

Abstract
It is well known that an amino acid can be encoded by more than one codon, called synonymous codons. The preferential use of one particular codon for coding an amino acid is referred to as codon usage bias (CUB). A quantitative analytical method, CUB and a related tool, Codon Adaptative Index have been applied to comparatively study whole genomes of a few pathogenic Trypanosomatid species. This quantitative attempt is of direct help in the comparison of qualitative features like mutational and translational selection. Pathogens of the Leishmania and Trypanosoma genus cause debilitating disease and suffering in human beings and animals. Of these, whole genome sequences are available for only five species. The complete coding sequences (CDS), highly expressed, essential and low expressed genes have all been studied for their CUB signature. The codon usage bias of essential genes and highly expressed genes show distribution similar to codon usage bias of all CDSs in Trypanosomatids. Translational selection is the dominant force selecting the preferred codon, and selection due to mutation is negligible. In contrast to an earlier study done on these pathogens, it is found in this work that CUB and CAI may be used to distinguish the Trypanosomatid genomes at the sub-genus level. Further, CUB may effectively be used as a signature of the species differentiation by using Principal Component Analysis (PCA).
Abbreviations
CUB - Codon Usage Bias, CAI - Codon Adaptative Index, CDS - Coding sequences, t-RNA - Transfer RNA, PCA - Principal Component Analysis.
Keywords: Codon Usage Bias, Codon Adaptation Index, Essential genes, Highly expressed genes, Translational selection, t-RNA
Background
Protozoan parasites from the order of Kinetoplastida cause diseases in humans as well as animals. Eleven genus of Trypanosomatidae have 565 species. Out of these, only Leishmania and Trypanosoma cause disease in humans. Trypnosoma brucei, Trypanosoma cruzi and various species of Leishmania are responsible for African Trypanosomiasis, Chagas disease and Leishmaniasis, devastating diseases that afflict millions of victims, largely in the tropical regions of the globe. So a comprehensive study of these genomes is expected to generate better comparative protein studies which may lead to more efficient drug designing. These single cell eukaryotes occupy a very ancient position on the evolutionary tree which diverged from the ancestor of the main eukaryotic branch [1, 2]. Trypanosomatids have unique mechanisms for gene expression, such as poly-cistronic transcription, trans-splicing, and the involvement of Pol I in the synthesis of mRNA and RNA editing [3].
Comparative genomics provides a gateway to understand the evolutionary and functional relations between different species. Codon usage bias (CUB) is one of the useful analytical methods for comparative genomics. Codon usage bias is needed for the selection of translational efficiency and hence gene expression. Several steps in the gene expression process may be modulated, including transcription, RNA splicing, translation, and post-translational modification of a protein. More than one triplet of nucleotides (codon) may code an amino acid. These codons are known as synonymous codons. The unequal usage of a particular codon is termed codon usage bias. [4]. The analysis of CUB may aid in understanding the expression of genes and pathogenesis of any organisms and allow re-engineering of the target genes to improve their expression for gene therapy [5]. The Codon Adaptation Index (CAI) value is useful in the study of gene expression level of a particular gene in an organism. The value of CAI nay be used to study of the gene expression level within species, the level of heterogeneous gene expression, and comparison of the codon usage in different organisms and identification of the protein coding reading frames [6].
David Horn [7] had studied the codon usage pattern of some tandem highly expressed genes of Trypanosomatids. He found the codon usage patterns of L.major, T.bruei and T. cruzi to be similar. Since one cannot determine the behavior of the entire genome on the basis of a particular group of genes, this study follows a more general approach. This is now possible due to more data for the coding sequences (CDS) and whole genome sequences being available.
Generally three basic questions are addressed in this research work. First-is the codon distribution of all CDSs conserved among species or not; second-is the translational selection in Trypanosomatids on the basis of cognate copy number of t-RNA or not; and third-is the distribution of codon usage and amino acid frequency for CDSs, highly expressed genes, low expressed genes and essential genes in Trypanosomatids similar or not.
It is found that the pattern of codon usage bias is more or less similar between all highly expressed, less expressed genes and complete CDS for a given genome, but the difference of CUB of different genomes is enough to distinguish the genomes at the sub- genus level. Principal Component Analysis (PCA) was used to compare the CUB of all five Trypanosomatid species, with Plasmodium vivax, an Apicomplexan genome taken as an outlier with GC% value close to that of T. brucei. The resulting plot clearly differentiates CUB signature of L.braziliensis from that of L.major, and T.brucei and T.cruzi are also shown well-separated as different species. However L.major Friedlin and L.infantum, the old world species, that have 92 % identity even at amino acid level [8] cannot be distinguished. This behavior is as expected and they may be treated as a single species.
Methodology
We have chosen five species to work on, namely- Leishmania major Friedlin, Leishmania infantum JPCM5, Leishmania braziliensis MHOM, Trypanosoma brucei brucei 927 and Trypanosoma cruzi CL Brener. We have also taken Plasmodium vivax as as outlier genome for PCA calculation.
Materials:
All the coding sequences for generating the codon usage table are obtained from NCBI (http: //www.ncbi.nlm.nih.gov/) and TriTrypDB (http://tritrypdb.org/tritrypdb/). The numbers of CDSs analyzed are 8102 in L.major, 7932 in L.infantum, 7834 in L.braziliensis, 8663 in T. brucei and 8990 CDSs of T.cruzi (see Table 1). The frequency of codons is calculated using CALCULATION OF PARAMETERS server (http://genomes.urv.cat/CAIcal). Only those sequences which are less than ten thousand nucleotides are considered because longer sequences contain repetitive regions with an in-built bias, and therefore are excluded from this study [9]. The DNA sequences of some factors which play a central role in replication and transcription process were downloaded from (http: //www.ncbi.nlm.nih.gov/)
Methods:
t-RNA sequences of Trypanosomatids are obtained from GeneDB (ttp://www.genedb.org). Anti-codons are predicted by using TFAM Webserver 1.3 (http://tfam. lcb.uu.se/) for analyzing the copy number of t-RNA of the corresponding codon [10].). The codon adaptation index (CAI) is used to predict gene expression levels and their value calculated by CAI CAL (CAI) (http://genomes.urv. cat/CAIcal/) for these factors. Codon frequency for highly expressed genes and low expressed genes are calculated by CODONW 1.3 (John Peden and fttp://molbiol.ox.ac.uk/cu/codonW.tar.Z/).
PCA is a useful statistical technique that finds patterns in data of high dimensionality. It reduces the variables by using suitable coordinate transformations without losing relevant information. Codon usage of sixty one codons (excluding the three stop codons) of the above five species and P.vivax have been taken as input variables in PCA analysis by SPSS 16.0.
Results & Discussion
Codon usage bias analysis:
The genome size of Trypanosomatids is very large, average genome size in four species is 30 Mb but T.cruzi has a larger size of about 90 Mb Table 1 (see supplementary table) because 50% of its genome contains repetitive elements [11]. Overall codon frequencies of Trypanosomatids are listed in Table 2 (see supplementary table). The dominant codons referred to in Table 2 are those that are at least 1.25 times more frequent than the expected frequency of synonymous codons that code a specific amino acid. High codon usage bias in Leishmania species and comparatively lesser codon usage bias in Trypanosoma species are seen (Figure 1). Bias of cognate codon of the Trypanosoma species are not conserved as much as in the Leishmania species.
Figure 1.
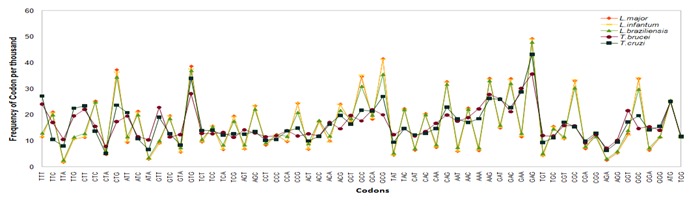
Plot of frequency of codon usage per thousand in all CDSs of length of less than ten thousands in L.major Freidlin(Orange), L.infantum JPCM5(Yellow), L.braziliensis MHOM(Green), T.brucei 927(Brown) and T.cruzi CL Brener (Blue).
Translational selection in Trypanosomatids:
Transfer RNA plays a very important role in decoding the genome in protein synthesis. All amino acids are coded by the three letter code of t-RNA called an anticodon. It is usually composed of adenosine (A), guanosine (G), cytidine (C) and uridine (U) [12]. Specific t-RNAs are responsible for encoding the multiple codons that differ only in the third position and give rise to the wobble hypothesis [13]. In Trypanosomatids t-RNA genes are present only for 43 to 45 codons (Table 1). The total copy numbers of t-RNAs show variability in the Trypanosomatids. There are 66 in L. infantum, 66 in L. braziliensis and 64 in T.brucei. But the number of t-RNAs is 83 in L. major and 115 in T.cruzi.
Of the four nucleotides in any DNA sequence, C and T are pyrimidines and A and G are purines. There are three different types of wobbling possible of an anticodon in the process of translation. Case 1 is when wobbling occurs between pyrimidine and pyrimidine; Case 2 occurs when wobbling is between purine and purine; Case 3 involves wobbling between pyrimidine and purine. About 13-16 codon pairs follow Case 1. The preponderance of pyrimidines at the third position of a codon as observed by Wald is seen in this study also [14].
It is noteworthy that only three types of wobbling codon pairs are observed out of eight possiblities of wobbling between pyrimidine and purine the details of which are given in (Table 2).
Comparison between codon usage pattern of complete CDSs and other genes:
Highly expressed genes tend to use only a limited number of codons and display a high codon bias due to translational selection [15]. High value of CAI of any gene represents higher expression of that gene. The value of 1.0 for CAI indicates the maximum usage of these codons and lower value shows the usage of less preferred codons [6]. The CAI is used to find out whether the expression of selected genes is low or high. The codon usage of some replication and transcription factors which are coded by essential genes is also analyzed. The study of essential genes is helpful in drug designing because these genes are important for the survival of organisms.
CUB largely reflects the differences in the selection pressure on synonymous codons and this selection pressure leads to a substitution in nucleotides composing the codons. This nucleotide substitution is inversely proportional to the degree of codon bias [16]. The rate of substitution is less in coding sequences as compared to non-coding regions. It is still lesser in highly expressed genes [17]. Codon usage frequencies of different types of genes are shown in (Figure 2). All CDS and essential genes show similar frequency of codons but highly and less expressed genes show different result in both Leishmania and Trypanosoma species. In Leishmania the frequencies of codons of highly expressed genes are higher than the value for overall CDSs [6].
Figure 2.
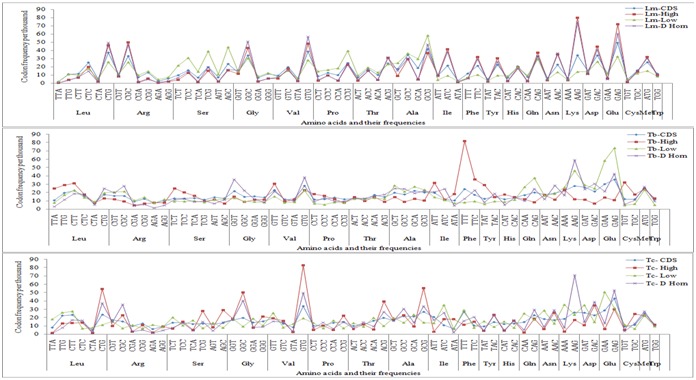
Plot of frequency of codon usage per thousand for different types of genes in Leishmania major(Lm), Trypanosoma brucei(Tb) and Trypanosoma cruzi(Tc).
Amino acids frequencies in all CDS and other genes:
Amino acid frequency is defined as the total frequency of their respective codons. It is almost the same in both the genus but that of Proline(P), Alanine(A), Asparagine(N), Lysine(L) and Glutamic acids are observed to vary. The frequencies are higher for Proline(P) and Alanine(A) in the Leishmania as compared to the Trypanosoma. But frequencies of Asparagine (N), Lysine (K) and Glutamic acid (E) in the Trypanosoma species are higher than in the Leishmania species (Figure 3). The frequencies of amino acids have similar distribution in CDSs and essential genes.
Figure 3.
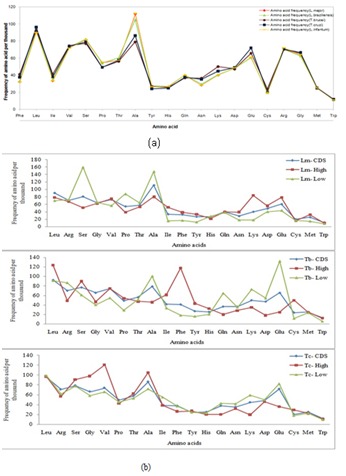
a) Plot of frequency of amino acids versus amino acids in L.major Freidlin(Orange), L.infantum JPCM5(Yellow), L.braziliensis MHOM(Green), T.brucei 927(Brown) and T.cruzi CL Brener (Blue); b) Plot of frequency of amino acid per thousand for different types of genes in Leishmania major(Lm), Trypanosoma brucei(Tb) and Trypanosoma cruzi(Tc).
At the level of properties, the amino acids are divided into four classes, hydrophobic, charged, polar/uncharged and special amino acids [18]. Charged and Polar amino acids may be grouped together as hydrophilic and are shown as such in Table 3 (see supplementary table). Frequencies of hydrophilic amino acids are slightly higher than hydrophobic amino acids in all chosen Trypanosomatids.
Principal Component Analysis is a very useful method for multi-variate analysis. There are 61 variables for the six species chosen. The method uses coordinate transformations to reduce the components to a few principal ones. The first component, PC1 and the second component, PC2 have subsumed 98.5% property of whole 61 variables in every species. The role of PC1 may be deduced to be related to the GC% of the CDS since the CUB values of outlier genome of P. vivax lies close to that of the T.brucei as expected and shown in (Figure 4), and GC% is found to be one of the controlling parameters shaping CUB [6].
Figure 4.
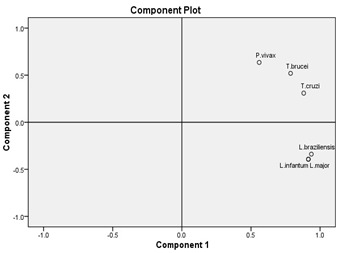
PCA plot for six species by using codon frequency per thousand.
Conclusions
t-RNA abundance, shared specific mutational bias, gene expression level, amino acid composition and GC composition are different factors influencing codon usage bias [19–19].The occurrence of codon usage varies from species to species. The codon usage bias is more conserved in the Leishmania than in the Trypanosoma. There is a significant variation in overall trends between T. brucei and T.cruzi. It is probably due to at least 50% of the T. cruzi genome being composed of repeats, consisting mostly of large gene families of surface proteins, retro-transposons, and subtelomeric repeats [24]. The two species of Trypanosoma genus T.brucei and T.cruzi belongs to two sub- genus Trypanozoon and Schizotrypanum. It is found in this study that the two sub- genus are differentiated at every level of analysis. The use of PCA identified the differences at species level.
Analysis of amino acids cannot give information about mutational selection because it may be possible that the amino acids will not change due to mutation in one nucleotide. Mutation in third position of codon cannot change the amino acid but mutation at the first and second position may change the amino acid. Because of the high degree of conservation at the amino acid level, we may conclude that for Leishmania, mutation occurs only at the third or synonymous position of codons. But in the Trypanosoma species studied here a mutation in the first and second position of the codon changes the amino acid. Therefore we can say that difference in frequency of these amino acids in Trypanosoma is the result of mutation of first position on that cognate codon. For example, lysine frequency is high in T.brucei but frequency of glutamic acid is high in T.cruzi. Lysine is encoded by AAA and AAG, and glutamic acid by GAA and GAG, showing that a mutation of first position of the codon can change the amino acid. It is affirmed that difference in frequency of these amino acids in Trypanosoma is the result of mutation of the nucleotide in the first position of that cognate codon (Table 2).
If the frequency of a particular codon in a genome is very high, then the copy number of the corresponding t-RNA is also high. A direct correlation in these variables is found (result not shown). In this study it is found that few codons of Trypanosomatids are highly preferred but their cognate t-RNA is absent. This indicates that wobbling plays a very important role in translational selection but it may also be possible that the data is incompletely represented or the database used is not comprehensive. The positive correlation between amino acid and t-RNA shows that translational selection is a major force affecting codon usage bias in Trypanosomatids.
The essential genes of Leishmania and Trypanosoma are homologs. But codon preference in essential genes of Leishmania is different from that of Trypanosoma. This is found to be primarily due to the difference in GC% (PCA result).The codon usage table is used as a reference table for calculation of CAI and estimating the gene expression from it. In other words, preferred codons of all essential genes and highly expressed genes are not similar in different species of Trypanosomatids, exception being L.major and L.infantum. Sequence conservation between Leishmania species is high, the average amino acid identity between L. major and L.infantum is 92%, and the average nucleotide identity is 94% [8].The study here also confirms that the variation of all properties including the two principal components in the PCA for L.major and L.infantum are almost identical. For all intents and purposes they can be taken to be one genome. All CDSs, essential genes, highly expressed genes and less frequently expressed genes have been analyzed and it is clear that the result is different from the previous study of D. Horn [7] and his deductions. It is confirmed that the CUB or codon usage bias may be used as a tool for detecting differences at the species level, and is therefore a useful tool for comparative genomics.
Supplementary material
Acknowledgments
MR gratefully acknowledges the award of Junior Research Fellowships from the Council for Scientific and Industrial Research, India, which aided in conducting this study.
Footnotes
Citation:Rashmi & Swati, Bioinformation 9(18): 912-918 (2013)
References
- 1.Landfear SM. PNAS. 2003;100:7. [Google Scholar]
- 2.Aguero F, et al. Genome Res. 2000;10:1996. [Google Scholar]
- 3.Calvillo SM, et al. J Biomed Biotechnol. 2010;2010:525241. doi: 10.1155/2010/525241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuglsang A. Mol Biol Evol. 2006;23:1345. doi: 10.1093/molbev/msl009. [DOI] [PubMed] [Google Scholar]
- 5.Lu H, et al. Acta Biochim Biophys Sin. 2005;37:1. doi: 10.1111/j.1745-7270.2005.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp PM, Li WH. Nucleic Acids Res. 1987;15:1281. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horn D. BMC Genomics. 2008;9:2. doi: 10.1186/1471-2164-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peacock CS, et al. Nature Genet. 2007;39:839. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puigbo P, et al. Biology Direct. 2008;3:38. doi: 10.1186/1745-6150-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taquist H, et al. Nucleic Acids Res. 2007;35:W350. doi: 10.1093/nar/gkm393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arner E, et al. BMC Genomics. 2007;8:391. doi: 10.1186/1471-2164-8-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agris PF. Nucleic Acids Res. 2004;32:223. doi: 10.1093/nar/gkh185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crick FHC. J Mol Biol. 1966;19:548. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 14.Wald N, et al. Nucleic Acids Res. 2012;40:7074. doi: 10.1093/nar/gks348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grantham R, et al. Nucleic Acids Res. 1981;9:43. doi: 10.1093/nar/9.1.213-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharp PM, Li WH. Nucleic Acids Res. 1986;14:7737. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li WH, et al. Nature. 1981;292:237. doi: 10.1038/292237a0. [DOI] [PubMed] [Google Scholar]
- 18.Umbarger HE. Annu Rev Biochem. 1978;47:532. doi: 10.1146/annurev.bi.47.070178.002533. [DOI] [PubMed] [Google Scholar]
- 19.Wan XF, et al. BMC Evol Bio. 2004;4:19. doi: 10.1186/1471-2148-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sueoka N, Kawanishi Y. Gene. 2000;261:53. doi: 10.1016/s0378-1119(00)00480-7. [DOI] [PubMed] [Google Scholar]
- 21.Sueoka TK, et al. Gene. 1999;238:59. doi: 10.1016/s0378-1119(99)00296-6. [DOI] [PubMed] [Google Scholar]
- 22.Black WJ, et al. Nature. 2003;422:633. [Google Scholar]
- 23.Ikemura T. Mol Biol Evol. 1985;2:13. doi: 10.1093/oxfordjournals.molbev.a040335. [DOI] [PubMed] [Google Scholar]
- 24.El-Sayed NM, et al. Science. 2005;309:409. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.