

Abstract
There is considerable interest in the regulation of sensorimotor gating, since deficits in this process could play a critical role in the symptoms of schizophrenia and other psychiatric disorders. Sensorimotor gating is often studied in humans and rodents using the prepulse inhibition of the acoustic startle response (PPI) model, in which an acoustic prepulse suppresses behavioral output to a startle-inducing stimulus. However, the molecular and neural mechanisms underlying PPI are poorly understood. Here, we show that a regulatory pathway involving protein phosphatase 2A (PP2A), glycogen synthase kinase 3 β (GSK3β), and their downstream target, the M-type potassium channel, regulates PPI. Mice (Mus musculus) carrying a hypomorphic allele of Ppp2r5δ, encoding a regulatory subunit of PP2A, show attenuated PPI. This PPP2R5δ reduction increases the phosphorylation of GSK3β at serine 9, which inactivates GSK3β, indicating that PPP2R5δ positively regulates GSK3β activity in the brain. Consistently, genetic and pharmacological manipulations that reduce GSK3β function attenuate PPI. The M-type potassium channel subunit, KCNQ2, is a putative GSK3β substrate. Genetic reduction of Kcnq2 also reduces PPI, as does systemic inhibition of M-channels with linopirdine. Importantly, both the GSK3 inhibitor 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)1H-pyrrole-2,5-dione (SB216763) and linopirdine reduce PPI when directly infused into the medial prefrontal cortex (mPFC). Whole-cell electrophysiological recordings of mPFC neurons show that SB216763 and linopirdine have similar effects on firing, and GSK3 inhibition occludes the effects of M-channel inhibition. These data support a previously uncharacterized mechanism by which PP2A/GSK3β signaling regulates M-type potassium channel activity in the mPFC to modulate sensorimotor gating.
Introduction
Prepulse inhibition of the acoustic startle response (PPI) is a neurobehavioral phenomenon by which a low-intensity stimulus, presented <100 ms before a high-intensity acoustic stimulus, “gates” or attenuates startle reactivity (Hoffman and Searle, 1965; Ison and Hammond, 1971). PPI deficits have been reported in patients with schizophrenia (Braff and Geyer, 1990; Cadenhead et al., 1993; Perry and Braff, 1994), obsessive-compulsive disorder (Hoenig et al., 2005), and autistic spectrum disorders (McAlonan et al., 2002; Perry et al., 2007), and may be elicited pharmacologically in rodents using drugs that affect dopaminergic (Geyer et al., 2001), serotonergic (Schmidt et al., 1995), NMDA/glutamatergic (Ossowska et al., 1999, 2000), or cholinergic (Jones and Shannon, 2000) systems. It has been proposed that PPI normally acts as a filtering mechanism to modulate responses to nonsalient stimuli (Braff et al., 1995) and deficits in PPI may contribute to the positive symptoms of schizophrenia.
The WNT (wingless) signaling pathway, originally studied for its role in development (Siegfried and Perrimon, 1994), has recently been investigated for its potential role in neurological and psychiatric disorders. In particular, glycogen synthase kinase 3 β (GSK3β) has been implicated in Parkinson's disease, Alzheimer's disease, and mood disorders (Koo and Kopan, 2004; O'Brien et al., 2004; Gould et al., 2006; Prickaerts et al., 2006; Aghdam and Barger, 2007; Rowe et al., 2007). GSK3β activity is inhibited by phosphorylation of serine 9, which is regulated in turn by the upstream signaling molecules, protein phosphatase 2A (PP2A) and protein kinase B (also called AKT) (Cross et al., 1995; Ivaska et al., 2002; Lin et al., 2007); GSK3β is also a direct target of inhibition by lithium (Klein and Melton, 1996), one of the most widely prescribed drugs for the treatment of mood disorders (Quiroz et al., 2004).
Several lines of evidence suggest that GSK3β may also be involved in sensorimotor gating. Chronic administration of antipsychotics (clozapine or risperidone) increases GSK3β expression in multiple brain regions of the rat (Alimohamad et al., 2005a, 2005b), although these same drugs can inhibit GSK3β activity when administered acutely (Li et al., 2007). Conversely, certain psychosis-inducing drugs (amphetamine, d-lysergic acid diethylamide, or phencyclidine) that attenuate PPI in rodents also inhibit GSK3β activity (Svenningsson et al., 2003). Additionally, GSK3β activity is reduced in postmortem brain tissue from schizophrenic patients (Kozlovsky et al., 2001, 2004); GSK3β activity in frontal cortex, a region central to the neurocircuitry modulating PPI (Swerdlow et al., 2001), is positively correlated with PPI among inbred strains of mice (Amar et al., 2004), and mutation of the Drosophila GSK3 homolog, shaggy, results in reduced olfactory startle habituation (Wolf et al., 2007). These data suggest that GSK3β normally facilitates sensorimotor gating, but a causal link between GSK3β activity and PPI modulation has not yet been established, nor have the underlying cellular mechanisms been described.
Recently, the M-type potassium channel subunit gene, Kcnq2, has been identified as a putative GSK3β substrate (Borsotto et al., 2007). Loss-of function mutations in Kcnq2 result in a mild form of epilepsy (Charlier et al., 1998; Singh et al., 1998; Cooper et al., 2000; Mulley et al., 2003) and a mutation in the phosphatidylinositol 4-phosphate 5-kinase IIα (PIP5K2A) gene, which leads to reduced M-channel activity, has been linked to schizophrenia (Fedorenko et al., 2008). These data suggest that neuronal hyperexcitability resulting from impaired M-channel function may lead to sensorimotor gating deficits. We therefore hypothesized that GSK3 activity positively regulates M-channel activity within the medial prefrontal cortex (mPFC) to facilitate PPI, which we have investigated using a combination of genetic, behavioral, pharmacological, and electrophysiological approaches.
Materials and Methods
Generation of Ppp2r5δ gene trap mice.
C57BL/6J-derived blastocysts were injected with Ppp2r5δGt embryonic stem cells, obtained from Bay Genomics (data and reagents now available from the International Gene Trap Consortium at http://www.genetrap.org/), according to standard protocols (Hogan, 1994). The RRK451 gene trap line was generated on a 129/OlaHsd (agouti) background. We crossed several chimeras to wild-type C57BL/6J mice and confirmed germ line transmission of the targeted allele in progeny by identifying agouti pups. Backcrosses of the resulting F1 mice (Ppp2r5δGt129/+B6 × C57BL/6J) for three generations (N4) were established to generate Ppp2r5δGt/+ heterozygous and wild-type controls for behavioral studies.
Genotypic analysis.
Mice were genotyped using DNA isolated from tail biopsies using standard protocols. For Ppp2r5δGt and Kcnq2Gt lines, the following primers were used to amplify a fragment of the gene trap insertion (forward, 5′-TTATCGATGAGCGTGGTGGTTATGC-3′; reverse, 5′-GCGCGTACATCGGGCAAATAATATC-3′). For the Gsk3β(tm1JW) line, mice were genotyped by multiplexing three primers to amplify mutant and wild-type-specific products: (1) 5′-CCAGTCACAAATCGTACTGC-3′, (2) 5′-AACCACAGTAGTGGCAACTC-3′, and (3) 5′-CGTGCTACTTCCATTTGTCACG-3′.
Quantitative RT-PCR.
Total RNA was isolated from whole-brain tissue using Trizol reagent (Invitrogen) according to the manufacturer's instructions and was treated with RNase-free DNase (Promega) to remove genomic DNA contamination. cDNA was synthesized from 1 μg of total RNA using reverse transcription reagents from Applied Biosystems. After synthesis, cDNA was diluted 1:10 in water. TaqMan QPCR was performed using standard thermal cycling conditions on an ABI PRISM 7900 Sequence Detection System (Applied Biosystems). Amplification reactions contained 5 μl of cDNA template, 1× Universal PCR Master Mix, 100 nm each of forward and reverse primers, and 200 nm FAM-labeled probe in a final volume of 10 μl. The Kcnq2 probe and primers set was Applied Biosystems catalog #Mm00440084. Rodent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe and primers (Applied Biosystems) were used as a control for the PCRs. Data were analyzed using the comparative Ct method (Applied Biosystems User Bulletin 2).
Western analysis.
Protein extracts were isolated from whole-brain tissue in radioimmunoprecipitation assay buffer containing phosphatase inhibitor (Sigma-Aldrich) using standard techniques. Ten microgram protein samples were electrophoresed on NuPAGE 4–12% SDS-polyacrylamide gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Invitrogen). Primary antibodies included goat anti-PPP2R5δ (1 μg/ml; Abcam), mouse anti-GSK3β (1:5000; BD Biosciences), rabbit anti-phospho-GSK3β (Ser9) (1:1000; Cell Signaling Technology), rabbit polyclonal to GSK3α (1:1000; Abcam), rabbit anti-phospho-GSK3α (Ser21) (1:1000; Abcam), rabbit anti-KCNQ2 (1:500; Alomone Labs), and mouse anti-GAPDH (1:5000; Thermo Fisher Scientific). Western blots were incubated with either HRP-linked donkey anti-rabbit or HRP-linked sheep anti-mouse secondary antibody (1:5000; GE Healthcare) and processed with ECL Plus Western Blotting Detection System (GE Healthcare). Chemiluminescence was visualized and quantified using the Storm 660 PhosphorImager system (Molecular Dynamics).
Subjects.
All animal protocols were approved by the Ernest Gallo Clinic and Research Center institutional animal care and use committee. Ppp2r5δGt mice were backcrossed to C57BL/6J for three to five generations before behavioral testing. Gsk3β(tm1Jrw) mice, kindly provided by Dr. James Woodgett (University of Toronto, Toronto, Ontario, Canada), were maintained on a C57BL/6J background as previously described (Hoeflich et al., 2000). Kcnq2(tm1Dgen) mice were obtained from The Jackson Laboratory, where they had been crossed to C57BL/6J for more than five generations. Male mice for behavioral experiments were bred in house and were 8–10 weeks of age at time of testing, with the exception of the Ppp2r5δGt and Kcnq2(tm1Dgen) experiments for which male and female behavioral data were combined. For each line, heterozygous mutants were compared with wild-type littermate controls (N = 16 Ppp2r5δGt/+ vs 24 wild-type mice; 10 Gsk3β(tm1Jrw)/+ vs 7 wild-type mice; 20 Kcnq2(tm1Dgen)/+ vs 20 wild-type mice). For the pharmacology and cannulation studies, male C57BL/6J mice (The Jackson Laboratory), 8–10 weeks of age, were housed four or five per cage (29 × 18.5 × 13 cm) in 12 h light/dark cycles and were acclimated to the colony for at least 2 weeks before behavioral testing.
Drug administration.
Drug-naive mice were divided into four treatment groups (nine mice per group), each receiving two intraperitoneal injections (in a volume of 10 ml/kg) consisting of the following: group 1, 10 mg/kg 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)1H-pyrrole-2,5-dione (SB216763) (Tocris Bioscience) in 5% Tween 80/0.9% NaCl and 5 mg/kg linopirdine (Sigma-Aldrich) dissolved in a minimal volume of 0.1N HCl and diluted in 0.9% NaCl; group 2, 10 mg/kg SB216763 in 5% Tween 80/0.9% NaCl and 0.1N HCl/0.9% NaCl; group 3, 5 mg/kg linopirdine in 0.1N HCl/0.9% NaCl and 5% Tween 80/0.9% NaCl; or group 4, 0.1N HCl/0.9% NaCl and 5% Tween 80/0.9% NaCl. We attempted to assign animals to each experimental group with a balanced design such that littermates were distributed among the experimental groups. Drug doses used in these studies were determined empirically as well as based on doses reported in the literature to affect behavior in rodents (Beaulieu et al., 2004; Otto et al., 2004). Treated mice were subjected to a single PPI test session 15 min after injection. We should note that stress can affect PPI performance. The animals in our experiments were not preexposed to the startle apparatus before PPI testing. However, to our knowledge, an effect of stress on PPI has primarily been shown to result from chronic stress during the postnatal and/or adolescent period in rodents (for review, see Van den Buuse et al., 2003). In addition, PPI in rodents tested multiple times show that PPI is remarkably consistent and reproducible (Willott et al., 2003) (our unpublished observations), suggesting that the acute stress of the assay was not sufficient to significantly affect PPI.
For brain infusions, 10 μm SB216763 or 10 μm linopirdine in 10% DMSO/90% polyethylene glycol 300 (PEG300) was administered bilaterally in a volume of 0.5 μl per side. The 10% DMSO/90% PEG300 (0.5 μl) was administered as vehicle to control animals; N = 8 SB216763-treated versus 8 vehicle-treated mice and 11 linopirdine-treated versus 14 vehicle-treated controls.
Behavioral testing.
Ppp2r5δGt/+, Gsk3β(tm1JW)/+, Kcnq2(tm1Dgen)/+, and wild-type control mice were first tested for PPI and were naive to the test apparatus at the time of testing. Three weeks later, the same group of animals was tested for startle reactivity. Both PPI and acoustic startle response (ASR) were measured during the light phase using the SR-Lab System (San Diego Instruments). During all testing, background was set at 70 dB white noise. The PPI test consisted of a 5 min acclimation period, followed by seven trial types presented six times each in pseudorandom order. Trials consisted of a 20 ms prepulse, an 80 ms interval, and a 40 ms startle stimulus. Prepulse intensity was set at background, 74, 78, 82, 86, or 90 dB, and the startle stimulus was 120 dB. One trial type consisted solely of background noise to control for general movement of the subject. Trials were presented with a variable intertrial interval of 15–20 s. The entire PPI test lasted ∼18 min. At the onset of the startle stimulus, maximum velocity (Vmax) in arbitrary units of the startle platform was recorded every 1 ms for 65 ms. Vmax was averaged across trial type, with PPI calculated as the difference between Vmax after prepulse trials (Vmax-PP) and Vmax after startle stimulus alone trials (Vmax-ctl), and was expressed as a percentage of Vmax-ctl [PPI = ((Vmax-ctl) − (Vmax-PP)/(Vmax-ctl))*100%].
The ASR test consisted of eight trial types presented a total of five times each, in pseudorandom order. Trial types consisted of a single, 40 ms startle stimulus of 90, 95, 100, 105, 110, 115, or 120 dB. Trials were presented with a variable intertrial interval of 15–20 s. At the onset of the startle stimulus, maximum velocity (Vmax) in arbitrary units of the startle platform was recorded every 1 ms for 65 ms. Vmax was averaged across trial type.
Locomotor activity measurements were performed in Plexiglas locomotor activity chambers (43 × 43 cm; MED Associates), located in sound-attenuating cubicles equipped with a 2.8 W house light and exhaust fans that mask external noise. Activity chambers contain two sets of 16 pulse-modulated infrared photobeams on opposite walls to record x, y ambulatory movements, and are computer interfaced for data sampling at 100 ms resolution. To measure baseline activity and habituation to the chambers, mice were placed in activity chambers for 2 h. Locomotor activity was measured as mean distance traveled (in centimeters) ± SEM during each 10 min period for the duration of the experiment.
Stereotaxic surgery and drug infusion.
Before surgery, mice were anesthetized with ketamine (100 mg/kg, i.p.) and xylazine (10 mg/kg, i.p.). Animals were placed in a stereotaxic alignment instrument (David Kopf Instruments) specifically fitted for mice with a nose clamp adaptor and ear bars that allow leveling of the skull in the anterior–posterior and mediolateral planes. Two holes were drilled in the skull directly above the sites of implantation using a 0.033 inch diameter carbide drill bit. Coordinates for implantation were based on the mouse brain atlas relative to bregma: +2.0 mm anterior–posterior, ±0.5 mm mediolateral, 2.2 mm dorsoventral. Two additional holes were drilled in the skull caudal to the implantation holes using a 0.024-inch-diameter carbide drill bit and used to attach two zinc-plated self-tapping screws (Small Parts), which serve as a support scaffold for the cannulae. Custom-made bilateral guide cannulae (Plastics One) were slowly lowered into the brain to the proper dorsoventral coordinate and cannula base and screws were coated in dental acrylic (Henry Schein) to form a secure head cap. Once the dental acrylic was dry (∼10 min), the animal was removed from the stereotaxic instrument and custom-made dummy cannulae were inserted into the guide cannulae. Ten days after surgery, subjects underwent the microinjection procedure. Unanesthetized mice were gently restrained and dummy cannulae were removed from implanted guide cannulae and replaced with the injection cannulae. Injection cannulae were connected to PE-20 tubing and prefilled with 0.5 μl of drug or vehicle, and infusion was performed at a rate of 0.1 μl/min using an infusion pump (Harvard Apparatus). During the 5 min infusion procedure, the subject was returned to the home cage and allowed unrestrained mobility to minimize stress. After infusion, drug was allowed to diffuse for an additional 2 min before replacing the injection cannulae with dummy cannulae. Subjects were immediately transferred to the acoustic startle apparatus to assess PPI. Seven days after behavioral assessment, subjects were infused bilaterally with 0.5 μl of India ink and killed, and brains were removed for histological analysis to verify cannula placement.
Immunohistochemistry.
Wild-type (C57BL/6J) mice were deeply anesthetized with Euthasol (Virbac) and perfused with 0.9% NaCl, followed by 4% paraformaldehyde in phosphate buffer, pH 7.4. Brains were sectioned on a Vibratome (VT1000S; Leica Instruments) using coronal orientation. For immunostaining, free-floating sections containing mPFC were permeabilized with 50% ethanol for 20 min, rinsed in PBS, blocked with 10% normal donkey serum in PBS for 30 min, and incubated for 48 h at 4°C with a mixture of primary antibodies: monoclonal anti-GSK-3β (610201; 1:250; BD Biosciences), polyclonal rabbit anti-KCNQ2 antibody (ab22897; 1:500; Abcam), and polyclonal chicken anti-MAP2 antibody (ab15452; 1:250; Millipore). Sections were rinsed with PBS and incubated in 2% normal donkey serum for 10 min and incubated with secondary antibodies: Alexa Fluor 488-labeled donkey anti-rabbit, Alexa Fluor 594-labeled donkey anti-mouse (1:500; Invitrogen), and DyLight649-conjugated donkey anti-chicken (1:300; Jackson ImmunoResearch) for 4–6 h. After staining, sections were rinsed in PBS, mounted on gelatin-subbed slides, air-dried, and coverslipped using Vectashield mounting medium (Vector Laboratories). Images were acquired using a Zeiss LSM 510 META laser confocal microscope (Zeiss) using optimal factory recommended filter configurations. Stacks of confocal images were analyzed using colocalization plugins of the MBF ImageJ (McMaster University, Hamilton, Ontario, Canada) and the Imaris 6.0 software (Bitplane).
Slice preparation.
Adult mice were deeply anesthetized with 40 mg/kg pentobarbital (intraperitoneally) and perfused transcardially with ∼30 ml of chilled modified artificial CSF (aCSF) at a rate of ∼10 ml/min. The modified aCSF for perfusion contained the following (in mm): 225 sucrose, 119 NaCl, 2.5 KCl, 1.0 NaH2PO4, 4.9 MgCl2, 0.1 CaCl2, 26.2 NaHCO3, 1.25 glucose, 1 ascorbic acid, and 3 kynurenic acid. The brain was removed rapidly, and coronal slices (200 μm) containing the prelimbic cortex were cut in this same modified aCSF. Slices recovered at 32°C in carbogen-bubbled aCSF (126 mm NaCl, 2.5 mm KCl, 1.2 mm NaH2PO4, 1.2 mm MgCl2, 2.4 mm CaCl2, 18 mm NaHCO3, 11 mm glucose, with pH 7.2–7.4 and milliosmolarity, 301–305), with 1 mm ascorbic acid added just before the first slice. Brain slices recovered for 45 min to 6 h before use in experiments. During experiments, slices were submerged and continuously perfused (∼2 ml/min) with carbogen-bubbled aCSF warmed to 31–32°C, and supplemented with picrotoxin (50 μm, to block GABAA receptors) and DNQX (10 μm, to block AMPA-type glutamate receptors). Recordings were combined from regular spiking and intrinsic bursting neurons, identified as previously described (Cao et al., 2009). Intermediate neurons were not studied because of their low abundance and their lack of accommodation (data not shown).
In vitro electrophysiology.
All electrophysiology experiments were performed using whole-cell recording and visualized infrared–differential interference contrast with 2.5–3.5 MΩ electrodes. The internal solution was KMeS (potassium methanesulfonate) based, containing the following (in mm): 130 mm KOH, 105 mm methanesulfonic acid, 17 mm HCl, 20 mm HEPES, 0.3 mm EGTA, 2.8 mm NaCl, 2.5 mg/ml MgATP, 0.25 mg/ml GTP, pH 7.2–7.4, 275–285 mOsm. Electrical signals were recorded using Clampex 9.2 or 10.1 and an Axon 700A or 700B patch amplifier (Molecular Devices). Current-clamp data were acquired at 20 kHz and filtered at 2 kHz. After breaking into a neuron, the resting membrane potential was set to −75 to −70 mV by injecting DC current through the patch amplifier. To measure firing, current steps were applied using a patch amplifier in current-clamp mode, and a series of seven to eight current steps (800 ms duration, 20 pA apart) were applied every 30 s, where the minimum current amplitude was set for each cell so that the first step was just subthreshold for spike firing. Depolarizing steps were alternated with a 33.3 pA hyperpolarizing pulse to examine the input resistance. Voltage values were not corrected for the liquid junction potential, estimated to be 10 mV. Bridge balance was used to compensate 60–80% of the series resistance.
Statistical analysis.
Data were analyzed using SigmaStat 3.1 software (Systat Software) with appropriate post hoc comparisons performed as indicated by SigmaStat. Statistical tests were performed as follows: PPI data and baseline startle reactivity data were analyzed by two-way repeated-measures ANOVA for genotype by prepulse intensity or genotype by startle reactivity, followed by Newman–Keuls post hoc comparison. Similarly, pharmacological data were analyzed by two-way repeated-measures ANOVA for treatment group by prepulse intensity followed by Newman–Keuls post hoc comparison. mRNA and protein quantification in Ppp2r5δGt+/− and Kcnq2+/− mice was normalized to control mice and GAPDH signal and compared by Student's t test. Data are presented as mean ± SEM.
Results
Generation and characterization of Ppp2r5δ gene trap mice
PPP2R5δ, also known as the B56δ regulatory subunit of PP2A (McCright et al., 1996; Janssens and Goris, 2001), interacts with catalytic and scaffolding subunits to form the PP2A holoenzyme. To generate mice in which the Ppp2r5δ gene has been disrupted, we obtained gene trap cell line RRK451 from Bay Genomics (http://www.genetrap.org/). This cell line contains a gene trap insertion in the second intron of the Ppp2r5δ gene and is predicted to result in a fusion transcript of exons 1 and 2 with the gene encoding β-galactosidase, thereby truncating the majority of the endogenous Ppp2r5δ transcript (Fig. 1A). Ppp2r5δ heterozygous gene trap mice (Ppp2r5δGt/+) were viable and normal in appearance, but we failed to recover homozygous Ppp2r5δGt mice, indicating that the gene trap allele is likely a null (or strongly hypomorphic) allele.
Figure 1.

Characterization of Ppp2r5δGt gene trap mice. A, Schematic of the Ppp2r5δ locus disrupted by gene trap insertion between exons 2 and 3 of the gene. Exon number and size of locus in kilobases are indicated. βgeo, β-Galactosidase/neomycin resistance markers within the gene trap insertion. B, C, Western blot analysis of brain lysates from Ppp2r5δGt/+ and wild-type control mice quantifying protein levels. “+” denotes wild-type mouse, and “−” denotes Ppp2r5δGt/+ mouse. PPPP2R5δ protein is reduced by ∼50% in Ppp2r5δGt/+ mice, whereas pGSK3β (Ser-9) levels are increased by ∼30%. Ppp2r5δ disruption does not appear to affect pGSK3α (Ser-21) phosphorylation or total amounts of GSK3α or GSK3β. D, E, Ppp2r5δGt/+ and Gsk3β(tm1JW)/+ mice show reduced PPI relative to wild-type controls. F, GSK3 inhibitor, SB216763 (10 mg/kg), attenuates PPI in wild-type mice when administered as a single, intraperitoneal injection. Drug was administered 15 min before behavioral testing. Error bars represent SEM. *p < 0.05.
To quantify the reduction in PPP2R5δ protein levels in Ppp2r5δGt/+ mice, we performed Western analysis on total brain lysates from Ppp2r5δGt/+ and wild-type mice using an antibody generated against a C-terminal specific peptide of the PPP2R5δ protein. PPP2R5δ levels were reduced in the heterozygous mutant by ∼50% (p < 0.05) (Fig. 1B,C). These data are consistent with the Ppp2r5δGt allele being null or strongly hypomorphic.
Regulatory subunits of PP2A confer tissue, temporal and substrate specificity to PP2A activity (Janssens and Goris, 2001). To determine whether PPP2R5δ regulates PP2A activity upstream of GSK3β in vivo, we assessed the phosphorylation state of GSK3β in brain tissue of Ppp2r5δGt/+ mice. GSK3β activity is inhibited by phosphorylation of serine-9 (Ser-9) (Cross et al., 1995). Using a GSK3β Ser-9 phosphospecific antibody, we observed a ∼30% increase in phosphorylation level of GSK3β in brain lysates from Ppp2r5δGt/+ mice (p < 0.05) (Fig. 1B,C), indicating that the PPP2R5δ subunit normally acts to positively regulate GSK3β activity in adult mouse brain. This effect appeared specific to GSK3β activity, as phosphorylation of GSK3α at Ser-21 was normal in the Ppp2r5δGt/+ mutants (Fig. 1B,C). We also failed to detect an effect of PPP2R5δ expression on the amount of total GSK3α or GSK3β protein, suggesting that compensatory changes in the levels of these proteins do not occur when one copy of the Ppp2r5δ gene is disrupted (Fig. 1B,C).
Genetic and pharmacological disruption of PP2A/GSK3β activity attenuates PPI
Ppp2r5δ expression in whole brain correlates positively with performance in the PPI assay among BXD (C57BL/6J × DBA/2J) recombinant inbred strains of mice (McCaughran et al., 1999) (http://www.genenetwork.org/). To determine whether disruption of the Ppp2r5δ gene affects sensorimotor gating, we examined whether PPI would be differentially affected in Ppp2r5δGt/+ mice and wild-type littermates. We observed a significant strain effect for PPI (F(1,38) = 9.6; p < 0.01), with Ppp2r5δGt/+ mice, showing a significant reduction in PPI at the 78, 82, 86, and 90 dB prepulse levels (Fig. 1D). In the absence of prepulse, Ppp2r5δGt/+ mice demonstrated normal startle reactivity to a range of startle intensities (F(1,24) = 0.50; p = 0.51), indicating that the PPI defect of Ppp2r5δGt/+ mice was not the result of impaired auditory or motor function. In addition, Ppp2r5δGt/+ mice showed normal locomotor activity levels (main effect of genotype: F(1,14) = 0.16; p = 0.70) and habituation to an open field (genotype by time interaction: F(1,11) = 0.69; p = 0.75).
Based on our biochemical data showing reduced GSK3β activity in Ppp2r5δGt/+ mice, we examined PPI in mice heterozygous for a null allele of Gsk3β, Gsk3β(tm1JW)/+ (Hoeflich et al., 2000). We observed a strain effect for PPI (F(1,15) = 4.8; p < 0.05) (Fig. 1E), with Gsk3β(tm1JW)/+ mice showing a significant reduction in PPI at the 74 dB prepulse level (p < 0.01) (Fig. 1E) relative to wild-type littermates. The attenuated PPI in Gsk3β(tm1JW)/+ mice was not associated with impaired baseline startle response (F(1,15) = 0.49; p = 0.49), locomotor activity (F(1,14) = 0.01; p = 0.93), or locomotor habituation (genotype by time interaction: F(1,10) = 0.86; p = 0.57). These data are consistent with PP2A regulating PPI, (at least) in part, through signaling via GSK3β.
Classical studies of PP2A and GSK3β function have established a role for these proteins in development (Siegfried and Perrimon, 1994). To determine whether the PPI impairment observed in Gsk3β(tm1JW)/+ mice was the result of an acute requirement for GSK3β or compensatory changes in the mutant, we tested the effect of acute, systemic administration of a pharmacological inhibitor of GSK3 activity on PPI. SB216763 inhibits both GSK3α and GSK3β by competing with ATP binding (Forde and Dale, 2007). SB216763 (10 mg/kg) robustly attenuated PPI in wild-type C57BL/6J mice, relative to vehicle-treated controls (strain effect for treatment group, F(1,16) = 9.9; p < 0.01, post hoc comparison, p < 0.05 at prepulse levels 78, 82, 86, and 90 dB) (Fig. 1F). These results demonstrate that PPI can be acutely modulated by inhibition of GSK3 activity.
Genetic and pharmacological disruption of KCNQ2/M-channel activity attenuates PPI
Kcnq2 encodes an M-type potassium channel (M-channel) protein and putative GSK3β substrate (Singh et al., 1998; Borsotto et al., 2007). Kcnq2 has previously been associated with bipolar disorder (Borsotto et al., 2007) and loss-of-function mutations in the gene are associated with benign neonatal familial convulsions, a mild form of epilepsy (Charlier et al., 1998; Singh et al., 1998; Cooper et al., 2000; Mulley et al., 2003). These observations suggest that neuronal hyperexcitability resulting from altered PP2A/GSK3β modulation of KCNQ2 channel activity might underlie the PPI defect of Ppp2r5δGt/+ and Gsk3β(tm1JW)/+ mutants and wild-type mice treated with SB216763. We obtained a Kcnq2 gene trap line, Kcnq2(tm1Dgen), derived from insertion of a gene trap vector in the first intron of the Kcnq2 gene (Fig. 2A) and predicted to result in a null allele. This mutation is homozygous lethal, with heterozygous animals being fertile and normal in appearance; the only reported behavioral phenotype of Kcnq2(tm1Dgen)/+ mice is a reduced threshold to pharmacologically induced seizures (http://www.informatics.jax.org/external/ko/deltagen/1112.html). Analysis of Kcnq2 transcript levels in brains from Kcnq2(tm1Dgen)/+ and control animals detected a 50% reduction in Kcnq2 mRNA in the heterozygous mice (p < 0.05) (Fig. 2B).
Figure 2.

M-type potassium channels regulate PPI. A, Schematic of the Kcnq2(tm1Dgen) locus. The gene trap insertion disrupts the Kcnq2 locus between exons 1 and 2. Exon number and size of locus in kilobases are indicated. βgeo, β-Galactosidase/neomycin resistance markers within the gene trap insertion. B, Kcnq2 mRNA is reduced in total brain tissue from Kcnq2(tm1Dgen)/+ mice by ∼50%. C, Kcnq2(tm1Dgen)/+ mice show reduced PPI relative to wild-type controls. D, Acute systemic administration of the M-channel blocker, linopirdine (5 mg/kg), attenuates PPI in wild-type mice when administered as a single, intraperitoneal injection. Drug was administered 15 min before behavioral testing. Coadministration of SB216763 (10 mg/kg) and linopirdine (5 mg/kg) conferred a similar effect on PPI as either drug alone. Error bars represent SEM. *p < 0.05.
We next tested whether genetic or pharmacological manipulations impairing M-channel function would alter PPI. Kcnq2(tm1Dgen)/+ mice had significantly reduced PPI relative to controls (main effect of genotype: F(1,38) = 7.1; p < 0.05; post hoc effect of genotype: p < 0.05 at 78, 86, and 90 dB) (Fig. 2C). Kcnq2(tm1Dgen)/+ mice showed no defects in overall ASR (F(1,18) = 0.18; p = 0.68), activity (F(1,14) = 0.04; p = 0.85), or habituation to an open field (genotype by time interaction: F(1,10) = 1.38; p = 0.19). We also tested the effects of acute pharmacological inhibition of M-channel function on PPI by treating wild-type C57BL/6J mice with a single intraperitoneal dose of the M-channel blocker linopirdine (5 mg/kg). Consistent with our results with the GSK inhibitor, SB216763, mice treated systemically with linopirdine showed a significant reduction in PPI relative to vehicle-treated controls (effect of drug: F(1,16) = 6.8; p < 0.05; post hoc effect of drug at 86 and 90 dB: p < 0.05) (Fig. 2D). Furthermore, treatment with both linopirdine and SB216763 reduced PPI to a similar extent as either drug alone (linopirdine and SB216763 vs vehicle: F(1,16) = 5.9; p < 0.05; post hoc effect of drug at 82 and 86 dB: p < 0.05; linopirdine and SB216763 vs SB216763 alone: F(1,16) = 0.64; p = 0.44; linopirdine and SB216763 vs linopirdine alone: F(1,16) = 0.02; p = 0.88) (Fig. 2D). Together, these data support the hypothesis that the acute inhibition of GSK3 with SB216763 decreases PPI by altering M-type potassium channel function.
Inhibition of GSK3 and M-channel activity within the mPFC attenuates PPI
Neural substrates underlying PPI involve descending projections from the forebrain that modulate the activity of pontine circuitry to control acoustic startle reactivity (Swerdlow et al., 2001). The mPFC likely plays a central role in the neurocircuitry controlling PPI, since it receives input from the mediodorsal thalamus and ventral tegmental area (VTA), and projects to the nucleus accumbens (NAc) as well as the VTA (Swerdlow et al., 2001). Because of the role of the mPFC in PPI and the observation that GSK3β activity in frontal cortex is positively correlated with PPI among inbred strains of mice (Amar et al., 2004), we investigated whether suppressing GSK3 and M-channel function locally within the mPFC alters PPI.
We first performed immunohistochemistry to determine whether GSK3β and KCNQ2 proteins are expressed in mPFC and colocalize in vivo. GSK3β and KCNQ2 showed overlapping expression in the mPFC, where both localized to MAP2-positive dendrites (Fig. 3A,B). Unbiased colocalization analysis and volume reconstruction performed on high-resolution confocal microscope image stacks confirmed colocalization between immunoreactivity for GSK3β and for KCNQ2 in dendrites.
Figure 3.
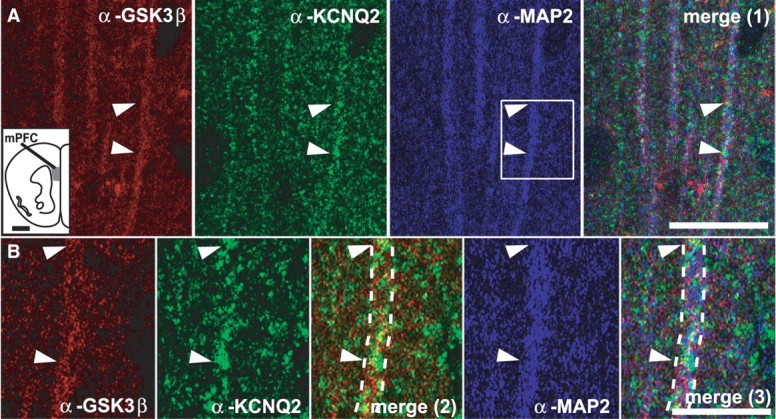
GSK3β and KCNQ2 proteins colocalize to mPFC. A, Triple-channel confocal images of mPFC. Individual apical dendrites of pyramidal cells were identified with the anti-MAP2 antibody (blue). Some dendrites were positive for both GSK3β (red) and KCNQ2 (green) indicating coexpression of both proteins in mPFC neurons (arrowheads point to same areas on all panels). Merge (1) panel shows overlay of anti-GSK3β, anti-KCNQ2, and anti-MAP2 staining in low-magnification panels. The inset shows the area of mPFC from which images were taken. B, Higher magnification images of areas shown in A, indicated in boxed region, illustrate colocalization of all three markers; the dashed lines outline the same dendrite fragment on all panels. Merge (2) panel shows overlay of anti-GSK3β and anti-KCNQ2 staining in high magnification panels; merge (3) panel shows overlay of anti-GSK3β, anti-KCNQ2, and anti-MAP2 staining in high-magnification panels. Scale bars: A, 20 μm; B, 5 μm.
To determine whether modulation of GSK3 activity in the mPFC is sufficient to alter PPI, we administered the GSK3 inhibitor SB216763 (5 pm) bilaterally into mPFC. GSK3 inhibition within the mPFC reduced PPI compared with vehicle-infused controls (F(1,14) = 5.0; p < 0.05; post hoc effect of drug at the 74, 78 and 86 dB levels, p < 0.05) (Fig. 4A). Placement of cannulae was verified by histological analysis demonstrating consistent targeting of the mPFC (primarily prelimbic and cortex areas) (Paxinos and Franklin, 2001) (Fig. 4C). These results demonstrate that inhibition of GSK3 in mPFC is sufficient to disrupt PPI.
Figure 4.

GSK3β and M-type potassium channels function in the mPFC to regulate PPI. A, PPI is reduced in wild-type mice administered SB216763 (5 pm) bilaterally in mPFC compared with vehicle-infused controls. B, PPI is reduced in wild-type mice administered linopirdine (5 pm) bilaterally in mPFC compared with vehicle-infused controls. C, Schematic depicting placement of cannulae targeting mPFC (between bregma +1.8 and bregma +2.8) in subjects from drug infusion experiments. Subjects were tested for PPI immediately after drug infusion. Error bars represent SEM. *p < 0.05.
We next focused on the mPFC as a candidate brain region in which the M-channel might also regulate PPI. If acute inhibition of GSK3 in the mPFC attenuates PPI by altering M-channel function, we would predict that acutely blocking M-channel activity in the mPFC would have a similar behavioral effect. Infusion of linopirdine (5 pm) bilaterally into the mPFC of wild-type mice reduced PPI relative to vehicle-infused controls (F(1,23) = 6.5; p < 0.05; post hoc effect of drug at 74 dB and 86 dB, p < 0.05) (Fig. 4B,C). Together, these results demonstrate that inhibition of GSK3 or M-channels within the mPFC is sufficient to disrupt PPI and support the hypothesis that GSK3 regulates PPI via modulation of M-channel activity within the mPFC.
Inhibition of GSK3 signaling in the mPFC alters M-type potassium channel activity
To better characterize the interaction of GSK3β and KCNQ in the mPFC, we performed in vitro brain slice electrophysiology in layer II/III prelimbic cortical pyramidal neurons from adult mice. Neurons were brought to a −75 to −70 mV resting potential by passage of DC current from the patch amplifier, and were depolarized by 800 ms current pulses. Changes in action potential (AP) firing with applied current were measured relative to rheobase, the first current step at which firing was evoked.
Inhibition of the M-current with linopirdine moderately enhanced firing of control mouse cortical neurons (Fig. 5A,B,D) (APs at the current step 100 pA greater than rheobase: baseline, 7.8 ± 0.6 APs; linopirdine, 9.6 ± 0.5 APs; n = 6). In contrast, after pretreatment with the GSK3 inhibitor SB216763, linopirdine had no effect on firing (Fig. 5A,B,D) (APs at the current step 100 pA greater than rheobase: baseline, 11.0 ± 0.8 APs; linopirdine, 11.1 ± 1.0 APs; n = 7). A two-way repeated-measures ANOVA showed a significant effect of group (F(1,25) = 4.912; p < 0.05), linopirdine (F(1,25) = 24.314; p < 0.001), and a significant group by linopirdine interaction (F(1,25) = 19.75; p < 0.001), and Tukey post hoc comparisons showed a significant effect of linopirdine in control neurons (p < 0.001), no effect of linopirdine in SB216763-treated cells (p > 0.05), a significant difference at baseline between control and SB216763-treated cells (p < 0.05), and no difference between control and SB216763-treated cells after linopirdine (p = 0.20). There was also no effect of SB216763 on the minimum current required to evoke an action potential (without SB216763, 91.1 ± 8.2 pA; with SB216763, 80.9 ± 17.7 pA; t(11) = 0.492; p > 0.05). Since linopirdine and SB216763 both increased firing, and linopirdine had no additional effect in SB216763-treated neurons, these results suggest that GSK3 inhibition reduced M-current function in mPFC neurons.
Figure 5.
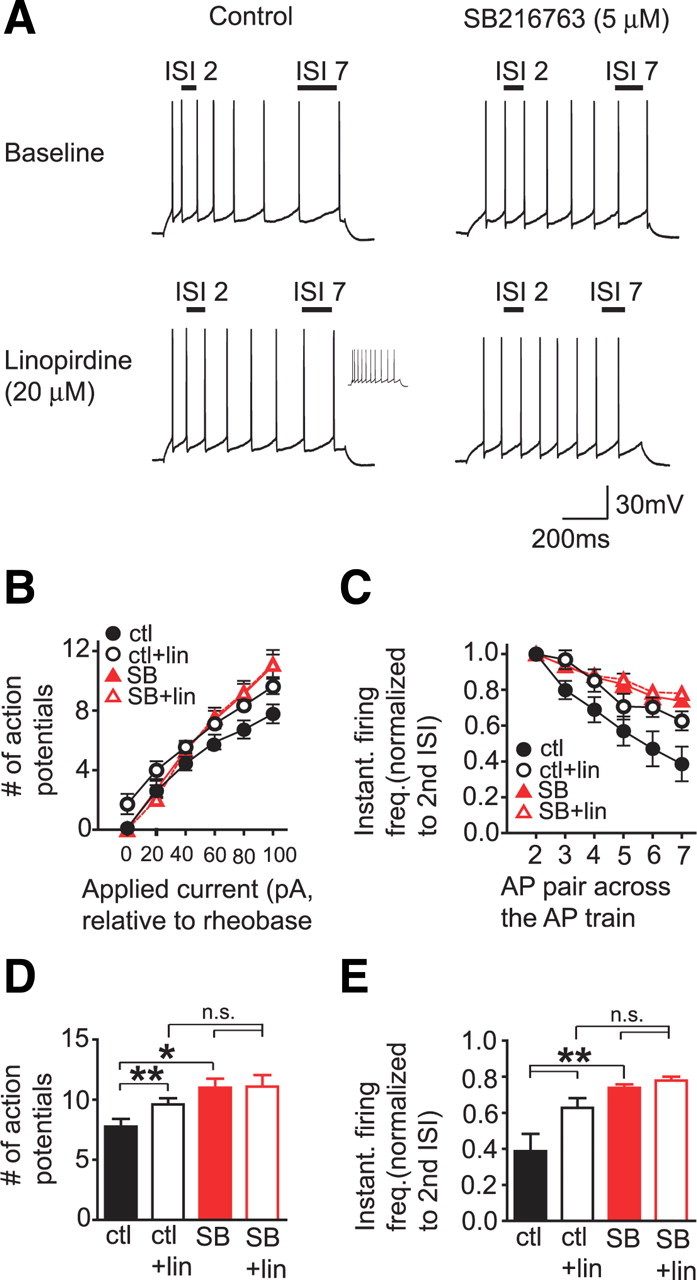
GSK3 and M-type potassium channel inhibition have similar effects on AP firing and accommodation in layer II/III prelimbic neurons in vitro. A, Example traces showing that linopirdine and SB216763 exposure reduce accommodation, measured at the current step at which eight APs were generated, and that linopirdine has no effect in SB216763-pretreated neurons. The inset for the control/linopirdine trace shows the increase in firing at the same current step shown for the control/baseline trace. Number of APs per current step was determined for each neuron in relation to rheobase, the minimum current required to evoke an AP in that neuron. B, Linopirdine and SB216763 enhance AP firing, and linopirdine has no effect on firing in SB216763-pretreated neurons. C, Linopirdine and SB216763 reduce accommodation, and linopirdine has no effect on accommodation in SB216763-pretreated neurons. Accommodation was assayed at the current step with eight APs and was determined by converting ISI values for each AP pair in an AP train to IFF, and then normalizing IFF values for each AP pair in the AP train to the IFF value for the second AP pair in the AP train. D, E, Bar graphs showing statistical differences between the APs generated at the 100 pA step above rheobase (D), and the IFF values for the seventh AP pair in the AP train (E). ctl, Control; freq., frequency; instant., instantaneous; lin, linopirdine; n.s., not significant, p > 0.05; SB, SB216763. *p < 0.05 or **p < 0.001.
Although linopirdine increased firing in control neurons, the effect was moderate relative to other brain areas such as the hippocampus (Aiken et al., 1995; Peters et al., 2005; Otto et al., 2006). Therefore, we next examined the effect of linopirdine and SB216763 on accommodation, a measure of the regularity of firing across an AP train, using methods adapted from Otto et al. (2002, 2004). Accommodation is an increase in the interspike interval (ISI) later in a train of APs relative to earlier in an AP train, and previous studies have shown that inhibition of the M-current reduces accommodation, making firing across the spike train more regular (Peters et al., 2005; Otto et al., 2006). To analyze accommodation, we converted the ISI for each AP pair in an AP train to an instantaneous firing frequency (IFF) and normalized the IFF values across the spike train to the second IFF of the spike train (the first IFF was not used because in some cells, the first two APs were very close together in a couplet). Analyses of accommodation were performed at a current step where eight APs were generated.
As shown in Figure 5, C and E, control mPFC neurons showed strong accommodation, indicated by higher normalized IFF values by the end of the AP train, and a reduction in accommodation by linopirdine (final IFF value in AP train: baseline, 0.39 ± 0.10; linopirdine, 0.63 ± 0.05). In contrast, neurons pretreated with SB216763 showed higher basal normalized IFF values, which were unaffected by linopirdine (final IFF value in AP train: baseline, 0.74 ± 0.02; linopirdine, 0.78 ± 0.02). A two-way repeated-measures ANOVA of the final normalized IFF in the AP train showed a significant effect of group (F(1,25) = 12.44; p < 0.01), linopirdine (F(1,25) = 36.16; p < 0.001), and a significant group by linopirdine interaction (F(1,25) = 18.29; p = 0.001), and Tukey's post hoc comparisons showed a significant effect of linopirdine in control neurons (p < 0.001), no effect of linopirdine in SB216763-treated cells (p > 0.05), a significant basal difference between control and SB216763-treated cells (p < 0.001), and no difference between control and SB216763-treated cells after linopirdine (p > 0.05). These data are in agreement with the firing rate results above and show that GSK3 inhibition occludes the effect of M-current inhibition on accommodation, suggesting that GSK3 modulates M-channel activity in vivo.
Discussion
We have identified a novel mechanism by which GSK3β regulation of the M-channel in the mPFC modulates sensorimotor gating, as assayed by PPI. Genetic knockdown of PP2A reduces PPI and enhances phosphorylation of GSK3β at a site associated with inhibition of GSK3β activity. Genetic knockdown or pharmacological inhibition of GSK3β or the M-channel also reduces PPI. Importantly, inhibition of GSK3β or M-channels locally within the mPFC also significantly reduces PPI. These results suggest that GSK3β positively regulates M-channel function within the mPFC and that this interaction is critical for expression of PPI.
Our data demonstrate that haploinsufficiency for either Ppp2r5δ or Gsk3β in mice results in reduced PPI of the ASR. PPP2R5δ acts as a regulatory subunit of the holoenzyme PP2A, which has been reported to function in a signaling cascade upstream of GSK3β (Sutherland et al., 1993; Cross et al., 1995). Here, we show that Ppp2r5δGt/+ mice have increased (Ser-9)-pGSK3β levels in brain tissue, indicating that PPP2R5δ is a positive regulator of GSK3β activity in the brain, an observation consistent with our behavioral data.
Interestingly, we observe a more robust PPI defect in Ppp2r5δGt/+ mice than Gsk3β(tm1JW)/+ mice. One possible explanation could be if loss of PPP2R5δ altered GSK3α in addition to GSK3β activity, which could have an additive effect on PPI. However, we observed no alteration in levels of (Ser-21)pGSK3α in brains of Ppp2r5δGt/+ mice. Transgenic knock-in mice with a constitutively active allele of Gsk3β [S9A] show decreased levels of total GSK3α protein in striatum (Prickaerts et al., 2006), although GSK3β haploinsufficiency does not alter GSK3α levels (Hoeflich et al., 2000; O'Brien et al., 2004). We did not, however, detect any alteration in total GSK3α or total GSK3β protein levels in Ppp2r5δGt/+ mice, raising the possibility that PP2A signaling through additional substrates may contribute to PPI.
We also observed that PPI was acutely attenuated by the potent and highly selective GSK3 inhibitor SB216763, which is important for several reasons. First, it indicates that GSK3 activity acutely modulates PPI, and suggests that the blunted PPI in Gsk3β(tm1JW)/+ mice was independent of developmental defects. The greater effect on PPI produced by SB216763 compared with the phenotype of the Gsk3β(tm1JW)/+ heterozygous mutants suggests that PPI may be highly sensitive to GSK3 activity and that merely reducing total GSK3β levels by 50% is not sufficient to induce a robust attenuation of PPI. Consistent with this premise, TDZD-8 (4-benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione) (30 mg/kg), a less effective GSK3 inhibitor (Beaulieu et al., 2004), produced a small reduction in PPI that was not statistically significant (data not shown). Alternatively, compensatory changes in the Gsk3β(tm1JW)/+ mice during development may potentially account for the lack of a robust PPI phenotype in the mutants. We should also note that, in contrast to our data, other groups have failed to detect a PPI defect in Gsk3β(tm1JW)/+ mice (O'Brien et al., 2004; Bersudsky et al., 2008). Differences between our data and that of others may be attributed to differences in genetic background, sex, experimental protocol, and/or animal handling, all of which can affect responses in behavioral assays (Crabbe et al., 1999). In the study by O'Brien et al. (2004), mice had been subjected to open field and ASR tests before PPI testing, whereas our animals were naive to behavioral testing before the PPI assay. Subjects in the study by Bersudsky et al. (2008) were experimentally naive before PPI testing, however were maintained on a mixed genetic background (C57BL/6J × C57BL/6OlaHsd). The performance of Gsk3β(tm1JW)/+ mice in assays of “behavioral despair” or depression-like behavior has similarly not been replicated across laboratories (O'Brien et al., 2004; Bersudsky et al., 2008), indicating that Gsk3β(tm1JW)/+ mice may be particularly sensitive to variations in experimental condition (Beaulieu et al., 2008). Nonetheless, our pharmacological results support a role for GSK3β in regulating PPI.
How might PP2A/GSK3β signaling modulate PPI? Although many substrates of GSK3β have been identified, including the transcriptional activator β-catenin (Yost et al., 1996; Doble and Woodgett, 2003), the rapid time frame of our pharmacological findings (15 min after systemic treatment or immediately after drug infusion in cannulation experiments) makes the involvement of transcriptional changes unlikely. Instead, our results suggest that GSK3β modulates PPI through the M-type potassium channel. The M-type potassium channel protein KCNQ2 was identified recently as a putative substrate of PP2A and GSK3β (Borsotto et al., 2007). Moreover, a mutation in the PIP5K2A gene, which leads to reduced M-channel activity, has been linked to schizophrenia (Fedorenko et al., 2008), suggesting that impaired M-channel function may lead to sensorimotor gating deficits. Here, we showed that Kcnq2(tm1Dgen)/+ mice showed reduced PPI, an effect that was also seen in wild-type mice treated with the M-channel blocker linopirdine.
M-channels are voltage-gated potassium channels that are strongly activated on depolarization, repolarizing the cell, and potently regulating neuronal excitability (Brown and Adams, 1980; Brown, 1988; Jentsch, 2000). These properties make the involvement of M-channels an attractive mechanism for modulating PPI. Our in vivo behavioral and in vitro electrophysiological analyses both showed a similar pattern of results, where M-channel inhibition and GSK3 inhibition had similar effects on PPI expression and on mPFC firing and GSK3 inhibition prevented additional effects of M-channel inhibition. Based on these findings, we suggest a hypothesis in which, under normal conditions, cortical firing during the first acoustic stimulus leads to M-channel activation, which depresses cortical firing and behavioral activation in response to the second acoustic stimulus, 80 ms after the first (Fig. 6). With impaired M-channels, we propose there would be no suppression of firing and behavior during the second acoustic stimulus. Although there are few detailed studies of M-channel kinetic properties, especially in intact neurons in which voltage-clamp control is more challenging, a recent study suggests that M-channels can certainly be activated within the 80 ms interval used in our PPI studies (Hu et al., 2007). Determining with certainty the level of M-channel activation in vivo during the first acoustic stimulus would require analysis of firing changes with in vivo electrophysiology, which is beyond the scope of the present work, although other studies have indicated that mPFC neurons often show strong increases in firing to potent or novel stimuli (Pirch et al., 1992; Baeg et al., 2001; Jackson and Moghaddam, 2006). Interestingly, we did not observe enhanced acoustic startle reactivity in Kcnq2(tm1Dgen)/+ mice in the absence of prepulses, suggesting that M-channel-induced changes in neuronal excitability specifically after the prepulse stimulus may be critical for modulating the subsequent response to the startle stimulus.
Figure 6.

Proposed model of how M-channel inhibition in mPFC attenuates PPI. Under normal conditions, mPFC neuron in the PPI circuitry fires in response to the prepulse stimulus. M-channel activation renders the neuron less responsive to the subsequent startle stimulus and startle reactivity is gated. mPFC neurons in which M-channels have been inhibited, either through reduced PP2A/GSK3 signaling or direct inhibition of the M-channel, are hypothesized to fire normally (or with increased probability) in response to a prepulse stimulus but remain responsive to the startle stimulus. “B56δ” denotes the PP2A regulatory subunit, PPP2R5δ.
KCNQ2 forms heteromeric channels with KCNQ3 in the brain (Cooper et al., 2000; Hadley et al., 2003), the activity of which may be blocked by hyperphosphorylation (Nakajo and Kubo, 2005). In vitro evidence suggests that GSK3β and PP2A may have opposing actions on KCNQ2 phosphorylation, with GSK3β and PKA synergistically phosphorylating KCNQ2 and PP2A dephosphorylating KCNQ2 (Borsotto et al., 2007). If PP2A were to directly dephosphorylate KCNQ2 in vivo, we might predict that loss of the positive regulatory subunit (PPP2R5δ) of PP2A would result in hyperphosphorylation of KCNQ2 and inhibition of the channel, consistent with our behavioral observations in Ppp2r5δGt/+ and Kcnq2(tm1Dgen)/+ mutants and mice treated with linopirdine. However, this model fails to account for PP2A signaling upstream of GSK3β and our behavioral data showing that GSK3β inhibition reduces PPI. PP2A and GSK3β modulation of M-channel activity in vitro could be different from in vivo, depending on which PP2A regulatory subunits, scaffolding proteins, and/or other cofactors are present.
Importantly, we observed that inhibition of GSK3 or the M-current directly within the mPFC reduced PPI, suggesting that these currents in the mPFC represent a novel and critical mechanism that regulates PPI. In support of our behavioral data suggesting an important role for the mPFC in PPI, our whole-cell recordings from mPFC neurons showed that GSK3 and M-channel inhibition similarly altered firing rate and spike frequency accommodation. Furthermore, GSK3 inhibition occluded the ability of an M-channel blocker to alter firing, suggesting that GSK3 normally activates M-channels in mPFC neurons. These data are an important confirmation of our behavioral data, demonstrating that inhibition of GSK3 or M-channel activity systemically or within the mPFC is sufficient to impair PPI.
It has been proposed that reducing dopaminergic tone in PFC attenuates PPI by disinhibition of descending glutamatergic fibers, resulting in increased striatal DA release (Koch and Bubser 1994; Ellenbroek et al., 1996; Japha et al., 1999). Specifically, disrupting mPFC function by locally administering D1 and D2 antagonists (Ellenbroek et al., 1996) or 6-OHDA lesions (Bubser and Koch, 1994, Swerdlow et al., 2006) disrupt PPI, although NMDA antagonists in the mPFC do not inhibit PPI (Bakshi and Geyer, 1998). These data are consistent with a model of reduced dopaminergic function in mPFC contributing to schizophrenia. In addition, local administration of picrotoxin into the mPFC, which might be predicted to increase cortical activity, can also disrupt PPI (Japha and Koch, 1999). Thus, we hypothesize that pharmacological inhibition of GSK3 and M-channels in the mPFC disrupts PPI through enhanced and perhaps aberrant activation of the cortex.
Several groups have reported that systemic treatment with the D2 antagonist haloperidol inhibits GSK3β activity in frontal cortex (Li et al., 2007; Roh et al., 2007). However, systemic administration of haloperidol in wild-type mice typically enhances PPI, despite increasing extracellular dopamine in the nucleus accumbens (Hernandez and Hoebel, 1989; Moghaddam and Bunney, 1990; McCaughran et al., 1997; Ouagazzal et al., 2001), whereas infusion of the D2 agonist quinpirole directly into nucleus accumbens impairs PPI (Wan and Swerdlow, 1993). Although we have not investigated the role of DA signaling in the context of GSK3β/M-channel activity and PPI, our behavioral and electrophysiological data are consistent with a model in which inhibition of GSK3β in the mPFC inhibits M-channel activity, resulting in hyperactivation of glutamatergic projections to the NAc, presumably increasing accumbal DA and reducing PPI.
Our data provide, to our knowledge, the first in vivo evidence linking GSK3β with M-channel function. We identify the mPFC as a site in which GSK3β and M-channels play a critical role in modulating PPI in the mouse and describe a model that may be relevant for studying the pathophysiology associated with sensorimotor gating disorders.
Footnotes
This work was supported by the State of California for medical research on alcohol and substance abuse through University of California, San Francisco; National Institutes of Health (U.H.); and National Alliance for Research on Schizophrenia and Depression (D.K.). We thank Dr. J. R. Woodgett at the University of Toronto for the generous gift of Gsk3(βtm1JW)/+ mice, and F. Giorgetti, S. Taylor, M. J. Wallace, J. Connolly, and S. A. Taha for advice and technical assistance.
References
- Aghdam SY, Barger SW. Glycogen synthase kinase-3 in neurodegeneration and neuroprotection: lessons from lithium. Curr Alzheimer Res. 2007;4:21–31. doi: 10.2174/156720507779939832. [DOI] [PubMed] [Google Scholar]
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alimohamad H, Rajakumar N, Seah YH, Rushlow W. Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry. 2005a;57:533–542. doi: 10.1016/j.biopsych.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Alimohamad H, Sutton L, Mouyal J, Rajakumar N, Rushlow WJ. The effects of antipsychotics on beta-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J Neurochem. 2005b;95:513–525. doi: 10.1111/j.1471-4159.2005.03388.x. [DOI] [PubMed] [Google Scholar]
- Amar S, Jones BC, Nadri C, Kozlovsky N, Belmaker RH, Agam G. Genetic correlational analysis of glycogen synthase kinase-3 beta and prepulse inhibition in inbred mice. Genes Brain Behav. 2004;3:178–180. doi: 10.1111/j.1601-183X.2004.00065.x. [DOI] [PubMed] [Google Scholar]
- Baeg EH, Kim YB, Jang J, Kim HT, Mook-Jung I, Jung MW. Fast spiking and regular spiking neural correlates of fear conditioning in the medial prefrontal cortex of the rat. Cereb Cortex. 2001;11:441–451. doi: 10.1093/cercor/11.5.441. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, Geyer MA. Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci. 1998;18:8394–8401. doi: 10.1523/JNEUROSCI.18-20-08394.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Wetsel WC, Gainetdinov RR, Caron MG. Reply to Belmaker et al.: GSK3β haploinsufficiency results in lithium-like effects in the forced-swim test. Proc Natl Acad Sci U S A. 2008;105:E24. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersudsky Y, Shaldubina A, Kozlovsky N, Woodgett JR, Agam G, Belmaker RH. Glycogen synthase kinase-3beta heterozygote knockout mice as a model of findings in postmortem schizophrenia brain or as a model of behaviors mimicking lithium action: negative results. Behav Pharmacol. 2008;19:217–224. doi: 10.1097/FBP.0b013e3282feb099. [DOI] [PubMed] [Google Scholar]
- Borsotto M, Cavarec L, Bouillot M, Romey G, Macciardi F, Delaye A, Nasroune M, Bastucci M, Sambucy JL, Luan JJ, Charpagne A, Jouët V, Léger R, Lazdunski M, Cohen D, Chumakov I. PP2A-Bgamma subunit and KCNQ2 K+ channels in bipolar disorder. Pharmacogenomics. 2007;7:123–132. doi: 10.1038/sj.tpj.6500400. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry. 1990;47:181–188. doi: 10.1001/archpsyc.1990.01810140081011. [DOI] [PubMed] [Google Scholar]
- Braff DL, Swerdlow NR, Geyer MA. Gating and habituation deficits in the schizophrenia disorders. Clin Neurosci. 1995;3:131–139. [PubMed] [Google Scholar]
- Brown D. M-currents: an update. Trends Neurosci. 1988;11:294–299. doi: 10.1016/0166-2236(88)90089-6. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Bubser M, Koch M. Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacology (Berl) 1994;113:487–492. doi: 10.1007/BF02245228. [DOI] [PubMed] [Google Scholar]
- Cadenhead KS, Geyer MA, Braff DL. Impaired startle prepulse inhibition and habituation in patients with schizotypal personality disorder. Am J Psychiatry. 1993;150:1862–1867. doi: 10.1176/ajp.150.12.1862. [DOI] [PubMed] [Google Scholar]
- Cao XY, Xu H, Wu LJ, Li XY, Chen T, Zhuo M. Characterization of intrinsic properties of cingulate pyramidal neurons in adult mice after nerve injury. Mol Pain. 2009;5:73. doi: 10.1186/1744-8069-5-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY. Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci U S A. 2000;97:4914–4919. doi: 10.1073/pnas.090092797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Wahlsten D, Dudek BC. Genetics of mouse behavior: interactions with laboratory environment. Science. 1999;284:1670–1672. doi: 10.1126/science.284.5420.1670. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116:1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellenbroek BA, Budde S, Cools AR. Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience. 1996;75:535–542. doi: 10.1016/0306-4522(96)00307-7. [DOI] [PubMed] [Google Scholar]
- Fedorenko O, Strutz-Seebohm N, Henrion U, Ureche ON, Lang F, Seebohm G, Lang UE. A schizophrenia-linked mutation in PIP5K2A fails to activate neuronal M channels. Psychopharmacology. 2008;199:47–54. doi: 10.1007/s00213-008-1095-x. [DOI] [PubMed] [Google Scholar]
- Forde JE, Dale TC. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci. 2007;64:1930–1944. doi: 10.1007/s00018-007-7045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology. 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Gould TD, Picchini AM, Einat H, Manji HK. Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr Drug Targets. 2006;7:1399–1409. doi: 10.2174/1389450110607011399. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Hoebel BG. Haloperidol given chronically decreases basal dopamine in the prefrontal cortex more than the striatum or nucleus accumbens as simultaneously measured by microdialysis. Brain Res Bull. 1989;22:763–769. doi: 10.1016/0361-9230(89)90097-x. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hoenig K, Hochrein A, Quednow BB, Maier W, Wagner M. Impaired prepulse inhibition of acoustic startle in obsessive-compulsive disorder. Biol Psychiatry. 2005;57:1153–1158. doi: 10.1016/j.biopsych.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Hoffman HS, Searle JL. Acoustic variables in the modification of startle reaction in the rat. J Comp Physiol Psychol. 1965;60:53–58. doi: 10.1037/h0022325. [DOI] [PubMed] [Google Scholar]
- Hogan B. Manipulating the mouse embryo: a laboratory manual. Plainview, NY: Cold Spring Harbor Laboratory; 1994. [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison JR, Hammond GR. Modification of the startle reflex in the rat by changes in the auditory and visual environments. J Comp Physiol Psychol. 1971;75:435–452. doi: 10.1037/h0030934. [DOI] [PubMed] [Google Scholar]
- Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kähäri VM, Heino J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol Cell Biol. 2002;22:1352–1359. doi: 10.1128/mcb.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ME, Moghaddam B. Distinct patterns of plasticity in prefrontal cortex neurons that encode slow and fast responses to stress. Eur J Neurosci. 2006;24:1702–1710. doi: 10.1111/j.1460-9568.2006.05054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Japha K, Koch M. Picrotoxin in the medial prefrontal cortex impairs sensorimotor gating in rats: reversal by haloperidol. Psychopharmacology. 1999;144:347–354. doi: 10.1007/s002130051017. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jones CK, Shannon HE. Muscarinic cholinergic modulation of prepulse inhibition of the acoustic startle reflex. J Pharmacol Exp Ther. 2000;294:1017–1023. [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M, Bubser M. Deficient sensorimotor gating after 6-hydroxydopamine lesion of the rat medial prefrontal cortex is reversed by haloperidol. Eur J Neurosci. 1994;6:1837–1845. doi: 10.1111/j.1460-9568.1994.tb00576.x. [DOI] [PubMed] [Google Scholar]
- Koo EH, Kopan R. Potential role of presenilin-regulated signaling pathways in sporadic neurodegeneration. Nat Med. 2004;10:S26–S33. doi: 10.1038/nm1065. [DOI] [PubMed] [Google Scholar]
- Kozlovsky N, Belmaker RH, Agam G. Low GSK-3 activity in frontal cortex of schizophrenic patients. Schizophr Res. 2001;52:101–105. doi: 10.1016/s0920-9964(00)00174-2. [DOI] [PubMed] [Google Scholar]
- Kozlovsky N, Shanon-Weickert C, Tomaskovic-Crook E, Kleinman JE, Belmaker RH, Agam G. Reduced GSK-3beta mRNA levels in postmortem dorsolateral prefrontal cortex of schizophrenic patients. J Neural Transm. 2004;111:1583–1592. doi: 10.1007/s00702-004-0166-3. [DOI] [PubMed] [Google Scholar]
- Li X, Rosborough KM, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacol. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120:2935–2943. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, Simmons A, Sigmundsson T, Greenwood K, Russell A, Schmitz N, Happe F, Howlin P, Murphy DG. Brain anatomy and sensorimotor gating in Asperger's syndrome. Brain. 2002;125:1594–1606. doi: 10.1093/brain/awf150. [DOI] [PubMed] [Google Scholar]
- McCaughran J, Jr, Mahjubi E, Decena E, Hitzemann R. Genetics, haloperidol-induced catalepsy and haloperidol-induced changes in acoustic startle and prepulse inhibition. Psychopharmacology. 1997;134:131–139. doi: 10.1007/s002130050434. [DOI] [PubMed] [Google Scholar]
- McCaughran J, Jr, Bell J, Hitzemann R. On the relationships of high-frequency hearing loss and cochlear pathology to the acoustic startle response (ASR) and prepulse inhibition of the ASR in the BXD recombinant inbred series. Behav Genet. 1999;29:21–30. doi: 10.1023/a:1021433705004. [DOI] [PubMed] [Google Scholar]
- McCright B, Brothman AR, Virshup DM. Assignment of human protein phosphatase 2A regulatory subunit genes b56alpha, b56beta, b56gamma, b56delta, and b56epsilon (PPP2R5A-PPP2R5E), highly expressed in muscle and brain, to chromosome regions 1q41, 11q12, 3p21, 6p21.1, and 7p11.2 → p12. Genomics. 1996;36:168–170. doi: 10.1006/geno.1996.0438. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Bunney BS. Acute effects of typical and atypical antipsychotic drugs on the release of dopamine from prefrontal cortex, nucleus accumbens, and striatum of the rat: an in vivo microdialysis study. J Neurochem. 1990;54:1755–1760. doi: 10.1111/j.1471-4159.1990.tb01230.x. [DOI] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, Berkovic SF. Channelopathies as a genetic cause of epilepsy. Curr Opin Neurol. 2003;16:171–176. doi: 10.1097/01.wco.0000063767.15877.c7. [DOI] [PubMed] [Google Scholar]
- Nakajo K, Kubo Y. Protein kinase C shifts the voltage dependence of KCNQ/M channels expressed in Xenopus oocytes. J Physiol. 2005;569:59–74. doi: 10.1113/jphysiol.2005.094995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowska K, Pietraszek M, Wardas J, Nowak G, Wolfarth S. Influence of long-lasting administration of neuroleptics on cortical NMDA receptors and phencyclidine-induced deficit in the sensorimotor gating in rats. Pol J Pharmacol. 1999;51:49–53. [PubMed] [Google Scholar]
- Ossowska K, Pietraszek M, Wardas J, Nowak G, Zajaczkowski W, Wolfarth S, Pilc A. The role of glutamate receptors in antipsychotic drug action. Amino Acids. 2000;19:87–94. doi: 10.1007/s007260070037. [DOI] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, Wilcox KS, White HS. Mice carrying the szt1 mutation exhibit increased seizure susceptibility and altered sensitivity to compounds acting at the M-channel. Epilepsia. 2004;45:1009–1016. doi: 10.1111/j.0013-9580.2004.65703.x. [DOI] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci. 2006;26:2053–2059. doi: 10.1523/JNEUROSCI.1575-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouagazzal AM, Jenck F, Moreau JL. Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: a model for detecting antipsychotic activity? Psychopharmacology. 2001;156:273–283. doi: 10.1007/s002130100763. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Perry W, Braff DL. Information-processing deficits and thought disorder in schizophrenia. Am J Psychiatry. 1994;151:363–367. doi: 10.1176/ajp.151.3.363. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor gating deficits in adults with autism. Biol Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Pirch JH, Turco K, Rucker HK. A role for acetylcholine in conditioning-related responses of rat frontal cortex neurons: microiontophoretic evidence. Brain Res. 1992;586:19–26. doi: 10.1016/0006-8993(92)91366-m. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, Daneels G, Bouwknecht JA, Steckler T. Transgenic mice overexpressing glycogen synthase kinase 3β: a putative model of hyperactivity and mania. J Neurosci. 2006;26:9022–9029. doi: 10.1523/JNEUROSCI.5216-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz JA, Gould TD, Manji HK. Molecular effects of lithium. Mol Interv. 2004;4:259–272. doi: 10.1124/mi.4.5.6. [DOI] [PubMed] [Google Scholar]
- Roh MS, Seo MS, Kim Y, Kim SH, Jeon WJ, Ahn YM, Kang UG, Juhnn YS, Kim YS. Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp Mol Med. 2007;39:353–360. doi: 10.1038/emm.2007.39. [DOI] [PubMed] [Google Scholar]
- Rowe MK, Wiest C, Chuang DM. GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev. 2007;31:920–931. doi: 10.1016/j.neubiorev.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Sorensen SM, Kehne JH, Carr AA, Palfreyman MG. The role of 5-HT2A receptors in antipsychotic activity. Life Sci. 1995;56:2209–2222. doi: 10.1016/0024-3205(95)00210-w. [DOI] [PubMed] [Google Scholar]
- Siegfried E, Perrimon N. Drosophila wingless: a paradigm for the function and mechanism of Wnt signaling. Bioessays. 1994;16:395–404. doi: 10.1002/bies.950160607. [DOI] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J. 1993;296:15–19. doi: 10.1042/bj2960015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Carruthers R, Rachleff I, Wattler S, Nehls M, McKinzie DL, Fienberg AA, Nomikos GG, Greengard P. Diverse psychotomimetics act through a common signaling pathway. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology. 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Kuczenski R, Bongiovanni MJ, Neary AC, Tochen LS, Saint Marie RL. Forebrain D1 function and sensorimotor gating in rats: effects of D1 blockade, frontal lesions and dopamine denervation. Neurosci Lett. 2006;402:40–45. doi: 10.1016/j.neulet.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Van den Buuse M, Garner B, Koch M. Neurodevelopmental animal models of schizophrenia: effects on prepulse inhibition. Curr Mol Med. 2003;3:459–471. doi: 10.2174/1566524033479627. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Swerdlow NR. Intra-accumbens infusion of quinpirole impairs sensorimotor gating of acoustic startle in rats. Psychopharmacology. 1993;113:103–109. doi: 10.1007/BF02244341. [DOI] [PubMed] [Google Scholar]
- Willott JF, Tanner L, O'Steen J, Johnson KR, Bogue MA, Gagnon L. Acoustic startle and prepulse inhibition in 40 inbred strains of mice. Behav Neurosci. 2003;117:716–727. doi: 10.1037/0735-7044.117.4.716. [DOI] [PubMed] [Google Scholar]
- Wolf FW, Eddison M, Lee S, Cho W, Heberlein U. GSK-3/Shaggy regulates olfactory habituation in Drosophila. Proc Natl Acad Sci U S A. 2007;104:4653–4657. doi: 10.1073/pnas.0700493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]