and in the reagents used in these analyses.
Behavioral assays.
The climbing behavior of parkin mutants and β-amyloid transgenic flies was performed as described previously (Greene et al., 2003). Briefly, 20 flies were placed into the first chamber of a countercurrent apparatus, tapped to the bottom, and given 30 s to climb a distance of 10 cm. Flies that climbed 10 cm in 30 s were shifted to another chamber, and the experiment was repeated. After five successive trials, the number of flies in each chamber were counted, and the climbing index was calculated as the weighted average of the number of flies in each chamber divided by five times the number of flies in the assay.
Lifespan analyses were performed using groups of 10–20 age-matched flies. Flies were transferred to fresh standard food or supplemented food every 2 d, and the number of dead flies was recorded during each transfer. Transfers were continued until all flies died, and each experiment was performed in triplicate.
Eclosion rates were determined by crossing Elav–Gal4 females to UAS–polyQ78 males on standard food or on supplemented food. All crosses were performed using an equal number of age-matched flies. The mean number of adult flies obtained from three independent crosses per genotype was determined.
Caffeine and nicotine assay.
The concentrations of caffeine and nicotine in our coffee and tobacco extracts, respectively, were quantified by liquid chromatography–ion trap mass spectrometry. Briefly, extracts were first cleaned using a Sep-Pak C18 column then resuspended in water. Ten microliters of each extract were then injected onto a Bruker Esquire LC-Ion Trap mass spectrometer. Quantification was performed using a standard curve involving known quantities of commercially obtained samples of caffeine and nicotine. The identification of caffeine and nicotine in extracts was based on the liquid chromatography retention time of known standards, and the concentrations of these agents were determined using linear regression analysis of absorption integration of standard curves. All quantification experiments were performed in triplicate. Results of our analyses indicated that the concentration of caffeine in our coffee extract was 0.63 ± 0.1 mg/ml, and the concentration of nicotine in our tobacco extract was 0.3 ± 0.26 mg/ml.
Primary neuron culture.
Primary DA neuron cultures were prepared as described previously (Park and Lee, 2006; Park et al., 2007) and grown for 3 d. Decaffeinated coffee and nicotine-free tobacco extracts were added to cultures at a final dilution of 1:2000. After 6 d of exposure to coffee and tobacco extracts, DA neurons were detected and quantified relative to 4′,6′-diamidino-2-phenylindole-positive cells, as described previously (Park and Lee, 2006; Park et al., 2007).
Reverse transcription-PCR, Western blot analysis, and glutathione assay.
Total RNA was isolated from 20 heads using Trizol (Invitrogen). First-strand cDNA was prepared according to the protocol of the manufacturer (Ambion). PCR was performed using standard procedures and was performed in triplicate and normalized to ferritin as internal control.
Western blot analysis to detect GFP abundance in gstD1–GFP transgenic flies was performed by homogenizing flies in lysis buffer (150 mm TrisHCl, 50 mm NaCl, and 0.1% NP-40) plus protease inhibitors (Invitrogen) and subjecting the homogenate to SDS-PAGE on a 4–20% polyacrylamide gel. Proteins were subsequently transferred to polyvinylidene difluoride membrane and probed with a rabbit anti-GFP antiserum (Santa Cruz Biotechnology) and a mouse anti-actin antibody (Millipore Bioscience Research Reagents). Blots were developed using HRP-conjugated secondary antibodies (Sigma) and ECL Western Blotting Substrate (Pierce) according to the protocols of the manufacturer. Quantification of signal intensity was performed using NIH Image J software, and the resulting measurements from three different experiments were averaged.
Glutathione abundance in third-instar larvae was determined using a commercial glutathione detection kit (Invitrogen) according to the protocol of the manufacturer. The assay was repeated at least three times.
Results
To test the hypothesis that coffee and tobacco contain neuroprotective agents, we raised our fly PD models on food supplemented with increasing concentrations of a coffee or tobacco extract (for details about how the coffee and tobacco extracts were prepared, see Materials and Methods). Although most of the epidemiological data linking tobacco exposure to decreased PD risk involves smokers, several previous studies suggest that the use of smokeless tobacco also correlates with a reduced risk of PD (O'Reilly et al., 2005; Ritz et al., 2007), thus providing justification for the use of tobacco rather than an oxidized derivative of tobacco in our experiments. Our previous work has shown that significant degeneration occurs in the protocerebral posterior lateral 1 (PPL1) cluster of DA neurons in 20-d-old α-synuclein transgenic flies and 20-d-old parkin mutants (Whitworth et al., 2005; Trinh et al., 2008). We detected a dose-dependent reduction in the degeneration of DA neurons in both of our fly PD models after their exposure to coffee or tobacco supplemented food (Fig. 1 and data not shown). Although all of the concentrations of coffee extract tested were found to be protective, we observed an attenuation of neuroprotection at the highest concentrations of tobacco extract tested, which correlated with an increased mortality rate, indicating a toxic effect of high concentrations of tobacco (data not shown). Exposure of wild-type (WT) flies to coffee and tobacco extracts had no effect on the number of DA neurons in the PPL1 cluster, suggesting that the rescue of neuron loss conferred by these agents in our PD models is not caused by excessive genesis of DA neurons during development (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The neuroprotective effects of coffee and tobacco also do not appear to result from an alteration in the eating behavior of flies in response to these chemical agents, because we detected no change in the quantity of food consumed by flies during inclusion of coffee and tobacco extracts in the fly food (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Moreover, these neuroprotective effects are not specific to the commercial sources of tobacco and coffee used in our experiments, because different commercial sources of tobacco and coffee conferred similar neuroprotective effects (data not shown).
Figure 1.

Coffee and tobacco are neuroprotective in both α-synuclein transgenic and parkin null mutant flies. A, The number of DA neurons in the PPL1 cluster of 20-d-old transgenic flies expressing the human α-synuclein protein after exposure to food containing the given concentration of coffee extract. B, The number of DA neurons in the PPL1 cluster of 20-d-old α-synuclein transgenic flies after exposure to food containing the given concentrations of tobacco extract. C, The number of DA neurons in the PPL1 cluster of 20-d-old parkin null flies after exposure to food containing the given concentration of coffee extract or tobacco extract. D, Representative images of PPL1 DA neurons in α-synuclein-expressing flies and parkin mutants after 20 d exposure to the indicated food supplement. Cof, Coffee; Tob, tobacco. Statistical tests were performed using Student's t test (*p < 0.01%; **p < 0.001%). The genotypes were as follows: TH-α-Syn, TH–Gal4 UAS–α-synuclein/TH–Gal4 UAS–α-synuclein; Park−/−, park25/park25.
We performed several experiments to test whether the neuroprotective effects of coffee and tobacco derive from caffeine and nicotine, respectively. First, we tested whether fly food supplemented with concentrations of caffeine and nicotine that approximate their concentrations in our coffee and tobacco extracts, respectively, were capable of conferring neuroprotection. Results of these experiments revealed no significant effects of caffeine or nicotine alone on the neurodegenerative phenotype of our PD models (Fig. 2). Although these findings suggest that caffeine and nicotine are not responsible for the neuroprotective effects of coffee and tobacco, respectively, an alternate interpretation of our findings is that the protective effects of caffeine and nicotine may require other components of coffee and tobacco, respectively. To distinguish between these models, we tested whether fly food supplemented with either decaffeinated coffee extract or nicotine-free tobacco extract were capable of providing neuroprotection. We found that decaffeinated coffee and nicotine-free tobacco extracts conferred neuroprotective properties that were indistinguishable from those of their caffeine and nicotine-containing counterparts, respectively (Fig. 3). These findings indicate that coffee and tobacco contain neuroprotective agents other than caffeine and nicotine, respectively.
Figure 2.
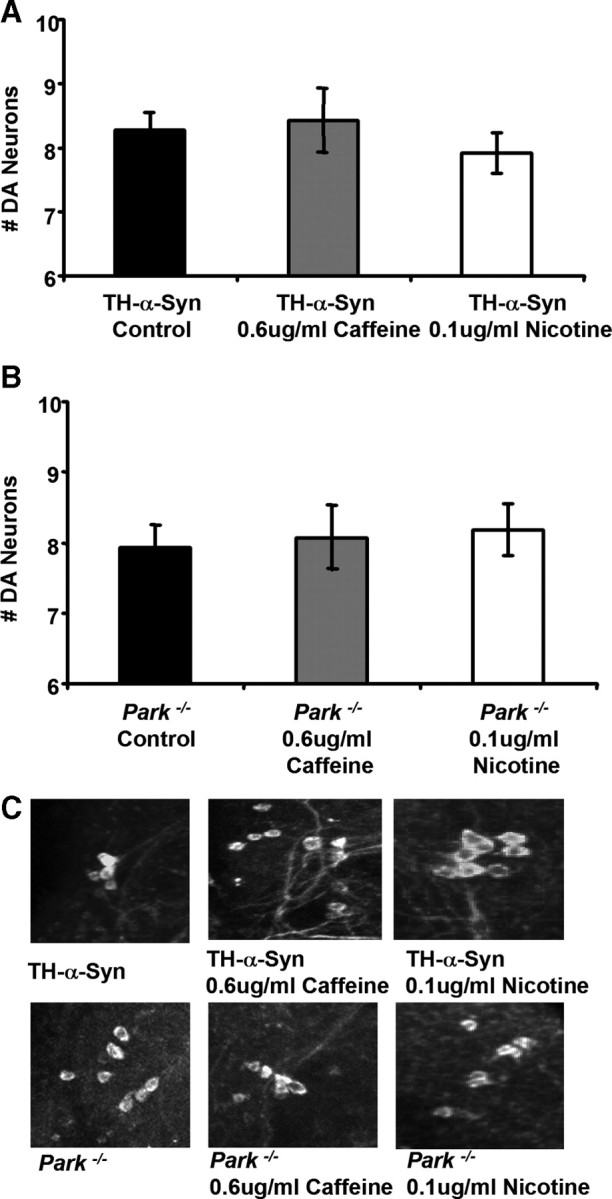
Caffeine and nicotine are not responsible for the neuroprotective properties of coffee and tobacco, respectively. A, The number of DA neurons in the PPL1 cluster of 20-d-old transgenic flies expressing the human α-synuclein protein after exposure to food containing pure caffeine (0.6 μg/ml) or nicotine (0.1 μg/ml) at concentrations that approximate their concentrations in our coffee and tobacco extracts, respectively. B, The number of DA neurons in the PPL1 cluster of 20-d-old parkin null mutants after exposure to food containing pure caffeine (0.6 μg/ml) or nicotine (0.1 μg/ml). C, Representative images of DA neurons in the PPL1 cluster of 20-d-old flies after exposure to food containing the given concentration of caffeine or nicotine. The genotypes were as follows: TH-α-Syn, TH–Gal4 UAS–α-synuclein/TH–Gal4 UAS–α-synuclein; Park−/−, park25/park25.
Figure 3.
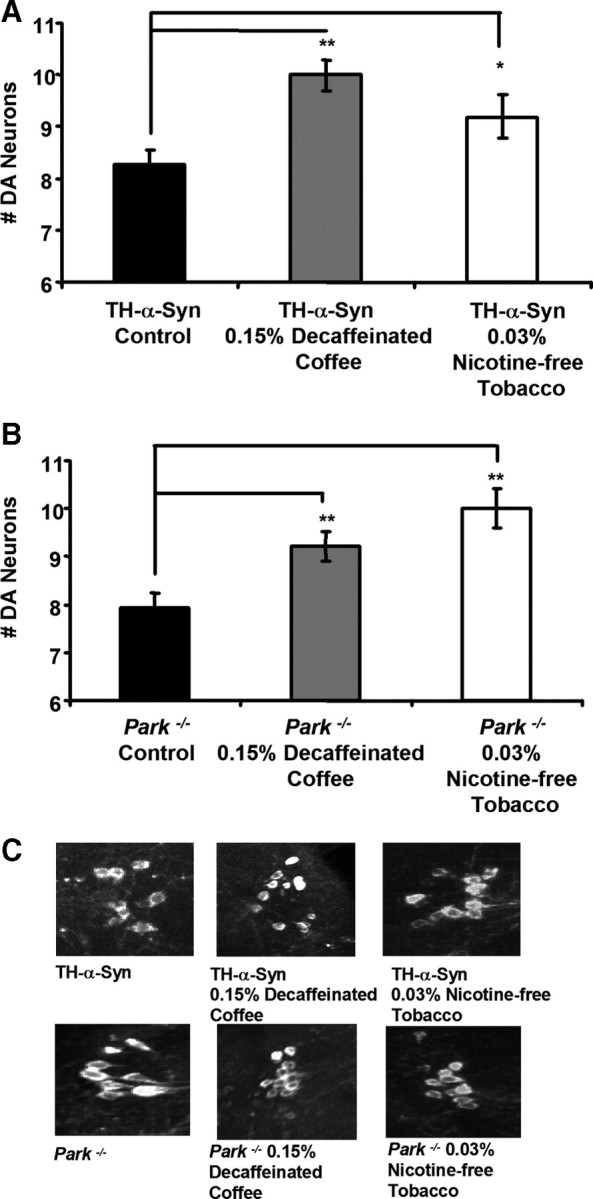
Decaffeinated coffee and nicotine-free tobacco confer neuroprotection in both α-synuclein transgenic and parkin null mutant flies. A, The number of DA neurons in the PPL1 cluster of 20-d-old transgenic flies expressing the human α-synuclein protein after exposure to food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. B, The number of DA neurons in the PPL1 cluster of 20-d-old parkin null mutants after exposure to food containing the given concentration of decaffeinated coffee or nicotine-free tobacco extract. C, Representative images of DA neurons in the PPL1 cluster of 20-d-old flies after exposure to food containing the given concentration of decaffeinated coffee or nicotine-free tobacco extract. Statistical tests were performed using Student's t test (*p < 0.01%; **p < 0.001%). The genotypes were as follows: TH-α-Syn, TH–Gal4 UAS–α-synuclein/TH–Gal4 UAS–α-synuclein; Park−/−, park25/park25.
Although we do not detect an effect of α-synuclein expression on fly behavior or lifespan, our previous work indicates that parkin mutants display a climbing defect and shortened lifespan (Greene et al., 2003). Thus, we tested whether the protective effects of decaffeinated coffee and nicotine-free tobacco would extend to the behavioral and lifespan phenotypes of parkin mutants. We found that decaffeinated coffee and nicotine-free tobacco partially rescued the climbing defect and shortened lifespan of parkin mutants (Fig. 4). The maximum lifespan of parkin mutants increased from 45 d on nonsupplemented food to 60 d on food containing decaffeinated coffee and to 57 d on food containing nicotine-free tobacco.
Figure 4.

Decaffeinated coffee and nicotine-free tobacco rescue the climbing and lifespan defects of parkin mutants. A, The climbing ability of 1-d-old parkin null mutants after exposure to food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. B, The survival rate of parkZ472/Z472 hypomorphic mutants on food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. Statistical tests were performed using Student's t test (*p < 0.05%). The genotype in A was Park−/− (park25/park25). The genotype in B was Park−/− (parkZ472/parkZ472).
To test whether the neuroprotective effects of coffee and tobacco require ingestion and possible metabolic conversion in the intestinal tract, we explored the effects of exposing a Drosophila cell culture model of α-synuclein toxicity to decaffeinated coffee or nicotine-free tobacco extracts. Previous work has shown that the expression of human α-synuclein in primary neuronal cultures from Drosophila embryos induces a progressive and selective loss of dopamine neurons over a period of several days (Park and Lee, 2006; Park et al., 2007). However, we found that supplementing the culture media with either a coffee extract or a tobacco extract completely suppressed the loss of TH-positive neurons induced by α-synuclein expression in this cell culture system (Fig. 5). These findings suggest that the neuroprotective components of coffee and tobacco act directly on DA neurons without previous metabolic conversion in the periphery.
Figure 5.
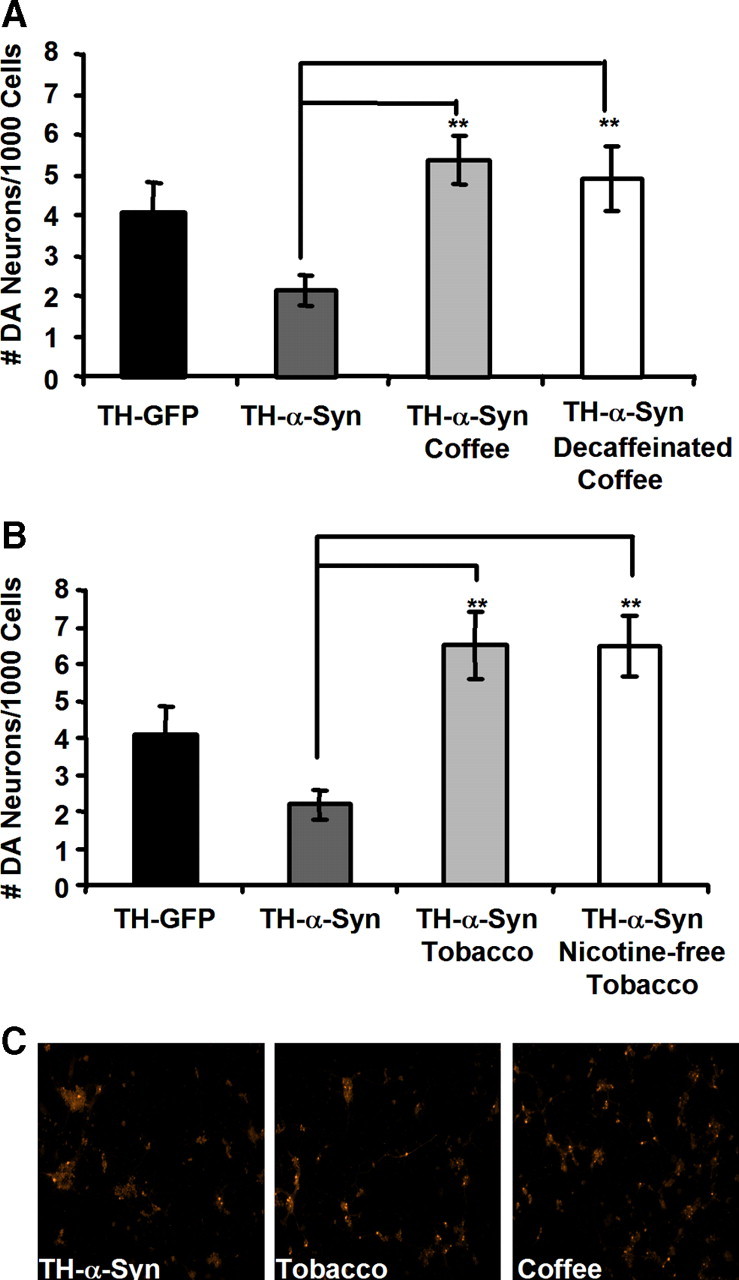
Coffee and tobacco confer neuroprotection in a cell culture model of PD. A, B, The number of DA neurons in primary embryonic neuronal cultures prepared from transgenic flies expressing α-synuclein after 3 d exposure to coffee or decaffeinated coffee (A), or tobacco or nicotine-free tobacco extracts (B). C, Representative images of primary embryonic DA neurons expressing human α-synuclein after treatment with the indicated extract. Coffee and tobacco extracts were added to cell culture media at a 1:2000 dilution. Statistical tests were performed using Student's t test (**p < 0.001%).
Recent work suggests that coffee exposure also correlates with a reduced risk of dementia and AD (Eskelinen et al., 2009), thus raising the possibility that the neuroprotective properties of decaffeinated coffee documented in our current work might also be relevant to AD. To test this hypothesis, we used an established Drosophila model of AD, involving transgenic expression of the human β-amyloid peptide in the brain, to examine the effects of decaffeinated coffee on AD pathogenesis (Finelli et al., 2004). We also tested the generality of our findings by exposing this same AD model as well as a transgenic model of polyglutamine disease (Warrick et al., 1998) to our nicotine-free tobacco and decaffeinated coffee extracts. We found that exposing the fly AD model to decaffeinated coffee or nicotine-free tobacco partially rescued the climbing defect and decreased lifespan that result from transgenic β-amyloid expression (Fig. 6). The maximum lifespan of β-amyloid transgenic flies increased from 55 d on nonsupplemented food to 65 d on food containing decaffeinated coffee and to 62 d on food containing nicotine-free tobacco. Moreover, decaffeinated coffee and nicotine-free tobacco both partially rescued the decreased eclosion rate and lifespan of polyglutamine transgenic flies (Fig. 6). The maximum lifespan of polyglutamine protein-expressing transgenic flies increased from 7 d on nonsupplemented food to 12 d on food containing decaffeinated coffee and to 10 d on food containing nicotine-free tobacco. Thus, our studies indicate that the protective effects of decaffeinated coffee and nicotine-free tobacco are not specific to PD pathogenesis but rather extend to a variety of different neurogenetic insults.
Figure 6.

Decaffeinated coffee and nicotine-free tobacco are protective in an Alzheimer's disease fly model and in a polyglutamine disease fly model. A, The climbing ability of 1-, 10-, and 20-d-old transgenic flies expressing the human β-amyloid peptide after exposure to food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. B, The survival rate of transgenic flies expressing the human β-amyloid peptide on food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. C, The relative number of transgenic flies expressing a human polyglutamine protein that survive to the adult stage of development after exposure to food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. D, The survival rate of transgenic flies expressing a human polyglutamine protein on food containing the given concentrations of decaffeinated coffee or nicotine-free tobacco extract. Statistical tests were performed using Student's t test (*p < 0.05%). The genotypes were as follows: Elav-Aβ42, Elav–Gal4/+; UAS–Αβ42/+; Elav-Q78, Elav–Gal4/+; UAS–polyQ78/+.
Our finding that the neuroprotective effects of decaffeinated coffee and nicotine-free tobacco are manifest in distinctly different models of neurodegeneration implicates a general neuroprotective pathway. One attractive candidate is the Keap1–Nrf2 pathway, which can be activated by a number of different chemical agents, including several known chemicals in coffee (Cavin et al., 2002; Yates and Kensler, 2007) and which we have shown previously can suppress the toxicity associated with parkin mutations and α-synuclein expression upon its induction in flies (Trinh et al., 2008). To test the hypothesis that the Keap1–Nrf2 pathway mediates the neuroprotective effects of coffee and tobacco, we performed several experiments. Previous work indicates that the Nrf2 transcription factor can bind to regulatory elements in genes encoding rate-limiting enzymes involved in glutathione biosynthesis and glutathione conjugation and promote their transcription (Dinkova-Kostova and Talalay, 2008). Thus, we tested whether coffee or tobacco extracts can influence glutathione abundance and whether these extracts can activate a reporter construct consisting of the gstD1 transcriptional regulatory sequence fused to the GFP coding sequence. We found that coffee and tobacco extracts significantly increased the glutathione abundance of WT flies treated with these extracts (Fig. 7). Similar increases in glutathione content were induced by treating animals with decaffeinated coffee and nicotine-free tobacco extracts (Fig. 7). We also found that coffee and tobacco extracts, as well as decaffeinated coffee and nicotine-free tobacco extracts, can activate a transgenic gstD1–GFP reporter construct (Sykiotis and Bohmann, 2008) that bears Nrf2 transcriptional activation binding sites (Fig. 7).
Figure 7.

Decaffeinated coffee and nicotine-free tobacco confer neuroprotection through an Nrf2-dependent mechanism. A, The glutathione abundance of WT animals raised on food supplemented with coffee, tobacco, decaffeinated coffee, or nicotine-free tobacco. B, The expression of GFP, relative to actin in transgenic flies bearing the Nrf2 reporter construct gstD1–GFP after exposure to food supplemented with coffee, tobacco, decaffeinated coffee, or nicotine-free tobacco. C, The abundance of the Cnc (the Drosophila Nrf2 homolog) transcript relative to ferritin in WT flies and in transgenic flies expressing a CncRNAi construct. D, The number of DA neurons in the PPL1 cluster of 20-d-old transgenic flies expressing the human α-synuclein protein and an RNAi construct targeting the Cnc transcript, after exposure to food containing coffee, tobacco, decaffeinated coffee, or nicotine-free tobacco extracts. Statistical tests were performed using Student's t test (*p < 0.05%). The genotypes were as follows: WT, W1118; gstD1-GFP, gstD1–GFP/gstD1–GFP; Elav-G, Elav–Gal4/+; Elav-CncRNAi, Elav–Gal4/+; UAS–CncRNAi/+; TH-CncRNAi, TH–Gal4/UAS–CncRNAi; TH-α-Syn; CncRNAi, TH–Gal4, UAS–α-synuclein/TH–Gal4, UAS–α-synuclein; UAS–CncRNAi/+.
The above findings suggest that decaffeinated coffee and nicotine-free tobacco extracts confer neuroprotection through an Nrf2-dependent mechanism. To conduct a more direct test of this hypothesis, we analyzed the neuroprotective effects of coffee, tobacco, decaffeinated coffee, and nicotine-free tobacco in flies with diminished Nrf2 activity. Because null mutations of the Drosophila Nrf2 homolog Cnc are inviable, we selectively expressed a CncRNAi construct in DA neurons using the TH–Gal4 driver and tested whether this manipulation attenuates the neuroprotective effects of our extracts. We found that expression of the CncRNAi construct significantly diminished the abundance of the Cnc transcript but did not detectably influence DA neuron integrity in WT flies or in α-synuclein transgenic flies. However, expression of the CncRNAi construct attenuated the neuroprotective effects of coffee, tobacco, decaffeinated coffee, and nicotine-free tobacco extracts in our α-synuclein transgenic model (Fig. 7), demonstrating that the neuroprotective effects of these extracts require the Keap1–Nrf2 pathway.
There are several chemical species in coffee that have been shown previously to activate the Keap1–Nrf2 pathway, including the sterol cafestol (Cavin et al., 2003). To test whether cafestol can activate the Nrf2 pathway in Drosophila and to identify suitable concentrations of cafestol sufficient for Nrf2 activation, we exposed transgenic gstD1–GFP reporter flies fly food supplemented with this chemical agent. When added to fly food at a concentration of 0.2 μg/ml, cafestol was found to significantly activate the gstD1–GFP reporter relative to controls (Fig. 8). We next tested whether the same concentration of cafestol can suffice to confer neuroprotection in our fly models of PD. Our experiments revealed that cafestol can confer neuroprotection in both α-synuclein transgenic and parkin null flies that is similar in magnitude to the neuroprotection conferred by our coffee extracts. This finding strongly suggests that cafestol is among the compounds in coffee that are responsible for the neuroprotective effects of decaffeinated coffee documented in our current work.
Figure 8.

The Nrf2 activator cafestol confers neuroprotection in Drosophila models of PD. A, The expression of GFP, relative to actin in transgenic flies bearing the Nrf2 reporter construct gstD1–GFP, after exposure to food supplemented with 0.2 μg/ml cafestol. B, The number of DA neurons in the PPL1 cluster of 20-d-old α-synuclein transgenic flies after exposure to food containing 0.2 μg/ml cafestol. C, The number of DA neurons in the PPL1 cluster of 20-d-old parkin null flies after exposure to food containing 0.2 μg/ml cafestol. Statistical tests were performed using Student's t test (*p < 0.05%). The genotypes were as follows: gstD1-GFP, gstD1–GFP/gstD1–GFP; TH-α-Syn, TH–Gal4 UAS–α-synuclein/TH–Gal4 UAS–α-synuclein; Park−/−, park25/25.
Discussion
In summary, our work suggests that the protective correlation associated with coffee and tobacco consumption and the incidence of PD, AD, and dementia in humans is a consequence of neuroprotective components in coffee and tobacco that activate the cytoprotective transcription factor Nrf2. Although our work indicates that the neuroprotective effects of coffee and tobacco do not derive from caffeine or nicotine, previous work has documented neuroprotective effects of these agents in toxin-induced models of PD (Kalda et al., 2006; Quik et al., 2008). There are three possible explanations for our inability to detect neuroprotective effects of nicotine and caffeine: the protective effects of nicotine and caffeine may be specific to toxin-induced cell death mechanisms; the molecular mechanisms by which nicotine and caffeine confer neuroprotection may not be conserved in Drosophila; and the quantities of coffee and tobacco extract that were sufficient for neuroprotection in our experiments may have contained inappropriate amounts of caffeine and nicotine, respectively, to allow detection of the neuroprotective effects of these agents, although we also tested both higher and lower concentrations of caffeine and nicotine than those shown in Figure 2 but were unable to document a protective effect of these agents at any of the concentrations examined (data not shown). However, regardless of the explanation for our inability to detect neuroprotective effects of nicotine and caffeine, an important conclusion of our current study is that Nrf2-activating compounds are at least part of the mechanism by which coffee and tobacco confer neuroprotection.
Our work also indicates that a known Nrf2-activator in coffee, cafestol, provides complete neuroprotection in both of our fly models of PD. Although this finding could ultimately lead to the development of a preventative therapy for PD, it is unclear that cafestol itself represents a good candidate for therapeutic intervention because cafestol also potently elevates serum cholesterol through an unknown mechanism (van Cruchten et al., 2010), and serum cholesterol levels correlate with heart disease risk (Stamler et al., 2000). However, it is possible that a structural analog of cafestol can be identified that retains the neuroprotective feature of cafestol but lacks its cholesterol-elevating property. Alternatively, a chemically distinct activator of Nrf2 might substitute for cafestol. Although there are several known Nrf2-activating compounds that could prove suitable (Eggler et al., 2008), including other known Nrf2-activating compounds in coffee (Cavin et al., 2003; Hwang and Jeong, 2008), a feature that is likely to prove instrumental in the efficacy of these compounds is their ability to cross the blood–brain barrier. Thus, the Nrf2-activating compounds in coffee and tobacco, which can presumably cross the blood–brain barrier and confer neuroprotection, given our current findings and previous epidemiological studies (de Lau and Breteler, 2006; Ritz et al., 2007), makes them particularly attractive therapeutic agents. Future studies involving fractionation of coffee and tobacco extracts and the use of Drosophila to assay these fractions should allow the identification of the full complement of neuroprotective Nrf2-activating compounds in coffee and tobacco. This knowledge could ultimately lead to the development of the first preventative therapy for PD, AD, and perhaps other neurodegenerative diseases.
Footnotes
This work was made possible by grants from the Michael J. Fox Foundation and National Institutes of Health Grant 5R21NS062985 (L.P.).
References
- Arias-Carrion O, Poppel E. Dopamine, learning, and reward-seeking behavior. Acta Neurobiol Exp (Wars) 2007;67:481–488. doi: 10.55782/ane-2007-1664. [DOI] [PubMed] [Google Scholar]
- Biskup S, Gerlach M, Kupsch A, Reichmann H, Riederer P, Vieregge P, Wullner U, Gasser T. Genes associated with Parkinson syndrome. J Neurol. 2008;255(Suppl 5):S8–S17. doi: 10.1007/s00415-008-5005-2. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Cavin C, Holzhaeuser D, Scharf G, Constable A, Huber WW, Schilter B. Cafestol and kahweol, two coffee specific diterpenes with anticarcinogenic activity. Food Chem Toxicol. 2002;40:1155–1163. doi: 10.1016/s0278-6915(02)00029-7. [DOI] [PubMed] [Google Scholar]
- Cavin C, Bezencon C, Guignard G, Schilter B. Coffee diterpenes prevent benzo[a]pyrene genotoxicity in rat and human culture systems. Biochem Biophys Res Commun. 2003;306:488–495. doi: 10.1016/s0006-291x(03)00994-x. [DOI] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52(Suppl 1):S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- Eggler AL, Gay KA, Mesecar AD. Molecular mechanisms of natural products in chemoprevention: induction of cytoprotective enzymes by Nrf2. Mol Nutr Food Res. 2008;52(Suppl 1):S84–S94. doi: 10.1002/mnfr.200700249. [DOI] [PubMed] [Google Scholar]
- Elbaz A, Tranchant C. Epidemiologic studies of environmental exposures in Parkinson's disease. J Neurol Sci. 2007;262:37–44. doi: 10.1016/j.jns.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16:85–91. doi: 10.3233/JAD-2009-0920. [DOI] [PubMed] [Google Scholar]
- Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer's Abeta42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S. Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol. 2003;54:618–627. doi: 10.1002/neu.10185. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang YP, Jeong HG. The coffee diterpene kahweol induces heme oxygenase-1 via the PI3K and p38/Nrf2 pathway to protect human dopaminergic neurons from 6-hydroxydopamine-derived oxidative stress. FEBS Lett. 2008;582:2655–2662. doi: 10.1016/j.febslet.2008.06.045. [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalda A, Yu L, Oztas E, Chen JF. Novel neuroprotection by caffeine and adenosine A2A receptor antagonists in animal models of Parkinson's disease. J Neurol Sci. 2006;248:9–15. doi: 10.1016/j.jns.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Hattori N, Kubo S, Sato S, Nishioka K, Hatano T, Tomiyama H, Funayama M, Machida Y, Mochizuki H. Progress in the pathogenesis and genetics of Parkinson's disease. Philos Trans R Soc Lond B Biol Sci. 2008;363:2215–2227. doi: 10.1098/rstb.2008.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly EJ, McCullough ML, Chao A, Henley SJ, Calle EE, Thun MJ, Ascherio A. Smokeless tobacco use and the risk of Parkinson's disease mortality. Mov Disord. 2005;20:1383–1384. doi: 10.1002/mds.20587. [DOI] [PubMed] [Google Scholar]
- Park SS, Lee D. Selective loss of dopaminergic neurons and formation of Lewy body-like aggregations in alpha-synuclein transgenic fly neuronal cultures. Eur J Neurosci. 2006;23:2908–2914. doi: 10.1111/j.1460-9568.2006.04844.x. [DOI] [PubMed] [Google Scholar]
- Park SS, Schulz EM, Lee D. Disruption of dopamine homeostasis underlies selective neurodegeneration mediated by alpha-synuclein. Eur J Neurosci. 2007;26:3104–3112. doi: 10.1111/j.1460-9568.2007.05929.x. [DOI] [PubMed] [Google Scholar]
- Quik M, O'Leary K, Tanner CM. Nicotine and Parkinson's disease: implications for therapy. Mov Disord. 2008;23:1641–1652. doi: 10.1002/mds.21900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz B, Ascherio A, Checkoway H, Marder KS, Nelson LM, Rocca WA, Ross GW, Strickland D, Van Den Eeden SK, Gorell J. Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol. 2007;64:990–997. doi: 10.1001/archneur.64.7.990. [DOI] [PubMed] [Google Scholar]
- Ross GW, Petrovitch H. Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson's disease. Drugs Aging. 2001;18:797–806. doi: 10.2165/00002512-200118110-00001. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson's disease. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Stamler J, Daviglus ML, Garside DB, Dyer AR, Greenland P, Neaton JD. Relationship of baseline serum cholesterol levels in 3 large cohorts of younger men to long-term coronary, cardiovascular, and all-cause mortality and to longevity. JAMA. 2000;284:311–318. doi: 10.1001/jama.284.3.311. [DOI] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell. 2008;14:76–85. doi: 10.1016/j.devcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh K, Moore K, Wes PD, Muchowski PJ, Dey J, Andrews L, Pallanck LJ. Induction of the phase II detoxification pathway suppresses neuron loss in Drosophila models of Parkinson's disease. J Neurosci. 2008;28:465–472. doi: 10.1523/JNEUROSCI.4778-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Cruchten ST, de Waart R, Kunne C, Hooiveld GJ, Boekschoten MV, Katan MB, Oude Elferink RP, Witkamp RF. Absorption, distribution and biliary excretion of cafestol, a potent cholesterol elevating compound in unfiltered coffees in mice. Drug Metab Dispos. 2010;38:635–640. doi: 10.1124/dmd.109.030213. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Paulson HL, Gray-Board GL, Bui QT, Fischbeck KH, Pittman RN, Bonini NM. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates MS, Kensler TW. Chemopreventive promise of targeting the Nrf2 pathway. Drug News Perspect. 2007;20:109–117. doi: 10.1358/dnp.2007.20.2.108343. [DOI] [PubMed] [Google Scholar]