

Abstract
Growing evidence indicates that the activity of infralimbic prefrontal cortex (IL) is critical for inhibiting inappropriate fear responses following extinction learning. Recently, we showed that fear conditioning and extinction alter the intrinsic excitability and bursting of IL pyramidal neurons in brain slices. IL neurons from Sprague Dawley rats expressing high fear had lower intrinsic excitability and bursting than those from rats expressing low fear, suggesting that regulating the intrinsic excitability and bursting of IL neurons would modulate fear expression. To test this, we combined patch-clamp electrophysiology, auditory fear conditioning, and IL infusions of M-type K+ channel modulators. Patch-clamp recordings from IL neurons showed that the M-type K+ channel blocker, XE-991, increased the number of spikes evoked by a depolarizing pulse and reduced the first interspike interval indicating enhanced bursting. To test whether pharmacological enhancement of IL excitability and bursting reduces fear expression and facilitates extinction, fear-conditioned rats were infused with XE-991 into IL before extinction training. XE-infused rats showed reduced freezing and facilitated extinction compared to vehicle-infused rats. The following day, recall of extinction memory was enhanced. Reducing IL excitability and bursting with the M-type K+ channel agonist, flupirtine, had the opposite effect. Flupirtine reduced IL spike count and bursting in brain slices. Fear-conditioned rats infused with flupirtine into IL before extinction showed significantly higher levels of freezing, indicating that stimulation of M-channels enhanced fear expression. Our findings suggest that the intrinsic excitability and bursting of IL neurons regulate fear expression even before extinction.
Introduction
Deficits in the mechanisms underlying fear extinction are thought to be the basis of certain anxiety disorders, such as posttraumatic stress disorder (PTSD) (Rothbaum and Davis, 2003; Milad et al., 2006). Inappropriate regulation of fear responses is proposed to be due in part to a hypoactive medial prefrontal cortex (mPFC) in PTSD patients (Shin et al., 2004; Milad et al., 2007). Studies done in rodents have strongly implicated the infralimbic subregion of the mPFC (IL) in inhibiting conditioned fear responses following extinction (Quirk et al., 2006). Disruption of IL function by using electrolytic lesions (Quirk et al., 2000; Lebrón et al., 2004) or pharmacological inactivation (Sierra-Mercado et al., 2006) before extinction training impaired subsequent recall of extinction memory. Intra-IL blockade of NMDA receptors (Burgos-Robles et al., 2007), protein kinases (Hugues et al., 2006), and protein synthesis (Santini et al., 2004; Mueller et al., 2008) also prevented extinction recall. Furthermore, potentiation of IL activity following fear extinction has been correlated with good recall of extinction (Herry and Garcia, 2002; Milad and Quirk, 2002; Barrett et al., 2003). Together, these findings suggest that extinction-induced plasticity in IL is required for recalling extinction memory.
Recently, we showed that fear conditioning reduced the intrinsic excitability of IL pyramidal neurons in brain slices and their ability to fire bursts of action potentials (Santini et al., 2008). In the same study, fear extinction increased IL excitability back to preconditioning levels and enhanced the burst firing of IL neurons. Furthermore, the first interspike interval in IL neurons was negatively correlated with fear behavior 24 h after extinction training. Together, these results suggest that the intrinsic excitability of IL neurons and their ability to fire bursts of action potentials controls fear expression. In line with this, burst firing in IL neurons in vivo shortly after extinction training correlates with good recall of fear extinction memory the next day (Burgos-Robles et al., 2007). Although these studies implicate IL excitability changes in modulating fear extinction, it remains to be determined whether changes in IL intrinsic excitability per se are sufficient to regulate fear expression and extinction. Thus, we hypothesize that pharmacological agents that directly enhance the intrinsic excitability and bursting of IL neurons will decrease fear expression and facilitate extinction.
In other brain structures, the M-type K+ channels modulate neuronal excitability and bursting (Aiken et al., 1995; Zaczek et al., 1998; Yue and Yaari, 2004; Vervaeke et al., 2006; Yoshida and Alonso, 2007). Since M-type K+ channels are active in the voltage range for action potential initiation, they regulate the dynamics of the neuronal firing (Rogawski, 2000; Hu et al., 2002; Peters et al., 2005). In hippocampal pyramidal cells, M-type K+ channels contribute to the afterhyperpolarization that occurs after single action potential and can, thereby, modulate the ability of neurons to fire bursts (Brown and Passmore, 2009). Therefore, to directly test whether IL excitability and bursting determines fear expression, we manipulated intrinsic excitability and bursting of IL neurons by blocking or enhancing the M-type K+ channels before extinction training.
Materials and Methods
Subjects.
The procedures were approved by the Institutional Animal Care and Use Committee of the Ponce School of Medicine in compliance with National Institutes of Health guidelines for the care and use of laboratory animals. Male Sprague Dawley rats (25–30 d postnatal) were transported from the Ponce School of Medicine colony to a satellite facility nearby where they were housed in transparent polyethylene cages inside a negative-pressure Biobubble (Colorado Clean Room). Rats were maintained on a 12/12 h light/dark schedule with ad libitum access to food (standard laboratory rat chow) and water.
Slice preparation.
Naive rats were deeply anesthetized with pentobarbital (150 mg/kg), and were perfused through the heart with ice-cold high sucrose solution: 252 mm sucrose, 2 mm KCl, 1.25 mm NaH2PO4, 3 mm MgSO4, 26 mm NaHCO3, 20 mm glucose, and 1 mm CaCl2. Brains were quickly removed and placed in ice-cold artificial CSF (ACSF) containing 126 mm NaCl, 3 mm KCl, 1.25 mm NaH2PO4, 1 mm MgSO4, 26 mm NaHCO3, 20 mm glucose, and 2 mm CaCl2 and bubbled with 95% O2 and 5% CO2. Coronal slices of the mPFC were cut at a thickness of 300 μm with a Vibratome 1000 Plus (Vibratome). Slices were incubated at room temperature in ACSF for at least 1 h before experiments. The NMDA receptor blocker MK-801 (10 μm) was added during the incubation of slices to increase neuronal survival (Schurr et al., 1995).
Slices were transferred to a submersion recording chamber mounted and perfused at 2–3 ml/min with room temperature ACSF. Neurons were visualized with infrared video microscopy using a 40× water-immersion objective on an upright E600FN microscope (Nikon Instruments). Whole-cell recordings were done with glass pipettes with a resistance of 3–5 MΩ when filled with an internal solution containing KCl (20), K-gluconate (115), HEPES (10), sodium phosphocreatine (10), biocytin (10), ATP (2), and GTP (3); pH was adjusted to 7.3 with KOH (290 mOsm).
Current-clamp recordings.
Whole-cell current-clamp recordings were obtained from the soma of mPFC pyramidal neurons located in layers II/III and V of IL. Cells were held in current-clamp mode at −60 mV and action potential discharges in response to the injection of depolarizing current pulses were recorded with a patch-clamp amplifier (MultiClamp 700A, Molecular Devices). Recordings were filtered at 4 kHz, digitized at 10 kHz, and saved to computer using pCLAMP9 (Molecular Devices). Membrane potentials were not corrected for the junction potential of 9 mV. The input resistance was measured from a 5 mV, 50 ms depolarizing pulse in voltage-clamp mode.
Morphological analysis.
Biocytin (5 mm) was included in the recording solution to label the neurons for post hoc morphological identification of IL pyramidal neurons. At the end of the electrophysiological recordings, the slices were fixed overnight in 4% paraformaldehyde. Neurons were subsequently visualized with a standard avidin–biotin peroxidase procedure (Vectastain ABC kit, Vector Laboratories) as previously described (Porter et al., 2001) and visualized with brightfield microscopy.
Behavioral apparatus.
Rats were fear conditioned, extinguished, and tested in a chamber of 25 × 29 × 28 cm with aluminum and Plexiglas walls (Coulbourn Instruments). The floor consisted of stainless-steel bars that could be electrified to deliver a mild shock. A speaker was mounted on the outside wall, and illumination was provided by a single overhead light. The chamber was situated inside a sound-attenuating box (Med Associates) with a ventilating fan, which produced an ambient noise level of 60 dB. The conditioned stimulus (CS) was a 4 kHz tone with duration of 30 s and an intensity of 80 dB. The unconditioned stimulus (US) was a 0.4 mA scrambled footshock, 0.5 s in duration, which coterminated with the tone during the conditioning phase. Between sessions, floor trays and shock bars were cleaned with soapy water and the chamber walls were wiped with a damp cloth. Behavior was recorded with digital video cameras (Micro Video Products).
Surgery.
Rats were anesthetized with ketamine and xylazine (10 ml/100 g) and placed in the stereotaxic apparatus. After anesthesia, the skin was retracted and holes were drilled in the skull. They were implanted with a single 26 gauge stainless-steel guide cannula (Plastics One) in the mPFC as described previously (Santini et al., 2004). Stereotaxic coordinates aiming toward the infralimbic cortex were 2.8 mm anterior, 1.0 mm lateral, and 4.1 mm ventral from bregma and toward the prelimbic cortex were 2.5 mm anterior, 0.5 mm lateral, and 3.2 mm ventral from bregma (Paxinos and Watson, 1986), with the cannula angled 11° toward the midline in the coronal plane. Rats were allowed 7 d to recover from surgery.
Drugs and infusion procedure.
Ten minutes before extinction training, 10,10-bis(4-pyridinylmethyl)-9(10H)anthracenone dihydrochloride (XE-991, 10 μm, Ascent Scientific) or 2-amino-6-[[(4-fluorophenyl)methyl] amino]-3-pyridinyl-carbamic acid ethyl ester maleate salt (flupirtine maleate, 30 μm, Sigma) was infused into the medial prefrontal cortex. XE-991 was dissolved in artificial CSF (ACSF-vehicle), and flupirtine was dissolved in 0.3% dimethylsulfoxide (DMSO-vehicle). For the infusions, cannula dummies were removed from guide cannulas and replaced with 33 gauge injectors, which were connected by polyethylene tubing (PE-20; Small Parts) to 5 μl syringes mounted in an infusion pump (Harvard Apparatus). Drugs were infused at a rate of 0.5 μl/min for 1 min.
Behavioral procedure.
On day 1, rats received 3 tone–shock pairings (conditioning phase). After matching for equivalent levels of freezing-conditioned rats were divided into the vehicle group (Veh) and the drug group (XE or Flu). On day 2, rats were infused with vehicle or drug followed by 12 tone-alone trials (extinction phase). On days 3–5, rats received two tone-alone trials in the same chamber to test for recall of extinction (test phase).
Open field.
Rats were tested for locomotion and anxiety in an open field (arena: 91.4 × 91.4 × 61 cm). To quantify locomotion, the total distance that the rats moved in the arena was calculated. To determine whether the drugs had any effect on general anxiety levels, the amount of time that the rats spent in the center (61 × 61 cm) of the arena was calculated. Both parameters were analyzed using an automated tracking software (EthoVision XT, Noldus Information Technology) by an observer blinded with respect to experimental group. Rats were given infusions of vehicle (n = 12), XE-991 (10 μm, n = 6), or flupirtine (30 μm, n = 6) 10 min before a 5 min session in the arena.
Statistical analysis.
The percentage of time spent freezing (Blanchard and Blanchard, 1972) was used as a measure of conditioned fear. Freezing is the cessation of all movements except respiration. The total time spent freezing during the 30 s tone was scored from videotape with a digital stopwatch by observers blinded with respect to experimental group. The electrophysiological data were analyzed using Clampfit (Molecular Devices). Student's t test or one-way ANOVA (STATISTICA, Statsoft) were used to analyze the behavioral and electrophysiological data. Following a significant main effect, post hoc tests were performed with Tukey HSD tests. Values are reported as the mean ± SEM.
Results
Blockade of M-type K+ channels increased the number of evoked spikes and burst firing in IL pyramidal neurons
Before testing whether the intrinsic excitability of IL neurons is a determinant of fear expression, we first assessed the effects of the M-type K+ channel blocker XE-991 on the membrane excitability of IL pyramidal neurons. At a concentration of 10 μm, XE-991 selectively blocks M-type K+ channels (Wang et al., 1998). To measure neuronal intrinsic excitability, we examined two parameters: number of evoked action potentials and the first interspike interval (ISI). Coronal slices of the mPFC containing IL were prepared and the intrinsic excitability of IL neurons was assessed with whole-cell patch-clamp recordings. To determine the effects of XE-991 on the relative excitability of IL neurons, we measured the number of action potentials elicited by a depolarizing current pulse. To standardize baseline firing across cells, the intensity of the current pulse was adjusted for each cell to evoke between three to four spikes. Bath application of 10 μm XE-991 caused a significant and persistent increase in the number of spikes evoked by a current pulse (Fig. 1A–C). One-way ANOVA revealed a significant main effect of drug (F(2,33) = 6.9153, p < 0.01). Post hoc comparisons indicated that XE-991 increased the number of evoked action potentials (XE = 153 ± 14% of baseline; p < 0.01). Figure 1D shows the distribution of spike count before and after XE-991. The effect of XE-991 did not reverse after 12 min of washout (washout = 143 ± 11% of baseline; p = 0.02) consistent with previously reported slow reversal of XE-991's effects after 30–40 min (Yue and Yaari, 2004).
Figure 1.

Blockade of M-type K+ channels with XE-991 increased the intrinsic excitability of IL pyramidal neurons. A, Trace examples showing the number of spikes evoked by a current pulse during baseline, perfusion with the M-type K+ channel blocker (XE-991, 10 μm), and washout. B, C, Time course and bar graph demonstrating that perfusion of XE-991 persistently increased the number of evoked spikes. D, Distribution of spike counts before and after bath applying XE-991. E, F, Example traces and bar graph showing that XE-991 decreased the ISIs independent of the number of evoked action potentials. (**p < 0.01; *p < 0.05).
To determine whether blockade of M-type K+ channels facilitated bursting firing in IL neurons, we assessed the effects of XE-991 on the first ISI independent of the number of evoked action potentials. We injected current at different intensities ranging from −20 pA to 340 pA before and after bath application of XE-991. Since the number of spikes can affect the ISI, the first ISI was compared in the traces that had the same number of evoked spikes (four spikes). Figure 1E shows examples of traces in which a depolarizing current step of 162 pA and 150 pA evoked four spikes before and after bath application of XE-991, respectively. In the presence of XE-991, the neuron discharged spikes with a shorter first ISI. Figure 1F shows that XE-991 significantly reduced the first ISI (XE: 70 ± 12% of baseline; t = 2.83, df = 7, p = 0.02) measured at traces showing four evoked spikes indicating that blockade of M-type K+ channels enhanced bursting in IL neurons.
XE-991 also depolarized the neurons (an average increase of 5 mV from the resting membrane potential; t = 6.39, df = 5, p < 0.01) and increased the input resistance (136.9% of baseline; t = 17.5, df = 5, p < 0.001) consistent with the closure of M-type K+ channels. In some neurons, M-type K+ channels contribute to the afterhyperpolarization following individual action potentials and to the medium afterhyperpolarization seen after a burst of action potentials (Gu et al., 2005). In IL neurons (Fig. 2A), XE-991 did not affect the afterhyperpolarizing potential following a burst of action potentials evoked by an 800 ms current pulse (mAHP; 121% of baseline; t = 1.6, df = 5, p = 0.16). In contrast, XE-991 reduced the afterhyperpolarizing potential following the first action potential (fAHP; 79% of baseline; t = 35, df = 5, p < 0.001) (Fig. 2B) consistent with the reduced first ISI. There was no significant difference in the fAHP measured after the second (105.8% of baseline; t = 0.62, df = 5, p > 0.05) or third (103% of baseline; t = 0.26, df = 5, p > 0.05) action potential. Together these findings indicate that blockade of M-type K+ channels enhances the intrinsic excitability and burst firing of IL pyramidal neurons. Therefore, blocking M-type K+ channels will allow us to enhance IL intrinsic excitability and burst firing to determine whether IL intrinsic excitability and burst firing modulates the expression of conditioned fear and fear extinction.
Figure 2.
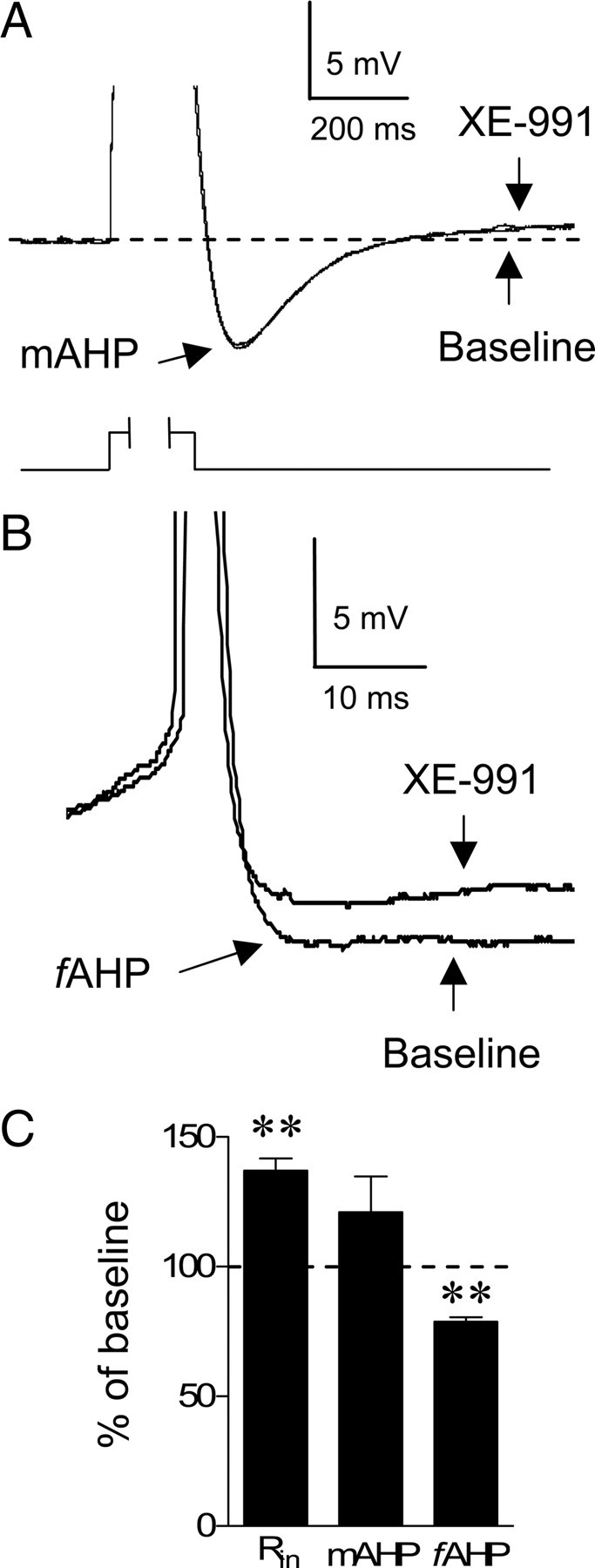
XE-991 reduced the afterhyperpolarizing potential following a single action potential but not a burst of action potentials. A, Superimposed average traces of the afterhyperpolarizing potential generated by a burst of action potentials (mAHP) evoked with an 800 ms current pulse before and after applying XE-991 showing that XE-991 had no effect on the mAHP. Responses to depolarization were truncated to emphasize the mAHP. B, Superimposed traces of the fAHP before and after applying XE-991. Action potentials were truncated for clarity. C, Bar graph showing the average effect of XE-991 on the input resistance (Rin), mAHP, and the fAHP. **p < 0.001.
Blockade of M-type K+ channels reduced fear expression and facilitated fear extinction
To determine whether IL excitability regulates fear expression, rats implanted with cannulas aimed toward the mPFC were fear conditioned to a tone (Fig. 3A). Following fear conditioning on day 1 (Fig. 3B), rats were matched and divided into two groups based on their levels of conditioned freezing during the last block of conditioning trials (vehicle: 66% ± 6.4, XE-991: 64% ± 7.0; t = 0.27, df = 21, p = 0.78). On day 2, rats received intra-IL infusions of either vehicle solution (n = 11) or XE-991 (10 μm, n = 12) 10 min before extinction training. XE-infused rats showed lower levels of conditioned freezing compared to vehicle-infused rats during the first extinction trial block, indicating that XE-991 reduced fear expression per se independent of facilitating extinction training. Repeated-measures ANOVA across the extinction phase on day 2 revealed a main effect of drug (F(1,21) = 6.26, p < 0.05). Post hoc comparisons indicated that XE-infused rats froze significantly less than vehicle-infused rats during the first four trial blocks (p < 0.05). On day 3, XE-infused rats still showed lower levels of conditioned freezing compared to vehicle-infused rats (t = 3.34, df = 21, p = 0.003). No significant difference was observed between the groups on day 5 (p > 0.05).
Figure 3.

Infusions of XE-991 into IL before, but not after, extinction training reduced fear expression and facilitated extinction. A, Diagram showing the location of the cannula tips in IL for the vehicle (white circles) and XE-991 (black circles) groups for the preextinction infusion experiment. B, Percentage freezing to the tone for vehicle-infused rats (n = 12) and rats infused with XE-991(10 μm, n = 12) into IL before extinction training. XE-infused rats showed reduced fear expression on days 2–4. C, Diagram showing the location of the cannula tips in IL for the vehicle (white circles) and XE-991 (black circles) groups for the postextinction infusion experiment. D, Percentage freezing to the tone for vehicle-infused rats (n = 8) and rats infused with XE-991 (10 μm, n = 12) into IL immediately after extinction training (day 2). No difference was observed between vehicle-treated and XE-treated rats in the levels of conditioned freezing on day 3. Arrows indicate the time of the infusion. (*p < 0.01).
Rats infused with XE-991 into IL showed decreased fear expression 24 h later. This could be explained by a fear expression deficit that facilitated recall of extinction the following day. Another plausible explanation is that blockade of M-type K+ channels in IL prevented consolidation of long-term extinction memory. To test this possibility, fear-conditioned rats were infused with vehicle (n = 8) or XE-991 (n = 12) into IL immediately after extinction training on day 2 (Fig. 3C). As shown in Figure 3D, rats infused with XE-991 after extinction showed similar levels of freezing to vehicle-treated rats on day 3 (vehicle: 40 ± 15%, XE-991: 20 ± 8%; t = 1.26, df = 18, p = 0.22), indicating that posttraining blockade of M-type K+ channels did not affect consolidation of fear extinction. Together, these findings suggest that enhancing IL excitability facilitates extinction recall by reducing fear expression during extinction training rather than by enhancing consolidation of extinction memory.
Since rats infused with XE-991 showed decreased freezing expression immediately after the infusion, it is possible that this effect was due to a drug-induced increase in locomotion independent of memory formation. To rule out this possibility, rats received intra-IL infusions of vehicle (n = 6) or XE-991 (n = 6) and were allowed to explore an open field. Blockade of M-type K+ channels had no effect on locomotor behavior, since there was no difference in the total distance that the rats moved across the open field (vehicle: 7968 ± 646 cm; XE: 7806 ± 433 cm; t = 0.21, df = 10, p = 0.84). In addition, rats infused with XE-991 spent similar amount of time in the center of the open field (vehicle: 3.4 ± 0.56 s; XE: 3.0 ± 0.54 s; t = 0.45, df = 10, p = 0.66). Together these results indicate that reduced freezing after blocking M-type K+ channels in IL is not attributable to changes in locomotion or general anxiety levels.
Stimulation of M-type K+ channels reduced the number of evoked spikes and burst firing in IL pyramidal neurons
The results described above show that blockade of M-type K+ channels in IL resulted in reduced conditioned freezing and facilitation of extinction. In the following experiments, we examined whether activation of M-type K+ channels would produce the opposite results, i.e., enhance fear expression and impair extinction. To test this, we used the M-type K+ channel agonist flupirtine to decrease neuronal excitability. Figure 4A–C show that bath application of flupirtine (30 μm) reduced the number of evoked action potentials in IL neurons. One-way ANOVA revealed a significant main effect of drug (F(2,21) = 35.92, p < 0.001), and post hoc comparisons indicated that flupirtine reduced the number of evoked action potentials (16 ± 3% of baseline; p < 0.001). This effect did not reverse during washout recordings (washout = 49 ± 12% of baseline; p = 0.01). Flupirtine also significantly increased the first interspike interval indicating that stimulation of M-type K+ channels reduced bursting firing in IL (Fig. 4D) (251 ± 63% of baseline; t = 2.79, df = 5, p = 0.03). Flupirtine also reduced the input resistance (87% of baseline; t = 8.2, df = 5, p = 0.001) consistent with opening of M-type K+ channels.
Figure 4.

Stimulation of M-type K+ channels with flupirtine reduced the intrinsic excitability of IL pyramidal neurons. A, Traces showing the number of spikes evoked by a current pulse during baseline, perfusion with the M-type K+ channel agonist (flupirtine, 30 μm) and washout. B, C, Time course and bar graph showing that flupirtine application decreased the number of evoked spikes in IL pyramidal neurons (n = 6). D, Flupirtine also increased the interspike intervals, consistent with reduced bursting in IL. (**p < 0.001).
Stimulation of M-type K+ channels in IL enhanced fear expression and delayed extinction
To determine whether reducing IL intrinsic excitability and bursting affects fear expression and extinction, we repeated the behavioral experiments and infused flupirtine (30 μm) into IL on day 2 before extinction training (Fig. 5A,B). Both groups of rats showed similar levels of conditioned freezing on day 1 (vehicle: 51% ± 4.2, Flu: 51% ± 5.1; p = 0.19). However, on day 2 Flu-infused rats showed significantly higher levels of conditioned freezing compared to vehicle rats, indicating that reducing IL excitability facilitated the expression of conditioned fear. Repeated-measures ANOVA across the extinction phase on day 2 revealed a main effect of group (F(1,30) = 5.9, p = 0.02) and a significant trial by group interaction (F(5,150) = 2.8, p < 0.05). Post hoc comparisons indicated Flu-infused rats froze significantly more than vehicle-infused rats during the first four trial blocks (p < 0.05). Although, no significant difference was observed between the groups on day 3, flupirtine significantly enhanced fear expression on days 4 and 5 (p < 0.05). Together these findings indicate that reducing the intrinsic excitability and bursting of IL neurons enhances the expression of conditioned fear.
Figure 5.

Infusions of flupirtine into IL before extinction training enhanced fear expression. A, Diagram showing the location of the cannula tips in IL for the vehicle (white circles) and flupirtine (black circles) groups. B, Freezing to the tone for vehicle-infused rats (n = 16) and rats infused with flupirtine (30 μm, n = 16). Flupirtine enhanced fear expression on days 2, 4, and 5. The arrow indicates the time of the infusion. *p < 0.05.
To test whether the effects of flupirtine were due to reduced locomotion or enhanced general anxiety independent of the conditioned fear memory, rats were infused into IL with vehicle (n = 6) or flupirtine (n = 6) and allowed to explore an open field for 5 min. There was no significant difference between the groups in terms of total distance moved (vehicle: 6750 ± 282 cm, Flu: 6293 ± 1204 cm; t = 0.37, df = 10, p = 0.72) or the amount of time that the rats spent in the center of the field (vehicle: 4.4 ± 1.3 s, Flu: 4.4 ± 1.4 s; t = 0.01, df = 10, p = 0.99). Therefore, flupirtine did not enhance fear expression by decreasing locomotion or anxiety levels.
Stimulation of M-type K+ channels in PL reduced fear expression, but had no effect on fear extinction
Since the inhibition of IL neuronal activity with tetrodotoxin (Sierra-Mercado et al., 2006) or the GABAA agonist muscimol (Laurent and Westbrook, 2009) did not increase fear expression, we were surprised to find that reducing IL neuronal excitability with flupirtine enhanced fear expression. Studies have shown that, in contrast to IL, the prelimbic subregion of the mPFC (PL) facilitates fear expression (Vidal-Gonzalez et al., 2006; Corcoran and Quirk, 2007; Burgos-Robles et al., 2009). Therefore, to rule out the possibility that flupirtine increased fear expression by affecting PL, we examined the effects of flupirtine on fear conditioning and extinction in an additional group of rats implanted with cannulas aimed toward PL (Fig. 6A). Rats were fear conditioned and, 24 h later, flupirtine (30 μm) was infused into PL before extinction training. As shown in Figure 6B, Flu-infused rats showed lower levels of conditioned freezing compared to vehicle-infused animals during the first two trials of the extinction session (vehicle: 80% ± 6.2, n = 6; Flu: 49% ± 10.6, n = 6; p < 0.05). However, both groups extinguished to the same levels by the end of the extinction phase. In addition, no difference between the groups was observed during recall of extinction the following day (vehicle: 57 ± 16%, Flu: 49 ± 17%; p = 0.13). Therefore, decreasing PL intrinsic excitability resulted in reduced fear expression but had no effect on extinction learning or recall.
Figure 6.

Infusions of flupirtine into the PL before extinction training reduced fear expression. A, Diagram showing the location of the cannula tips in PL for the vehicle (white circles) and flupirtine (black circles) groups. B, Freezing to the tone for vehicle-infused rats (n = 6) and rats infused with flupirtine (30 μm, n = 6). Flupirtine-infused rats showed reduced fear expression on day 2. No significant difference between the groups was observed on days 3–5. The arrow indicates the time of the infusion. *p < 0.05.
Discussion
Our results show that direct pharmacological manipulation of the intrinsic excitability and bursting of IL neurons modulates the expression of conditioned fear. Enhancing the excitability and bursting of IL neurons by blocking M-type K+ channels reduced fear expression and facilitated extinction recall. In contrast, stimulation of M-type K+ channels on IL neurons reduced neuronal excitability and bursting and enhanced the expression of conditioned fear. Thus, fear expression can be bidirectionally modified by manipulating the intrinsic excitability of IL neurons.
How can M-type K+ channels modulate fear expression? Consistent with results from the hippocampus (Yue and Yaari, 2004; Gu et al., 2005), blocking M-type K+ channels increased the number of evoked spikes in response to a depolarizing current pulse and shortened the first ISI indicating an enhanced ability to fire bursts of action potentials. We recently showed that neurons from fear extinguished animals, showing low fear expression at test, fired spikes with shorter first ISIs in brain slices (Santini et al., 2008). In line with this, IL bursting in vivo correlates with low fear expression (Burgos-Robles et al., 2007). Therefore, XE-991 may have decreased fear expression by increasing IL bursting during extinction. In addition, blocking M-type K+ channels depolarized the neurons, thereby bringing them closer to threshold, and increased the input resistance, which would enhance depolarization and summation of synaptic inputs. Since blocking M-channels in slices (Lampe et al., 1997; Fontán-Lozano et al., 2009) and in vivo (Fontán-Lozano et al., 2009; Song et al., 2009) lowers the threshold for inducing long-term potentiation, the enhanced IL excitability may have facilitated synaptic plasticity to increase the long-term stabilization of the extinction memory. Although the lack of effect of postextinction infusions of XE-991 on extinction recall suggests that XE-991 did not affect consolidation of fear extinction, the XE-mediated increase in IL excitability may need to occur during presentations of the tone to facilitate recall of extinction the following day. Consistent with this, previous studies have shown that brief electrical stimulation in IL during extinction facilitates extinction recall 24 h later only when the stimulation is given 0.1 s after tone onset (Milad and Quirk, 2002; Milad et al., 2004). Therefore, increasing IL excitability might facilitate long-term extinction memory only if the increase in excitability occurs during extinction training.
We observed that intra-IL blockade of M-type K+ channels with XE-991 reduced conditioned freezing even to the first tone-alone trial, before extinction learning. Since XE-991 increased the number of evoked spikes in response to a depolarizing current pulse and shortened the first ISI in IL neurons in slices, the infusion of XE-991 presumably increased the firing of IL neurons in response to synaptic inputs and enhanced bursting. Similarly, enhancing IL activity via electrical stimulation also reduced fear expression before extinction learning (Milad et al., 2004). Pharmacological or electrical stimulation of IL activity may reduce fear expression by activating glutamatergic projections from IL to GABAergic intercalated cells (Vertes, 2004; Gabbott et al., 2005; Likhtik et al., 2008), resulting in decreased activity in the central nucleus of the amygdala that controls fear expression (Quirk et al., 2003; Likhtik et al., 2005).
Enhancing M-type K+ channel activity with localized infusions of flupirtine into IL increased conditioned freezing to the first extinction tone. Since fear conditioning does not enhance the activity of IL neurons (Milad and Quirk, 2002), our results suggest that basal IL activity modulates conditioned fear. Consistent with this idea, we previously observed that after fear conditioning when fear expression is increased the intrinsic excitability and bursting of IL neurons is reduced (Santini et al., 2008). Although numerous studies indicate that IL plays a critical role in extinction recall (Quirk et al., 2006), our data suggest that IL activity is important for modulating conditioned fear even before fear extinction. In contrast to our results, infusion of tetrodotoxin (Sierra-Mercado et al., 2006) or muscimol (Laurent and Westbrook, 2009) into IL before fear extinction did not enhance fear expression. One possible explanation for this discrepancy that the concentrations of tetrodotoxin (63 μm) and muscimol (220 μm) infused into IL reached PL at concentrations sufficient to inhibit PL activity, which has been shown to be important for fear expression (Laurent and Westbrook, 2009). In brain slices, tetrodotoxin inhibits sodium channels at a concentration of 1 μm (Stuart et al., 1997) and 10 μm muscimol silences firing (Gribkoff et al., 2003). Therefore it is possible that, even after being diluted by diffusion away from the site of infusion, the tetrodotoxin and muscimol may have reached PL at a concentration sufficient to inhibit PL activity. We infused 30 μm flupirtine, which causes a submaximal activation of M-type K+ currents in brain slices (Wladyka and Kunze, 2006). Therefore, flupirtine would more quickly reach ineffective concentrations as it diffused from the infusion site, thereby restricting the region in which mPFC neurons were inhibited.
Recent work has shown that, in contrast to IL, PL facilitates fear expression (Vidal-Gonzalez et al., 2006; Corcoran and Quirk, 2007; Laurent and Westbrook, 2009). In further support of this, we observed that rats infused with flupirtine into PL showed lower levels of conditioned fear. This effect is likely due to the activation of M-type K+ channels in PL, which would hyperpolarize the cells and thereby reduce their excitability and prevent the sustained neuronal responses to the tone (Burgos-Robles et al., 2009). As flupirtine infused into PL and IL enhanced and reduced fear expression, respectively, these data further support the view that IL and PL play opposite roles during the expression of conditioned fear responses (Vidal-Gonzalez et al., 2006; Corcoran and Quirk, 2007). Inhibiting PL neuronal excitability with flupirtine parallels the findings of a recent study showing that inactivation of PL with muscimol reduced the expression of fear responses but had no effects on long-term fear extinction (Laurent and Westbrook, 2009).
The fact that pharmacological modulation of M-type K+ channels altered fear extinction does not mean that fear extinction induces changes in the expression of the M-type K+ channels. Our finding that extinction recall was enhanced 24 h later when the XE-991 should be no longer present suggests that a prolonged reduction in M-type K+ currents is not needed to enhance recall of extinction. Therefore, we propose that a temporary reduction in M-type K+ channel activity during extinction training is sufficient to facilitate extinction recall.
Since muscarinic receptors in other structures inhibit M-type K+ channels (Brown and Passmore, 2009) and acetylcholine modulates various types of learning (Power et al., 2003a,b; Boccia et al., 2009), during fear extinction cholinergic projections to IL may release acetylcholine to activate muscarinic receptors causing inhibition of M-type K+ channels. It is also possible that endocannabinoids produced in IL during extinction (Lin et al., 2008, 2009) may inhibit M-type K+ channels (Schweitzer, 2000; Kano et al., 2009). The inhibition of M-type K+ channels depolarizes the neurons and increases their membrane resistance to increase the probability that the IL neurons will fire spikes during the tone. Fewer active M-type K+ channels will also allow the neurons to fire bursts of action potentials, which will strengthen the activation of downstream targets such as the inhibitory neurons in the amygdala to reduce fear expression (Vertes, 2004; Gabbott et al., 2005; Likhtik et al., 2008). The enhanced IL bursting may also facilitate the potentiation of synaptic inputs necessary to suppress fear by enhancing calcium spikes in the IL dendrites (Schiller et al., 1998; Schiller and Schiller, 2001; Burgos-Robles et al., 2007).
Over the last decade, the interest in understanding the neurobiology of fear extinction has grown (Myers and Davis, 2002). It has been suggested that PTSD patients are deficient in their ability to form strong extinction memories (Charney and Deutch, 1996; Bechara et al., 1999; Rothbaum and Davis, 2003; Milad et al., 2006, 2008, 2009). Even though extinction-based exposure therapy is being used to treat PTSD patients, relapse accompanied by exaggerated fear responses is commonly observed (Pitman et al., 2001; van Minnen and Hagenaars, 2002; Davis et al., 2006; Rauch et al., 2006). M-type K+ channel antagonists might be used in combination with exposure therapy to reduce the inappropriate expression of fear responses observed in PTSD patients.
Footnotes
This work was supported by National Institutes of Health Grant S06-GM08239 and National Science Foundation Grant IOS 0842159 to J.T.P., and Research Center in Minority Institutions Grant 5G12RR003050. We thank Joel Nigaglioni and Ana V. López for assistance with the behavioral studies, and Drs. Gregory J. Quirk and Mohammed R. Milad for comments on the manuscript.
References
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett D, Shumake J, Jones D, Gonzalez-Lima F. Metabolic mapping of mouse brain activity after extinction of a conditioned emotional response. J Neurosci. 2003;23:5740–5749. doi: 10.1523/JNEUROSCI.23-13-05740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR, Lee GP. Different contributions of the human amygdala and ventromedial prefrontal cortex to decision-making. J Neurosci. 1999;19:5473–5481. doi: 10.1523/JNEUROSCI.19-13-05473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard DC, Blanchard RJ. Innate and conditioned reactions to threat in rats with amygdaloid lesions. J Comp Physiol Psychol. 1972;81:281–290. doi: 10.1037/h0033521. [DOI] [PubMed] [Google Scholar]
- Boccia MM, Blake MG, Baratti CM, McGaugh JL. Involvement of the basolateral amygdala in muscarinic cholinergic modulation of extinction memory consolidation. Neurobiol Learn Mem. 2009;91:93–97. doi: 10.1016/j.nlm.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J Neurosci. 2009;29:8474–8482. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney DS, Deutch A. A functional neuroanatomy of anxiety and fear: implications for the pathophysiology and treatment of anxiety disorders. Crit Rev Neurobiol. 1996;10:419–446. doi: 10.1615/critrevneurobiol.v10.i3-4.70. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Quirk GJ. Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci. 2007;27:840–844. doi: 10.1523/JNEUROSCI.5327-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Myers KM, Chhatwal J, Ressler KJ. Pharmacological treatments that facilitate extinction of fear: relevance to psychotherapy. NeuroRx. 2006;3:82–96. doi: 10.1016/j.nurx.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontán-Lozano A, Suárez-Pereira I, Delgado-García JM, Carrión AM. The M-current inhibitor XE991 decreases the stimulation threshold for long-term synaptic plasticity in healthy mice and in models of cognitive disease. Hippocampus. 2009 doi: 10.1002/hipo.20717. Advance online publication. Retrieved December 6, 2009. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Pieschl RL, Dudek FE. GABA receptor-mediated inhibition of neuronal activity in rat SCN in vitro: pharmacology and influence of circadian phase. J Neurophysiol. 2003;90:1438–1448. doi: 10.1152/jn.01082.2002. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol. 2002;545:783–805. doi: 10.1113/jphysiol.2002.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugues S, Chessel A, Lena I, Marsault R, Garcia R. Prefrontal infusion of PD098059 immediately after fear extinction training blocks extinction-associated prefrontal synaptic plasticity and decreases prefrontal ERK2 phosphorylation. Synapse. 2006;60:280–287. doi: 10.1002/syn.20291. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Lampe BJ, Gaskill JL, Keim SC, Brown BS. Linopirdine reduces stimulus intensity threshold for induction of long-term potentiation in the Schaffer collateral/CA1 pathway in rat hippocampal slices. Neurosci Lett. 1997;222:135–137. doi: 10.1016/s0304-3940(97)13347-x. [DOI] [PubMed] [Google Scholar]
- Laurent V, Westbrook RF. Inactivation of the infralimbic but not the prelimbic cortex impairs consolidation and retrieval of fear extinction. Learn Mem. 2009;16:520–529. doi: 10.1101/lm.1474609. [DOI] [PubMed] [Google Scholar]
- Lebrón K, Milad MR, Quirk GJ. Delayed recall of fear extinction in rats with lesions of ventral medial prefrontal cortex. Learn Mem. 2004;11:544–548. doi: 10.1101/lm.78604. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Popa D, Apergis-Schoute J, Fidacaro GA, Paré D. Amygdala intercalated neurons are required for expression of fear extinction. Nature. 2008;454:642–645. doi: 10.1038/nature07167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Mao SC, Chen PS, Gean PW. Chronic cannabinoid administration in vivo compromises extinction of fear memory. Learn Mem. 2008;15:876–884. doi: 10.1101/lm.1081908. [DOI] [PubMed] [Google Scholar]
- Lin HC, Mao SC, Su CL, Gean PW. The role of prefrontal cortex CB1 receptors in the modulation of fear memory. Cereb Cortex. 2009;19:165–175. doi: 10.1093/cercor/bhn075. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Milad MR, Vidal-Gonzalez I, Quirk GJ. Electrical stimulation of medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci. 2004;118:389–394. doi: 10.1037/0735-7044.118.2.389. [DOI] [PubMed] [Google Scholar]
- Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Milad MR, Orr SP, Lasko NB, Chang Y, Rauch SL, Pitman RK. Presence and acquired origin of reduced recall for fear extinction in PTSD: results of a twin study. J Psychiatr Res. 2008;42:515–520. doi: 10.1016/j.jpsychires.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M. Behavioral and neural analysis of extinction. Neuron. 2002;36:567–584. doi: 10.1016/s0896-6273(02)01064-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Ed 2. San Diego: Academic; 1986. The rat brain in stereotaxic coordinates. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Shin LM, Rauch SL. Investigating the pathogenesis of posttraumatic stress disorder with neuroimaging. J Clin Psychiatry. 2001;62(Suppl 17):47–54. [PubMed] [Google Scholar]
- Porter JT, Johnson CK, Agmon A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci. 2001;21:2699–2710. doi: 10.1523/JNEUROSCI.21-08-02699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power AE, Vazdarjanova A, McGaugh JL. Muscarinic cholinergic influences in memory consolidation. Neurobiol Learn Mem. 2003a;80:178–193. doi: 10.1016/s1074-7427(03)00086-8. [DOI] [PubMed] [Google Scholar]
- Power AE, McIntyre CK, Litmanovich A, McGaugh JL. Cholinergic modulation of memory in the basolateral amygdala involves activation of both m1 and m2 receptors. Behav Pharmacol. 2003b;14:207–213. doi: 10.1097/00008877-200305000-00004. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Paré D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci. 2003;23:8800–8807. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, González-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci. 2000;23:393–398. doi: 10.1016/s0166-2236(00)01629-5. [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Davis M. Applying learning principles to the treatment of post-trauma reactions. Ann N Y Acad Sci. 2003;1008:112–121. doi: 10.1196/annals.1301.012. [DOI] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Peña de Ortiz S, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J, Schiller Y. NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr Opin Neurobiol. 2001;11:343–348. doi: 10.1016/s0959-4388(00)00217-8. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Rigor BM. Protection by MK-801 against hypoxia-, excitotoxin-, and depolarization-induced neuronal damage in vitro. Neurochem Int. 1995;26:519–525. doi: 10.1016/0197-0186(94)00148-n. [DOI] [PubMed] [Google Scholar]
- Schweitzer P. Cannabinoids decrease the K(+) M-current in hippocampal CA1 neurons. J Neurosci. 2000;20:51–58. doi: 10.1523/JNEUROSCI.20-01-00051.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, Peters PM, Metzger LJ, Dougherty DD, Cannistraro PA, Alpert NM, Fischman AJ, Pitman RK. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61:168–176. doi: 10.1001/archpsyc.61.2.168. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Jr, Corcoran KA, Lebrón-Milad K, Quirk GJ. Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci. 2006;24:1751–1758. doi: 10.1111/j.1460-9568.2006.05014.x. [DOI] [PubMed] [Google Scholar]
- Song MK, Cui YY, Zhang WW, Zhu L, Lu Y, Chen HZ. The facilitating effect of systemic administration of Kv7/M channel blocker XE991 on LTP induction in the hippocampal CA1 area independent of muscarinic activation. Neurosci Lett. 2009;461:25–29. doi: 10.1016/j.neulet.2009.05.042. [DOI] [PubMed] [Google Scholar]
- Stuart G, Schiller J, Sakmann B. Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol. 1997;505:617–632. doi: 10.1111/j.1469-7793.1997.617ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Minnen A, Hagenaars M. Fear activation and habituation patterns as early process predictors of response to prolonged exposure treatment in PTSD. J Trauma Stress. 2002;15:359–367. doi: 10.1023/A:1020177023209. [DOI] [PubMed] [Google Scholar]
- Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem. 2006;13:728–733. doi: 10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Alonso A. Cell-type specific modulation of intrinsic firing properties and subthreshold membrane oscillations by the M(Kv7)-current in neurons of the entorhinal cortex. J Neurophysiol. 2007;98:2779–2794. doi: 10.1152/jn.00033.2007. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, Fisher BN, Rominger DH, Earl RA. Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther. 1998;285:724–730. [PubMed] [Google Scholar]