

Abstract
The need for rapid methods in order to precisely detect methicillin-resistant Staphylococcus aureus (MRSA) is extensively acknowledged. This study evaluated a quantitative real-time PCR assay targeting mecA (encoding high level resistance to methicillin) and femB (a specific genomic marker for S. aureus) genes to detect MRSA from broth culture, from serum seeded with MRSA and straight from the patient's serum. One hundred and thirty-five clinical isolates of MRSA strains and different species were utilised in this study. In addition, a pilot study with 9 patients' serum samples was performed. The sensitivity and specificity values for this assay were 99% and 100% respectively. The detection limit for this method was 1.23×102 CFU/ml from the serum seeded with MRSA cells and the limiting concentration of DNA for detection was 18 fg, which equates to 5.14 genomic DNA copies. In addition, this assay detected MRSA from patient's serum (7 out of 9) with sensitivity of 77.8%. Overall, the assay was rapid, efficient, sensitive and easy to perform.
Keywords: MRSA, real-time PCR, mecA, femB, Evaluation, Duplex, Sera
Background
MRSA has emerged as major cause of bloodstream infections associated with raised risk of nosocomial infection and an increase in morbidity, mortality and hospitalization costs [1, 2]. Recently MRSA infections have also appeared in community settings in many different countries [3, 4]. Although S. aureus is relatively easy to cultivate, conventional identification methods may yield false-positive or false-negative results [5]. Conventional methods require 48 h for the detection of MRSA, in spite of shorter detection times using the most recent generation of chromogenic selective agar media [6]. Furthermore, the sensitivity of blood cultures is markedly reduced if blood samples are obtained during antimicrobial treatment [7]. Therefore, several real-time PCR methods have recently been developed for detecting MRSA directly from clinical samples on the same day acquired [8, 9].
The purpose of this study was to investigate the effectiveness of a duplex real-time PCR assay using the TaqMan system in detecting MRSA from serum seeded with bacteria and patients' sera to assess the diagnostic efficacy.
Methodology
Patients and clinical samples:
A total of 100 clinical isolates of MRSA strains and 35 isolates of a number of different species collected that had been previously identified by Manchester clinical bacteriology laboratory staff in the MRI were utilised in this study. In addition serum samples were taken from nine different MRSA septicemic (deep-seated infection) patients to evaluate this duplex real-time PCR. These patients were enrolled in a clinical trial with NeuTec Pharma (Manchester, UK). Full ethical approval for analysis had been granted by the Northen and Yorkshire MREC with reference number 04/MRE03/33.
Taqman™ Duplex real-time PCR:
DNA was extracted from 200µl of each bacterial suspension and from each patient's serum following manufacturer's instruction according to Qiagen kit. The oligonucleotide primers and fluorescence-labelled hybridization probes for the mecA and femB genes sequences were selected by using Primer Express software (Applied Biosystems) Table 1 (see supplementary material). The mecA probe was labelled with the fluorophore FAM (6-carboxy fluorescein) and the femB probe labelled with the fluorophore VIC (Perkin-Elmer, Applied Biosystems, UK), The TaqmanTM probes were synthesized with the reporter dye covalently linked to the 5' ends, and the quencher dye, TAMRA (6-carboxy tetramethyl rhodamine) at the 3' ends. In addition, the 3' ends were phosphorylated to prevent probe extension.
After primer and probe optimisation had been performed, the amplification of both mecA and femB genes in a duplex PCR reaction was performed on all of the samples. A sufficient volume of PCR mastermix solution was made for the number of reactions to be tested comprising of, Taqman™ Universal mastermix, 4µM of mecA forward and reverse primers, 4µM of femB forward and reverse primers, 2µM of mecA and femB probes (Perkin-Elmer, Applied Biosystems, UK) and pharmacy grade sterile injectable water (Phoenix Pharmaceuticals) to give final concentrations of forward primer 200nM, reverse primer 200nM and probe 100nM. A 20µl volume of PCR mastermix was placed into a well on the Micro-Amp reaction plate (Perkin-Elmer, Applied Biosystems, UK) for each reaction to be tested. To this approximately 0.4µg of each sample of DNA extract was added giving a total reaction volume of 25µl. Each well was then capped using Micro-Amp optical cap strips (Perkin-Elmer, Applied Biosystems, UK) and the plate was loaded onto the PE-ABI 7700 automated Taqman™ system where amplification was performed using the following thermocycling parameters: one cycle for 2 min at 50°C, one cycle for 10 min at 95°C, 45 cycles for 15 sec at 95°C, and finally 45 cycles for 1 min at 60°C.
Data points collected at the end of each extension stage were analysed after thermal cycling. During the first 15 cycles, data was collected from all wells and the background fluorescence was determined. A threshold value was calculated as 10 SD above the mean background fluorescence. A sample was considered positive if the fluorescence signal crossed this threshold within 45 cycles; the cycle number at which this occurs was recorded as the Ct value.
After performing the duplex real-time PCR the amplification plots of both the dye layers VIC and FAM were examined to determine that both gene target sites had been amplified. This was verified by analysing the multi-component view which shows an increase in both dye layers as the reporter dye is released and the quencher dye fluorescence decreased (Figure 1).
Figure 1.
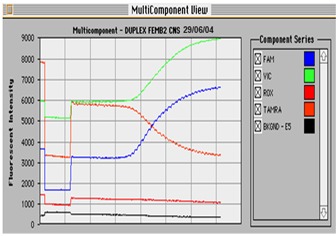
Multicomponent view of a Taqman amplification. The multicomponent view shows fluorescent intensity of background, passive dye (ROX), quencher dye (TAMRA) and reporter dyes FAM and VIC. During amplification digestion of the reporter dye from the 5'end of the probe results in an increase in fluorescent signal. As the reporter dye is released the quencher dye fluorescence decreases. This sample showed positive amplification of the gene targets mecA and femB and was therefore MRSA.
Detection limit of duplex real-time PCR :
DNA was extracted from three samples (200µl each) of an EMRSA 15 strain grown in broth culture. The concentration of DNA in each sample was determined by using Genequant II apparatus (Pharmacia Biotech) according to the manufacturer's direction for use. The known concentration of extracted DNA was serially diluted with PBS from10-1 to 10-7 final dilution factor for each sample. A 5µl aliquot from each dilution was used as template for duplex real-time PCR in order to determine the lowest amount of DNA that is able to be detected by this assay.
A single colony of MRSA was inoculated into 300µl of normal serum from a healthy individual and mixed well by vortex. Subsequently, serial dilutions from 10-1 to 10-7 using healthy human serum as diluent was performed. DNA was extracted from a 200µl aliquot of each dilution and then used as a template for amplification in duplex real-time PCR.:
Statistical analysis:
The number of genomic DNA copies for the different groups was compared using the Mann-Whitney test. Data was analyzed in SPSS. A P< 0.05 was considered to be statistically significant.
Results
Specificity and sensitivity of duplex real-time PCR:
This test amplified these genes with 99% (99 of 100) sensitivity and 100% (35 of 35) specificity. Only one strain was incorrectly identified by the duplex real-time PCR assay. This strain was confirmed by conventional methods as MRSA. However, this strain was found to be positive for femB and negative for mecA in this investigation Table 2 (see supplementary material).
Detection limit of duplex real-time PCR:
The assay used in the current study was able to detect a minimum of 1.23× 102 CFU/ml from serum seeded with MRSA cells, and the limiting concentration of DNA for detection was 18 fg, which equates to 5.14 genomic DNA copies(3.5 fg/cell) [10, 11]. In addition, this assay was found to be able to detect MRSA from patient's serum (7 out of 9). This gives a sensitivity of 77.8% for the assay as applied to patients' sera.
Discussion
Although there are numerous reports on PCR assays for detecting MRSA, the present study is believed to be the first to investigate the effectiveness of PCR in detecting MRSA directly from serum samples [12, 13]. It was found that the sensitivity and specificity of a PCR assay is dependent on target genes, primer sequences, PCR techniques, DNA extraction procedures, and PCR product detection methods. In addition, the extraction yield of target DNA is a critical factor in the PCR detection of bacteria in clinical specimens, particularly when only a few bacteria are expected to be in specimens [14, 15].
Detection of mecA in conjunction with femB as a second target has been reported for identification of MRSA [16, 17]. In the present study, the gene femB was selected as a gene target for MRSA confirmation. Primers and probes were chosen in a way so that they do not form considerable hair-pin or secondary structures and do not form stable dimers, which reduce the efficiency of PCR amplification. In order to confirm the identification of MRSA isolates, both mecA and femB genes targets should be amplified (positive), because these two targets exist as single copies within the genome, an extracted culture would contain equal numbers of both genes. This assay was optimised to amplify these two genes with equal efficiency. The use of the TaqmanTM ‘real time' PCR enabled the performance of each primer to be accurately assessed with respect to the accumulated fluorescent signal (ΔRn) and to the Ct value. All samples were identified correctly for both mecA and femB genes except one sample, which was identified as MRSA by conventional methods. After using this assay, this sample was found positive for femB and negative for mecA. Further conventional tests confirmed that this strain was MRSA, which was a borderline oxacillin resistant Staphylococcus aureus (BORSA). In this case, the mecA gene might be present or absent. BORSA isolates lacking the mecA gene have been reported to possess other mechanisms of resistance, such as the hyper-production of β-lactamase [18, 19].
It has been reported that one out of 327 CNS strains had the femB gene [20]. Jonas et al, also found that the product of the femB gene can be detected in Staphylococcus auricularis, which is a CNS species. Nonetheless, the habitat for this organism is the external auditory meatus of the ear and it does not appear to colonise the sites that are normally screened for MRSA. Therefore, the assay designed in the present study could be applied potentially in clinical laboratories to detect MRSA directly from clinical samples [16].
The low sensitivity of this assay in detecting MRSA from patients' serum might be because of the degradation of DNA due to the repeated freezing and thawing of the samples. This result is supported by the findings of a study in which the sensitivity of PCR was found to be affected by the duration of serum storage at -20°C, through the progressive degradation of frozen DNA. Moreover, repeated thawing of frozen samples may have played a role in the observed decrease in sensitivity as previously documented [21]. A mixture of mecA positive CNS and a BORSA isolate sample could be misdiagnosed by conventional PCR since both mecA and femB genes can be detected. However, this problem is less likely to occur using TaqMan real- time PCR as analysis of the amplification plots would show similar Ct values if the two genes were present in equal amounts. A mixed culture would be unlikely to contain equal amounts of both targets and therefore suspicion would be placed on such a result.
The system showed good specificity and sensitivity for the detection of MRSA without any requirement for isolation or enrichment of the bacteria. This reduced the number of sample preparation steps and the time consumed. This assay also eliminates the risk of contamination by sealing the micro-titre plates prior to amplification and without the need for further analysis. Although positive, PCR results always need confirmation with the culture in order to exclude false-positive results or to gain isolates for further testing (susceptibility, virulence factors), this assay could help to reduce the workload associated with MRSA surveillance programs. Moreover, this assay could minimize the nosocomial MRSA transmission by the well-timed detection of MRSA carriers which may be useful in the screening of some high-risk groups of patients. Focusing on the cost-effectiveness of rapid MRSA technology still remains a debated issue [22].
Financial Disclosure
The authors state no conflict of interest.
Supplementary material
Footnotes
Citation:Awad et al, Bioinformation 9(18): 896-900 (2013)
References
- 1.Laupland K, et al. J Infect Dis. 2008;198:336. doi: 10.1086/589717. [DOI] [PubMed] [Google Scholar]
- 2.Naber CK. Clin Infect Dis. 2009;48:S231. doi: 10.1086/598189. [DOI] [PubMed] [Google Scholar]
- 3.Fang H, et al. Clin Microbiol Infect. 2008;14:370. doi: 10.1111/j.1469-0691.2007.01941.x. [DOI] [PubMed] [Google Scholar]
- 4.Udo EE, et al. J Clin Microbiol. 2008;46:3514. doi: 10.1128/JCM.00966-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frebourg NB, et al. J Clin Microbiol. 1998;36:52. doi: 10.1128/jcm.36.1.52-57.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diederen BM, Kluytmans JA. J Infect. 2006;52:157. doi: 10.1016/j.jinf.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Grace CJ, et al. Clin Infect Dis. 2001;32:1651. doi: 10.1086/320527. [DOI] [PubMed] [Google Scholar]
- 8.Huletsky A, et al. Clin Infect Dis. 2005;40:976. doi: 10.1086/428579. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi N, et al. Diagn Microbiol Infect Dis. 2009;64:172. doi: 10.1016/j.diagmicrobio.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, et al. J Clin Microbiol. 1995;33:596. doi: 10.1128/jcm.33.3.596-601.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francois P, et al. J Clin Microbiol. 2003;41:254. doi: 10.1128/JCM.41.1.254-260.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang K, et al. J Clin Microbiol. 2008;46:1118. doi: 10.1128/JCM.01309-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Talib H, et al. BMC Microbiol. 2009;9:113. doi: 10.1186/1471-2180-9-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demeke T, Jenkins GR. Anal Bioanal Chem. 2010;396:1977. doi: 10.1007/s00216-009-3150-9. [DOI] [PubMed] [Google Scholar]
- 15.Demeke T, et al. J AOAC Int. 2009;92:1136. [PubMed] [Google Scholar]
- 16.Jonas D, et al. Eur J Clin Microbiol Infect Dis. 1999;18:643. doi: 10.1007/s100960050365. [DOI] [PubMed] [Google Scholar]
- 17.Klotz M, et al. J Clin Microbiol. 2003;41:4683. doi: 10.1128/JCM.41.10.4683-4687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartman BJ, Tomasz A. J Bacteriol. 1984;158:513. doi: 10.1128/jb.158.2.513-516.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown DF, et al. J Antimicrob Chemother. 2005;56:1000. doi: 10.1093/jac/dki372. [DOI] [PubMed] [Google Scholar]
- 20.Zeaiter Z, et al. J Clin Microbiol. 2002;40:1023. doi: 10.1128/JCM.40.3.1023-1030.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farnert A, et al. Trans R Soc Trop Med Hyg. 1999;93:50. doi: 10.1016/s0035-9203(99)90177-3. [DOI] [PubMed] [Google Scholar]
- 22.Sturenburg E. Ger Med Sci. 2009;6:Doc06. doi: 10.3205/000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.