

Abstract
Chemokines and their receptors function in migration and homing of cells to target tissues. Recent evidence suggests that cancer cells use a chemokine receptor axis for metastasis formation at secondary sites. Previously, we showed that binding of the chemokine CXCL12 to its receptor CXCR4 mediated signaling events resulting in matrix metalloproteinase-9 expression in prostate cancer bone metastasis. A variety of methods, including lipid raft isolation, stable overexpression of CXCR4, cellular adhesion, invasion assays, and the severe combined immunodeficient–human bone tumor growth model were used. We found that (a) CXCR4 and HER2 coexist in lipid rafts of prostate cancer cells; (b) the CXCL12/CXCR4 axis results in transactivation of the HER2 receptor in lipid rafts of prostate cancer cells; (c) Src kinase mediates CXCL12/CXCR4 transactivation of HER2 in prostate cancer cells; (d) a pan-HER inhibitor desensitizes CXCR4-induced transactivation and subsequent matrix metalloproteinase-9 secretion and invasion; (e) lipid raft–disrupting agents inhibited raft-associated CXCL12/CXCR4 transactivation of the HER2 and cellular invasion; (f) overexpression of CXCR4 in prostate cancer cells leads to increased HER2 phosphorylation and migratory properties of prostate cancer cells; and (g) CXCR4 overexpression enhances bone tumor growth and osteolysis. These data suggest that lipid rafts on the cell membrane are the key site for CXCL12/CXCR4–induced HER2 receptor transactivation. This transactivation contributes to enhanced invasive signals and metastatic growth in the bone microenvironment.
Introduction
CXCR4 is a seven-transmembrane trimeric G-protein–coupled receptor, and its only known ligand is CXCL12, an 11 kDa peptide. Epithelial tumor cells express CXCR4. CXCL12 is expressed locally in the microenvironment of common metastatic sites. Furthermore, binding of CXCL12 to CXCR4 has been shown to play a crucial role in site-specific metastasis to lymph nodes, lung, and bone (1). In prostate cancer, we and others showed CXCL12/CXCR4 signaling in tumor cells when in bone tissue (2–4). We also showed that CXCL12/CXCR4 interaction leads to mitogen-activated protein kinase and phosphoinositide 3-kinase/Akt–mediated MMP-9 expression, migration, and invasion of prostate cancer cells (2).
In general, G-protein–coupled receptors can transactivate HER family members either by ectodomain shedding of membrane-bound HER family receptor ligands by members of the ADAM (a Disintegrin and metalloprotease) family of proteases (5) or by intracellular phosphorylation of HER family members involving Src kinase (6). In breast cancer cells, CXCL12/CXCR4 interaction activates HER2 in a Src kinase-dependent mechanism (7). In ovarian cancer cells, CXCL12/CXCR4 interaction also activates epidermal growth factor receptor (EGFR), which leads to both mitogen-activated protein kinase and Akt activation (8). Whether CXCL12/CXCR4 activates HER2 in prostate cancer is unknown.
Aggressive cancer phenotypes are also promoted by tyrosine kinase growth factor receptor signaling. On a cellular level, HER2 expression in carcinoma cells is known to cause migration and invasion (9–11). In breast cancer, therapeutic targeting of HER family receptor signaling has been widely successful (12). In prostate cancer, HER2 expression has been associated with androgen-independent androgen receptor signaling (13, 14) and poor survival in androgen-independent disease (15, 16). Despite the importance of HER2 signaling in prostate cancer cells, limited success has been reported with targeting HER kinase receptors in prostate cancer patients (17). This lack of clinical response with single-agent HER2 or EGFR tyrosine kinase therapies with prostate cancer patient populations prompts usage of multidrug combinations to target multiple members of growth factor receptors and/or their regulating signaling partners in clinical trails. Toward this end, a recent clinical trial with pertuzumab, which targets the dimerization process of HER molecules, shows a delayed progression of disease in patient populations who were shown to be androgen independent and failed previous chemotherapy treatment regimens (18). In addition, a potential relationship between CXCL12/CXCR4 interaction and tyrosine kinase growth factor signaling in prostate cancer cells has not been described.
Lipid rafts are specialized microdomains located within the plasma membrane enriched with cholesterol, sphingolipids, and various cell signaling proteins. G-protein–coupled receptors and trimeric G-proteins are associated with lipid rafts where they initiate signal transduction (19). In prostate cancer cells, we showed that CXCR4 is associated with lipid rafts (2). HER family receptors have also been shown to be associated with lipid rafts (20–22). Herein, we hypothesized that CXCL12/CXCR4 interaction leads to HER2 activation in prostate cancer and that this transactivation occurs in lipid rafts. Moreover, we wished to determine the role of Src kinase inhibitor and lipid raft–destabilizing agents in this transactivation process in prostate cancer cells. CXCL12/CXCR4/HER2 signaling pathway in lipid rafts may give prostate cancer a growth advantage in the bone microenvironment.
Results
CXCL12/CXCR4 Cross Talk with HER2 in Lipid Rafts of Prostate Cancer Cells
CXCR4 was shown to associate with the lipid rafts in prostate cancer cells and its only known ligand, CXCL12, is highly expressed in bone tissue cells. CXCL12/CXCR4 signaling events have been shown to participate in bone metastasis. To further understand the lipid raft–associated CXCR4 signaling events contributing to bone metastasis, we studied the potential transactivation of HER2 in lipid rafts of prostate cancer cells.
Western blot analysis of biotinylated cells was done with streptavidin-agarose bead–captured complexes from PC-3 and C4-2B cells. Biotinylated HER2 and CXCR4 proteins ran at higher molecular weight than cellular HER2 and CXCR4 (Fig. 1A, lane 3), suggesting that HER2 and CXCR4 on cell surface were likely biotinylated in prostate cancer cells. Immunoblot analysis was applied to lipid raft fractions that were isolated using successive detergent solubilization of biotinylated cells. The data showed that both CXCR4 and HER2 were localized at cell surface in lipid raft domains and nonrafts in biotinylated prostate cancer cells (Fig. 1B). Quantitation of band intensities from multiple independent cell fractionation studies show that CXCR4 was highly localized in lipid rafts on cell surface in both PC-3 and C4-2B cells. HER2, on the contrary, mainly localized to nonrafts in both cells, but HER2 association with lipid rafts were higher in C4-2B cells than in PC-3 cells. We used sucrose density gradient ultracentrifugation of prostate cancer cell lysates as a complementary method to isolate rafts. Some CXCR4 and HER2 was trapped in the heavy cellular sediment at the bottom of the tube (Fig. 1D), but we also found that CXCR4 and HER2 localized to lighter fractions at the interface between the 5% and 30% sucrose layers. The lipid raft marker Gi3α also localized to these lighter fractions. Together, these data confirmed that CXCR4 and HER2 are found in rafts.
FIGURE 1.

CXCR4 and HER2 localize to lipid rafts of prostate cancer cells. A. PC-3 and C4-2B cells were biotinylated (lane 2) or not biotinylated (lane 1) as described in Materials and Methods. Total cell lysates were prepared, biotinylated proteins were captured with streptavidin agarose beads, and these complexes were immunoblotted with anti-HER2 and anti-CXCR4 antibodies. Total cell lysates from PC-3 and C4-2B cells (lane 3) were run as control for Western blot analysis. B. PC-3 and C4-2B cells were biotinylated (Biotin) or not biotinylated (No biotin) as described in Materials and Methods. Lipid raft fractions that are Triton X insoluble (I fraction) and cell surface proteins that are Triton X soluble (S fraction) were isolated from prostate cancer cells using a successive detergent solubilization technique. Biotinylated proteins in these fractions were captured with streptavidin agarose beads. Biotin-streptavidin complexes were immunoblotted with anti-HER2 and anti-CXCR4 antibodies. C. The pixel intensities from HER2 and CXCR4 bands were quantitated from insoluble and soluble fractions isolated from four independent PC-3 cell and three independent C4-2B cell fractionation studies. D. Total cellular proteins were isolated from PC-3 and C4-2B cells by homogenization and subjected to sucrose gradient ultracentrifugation. Fractions were collected from top to bottom, and Western blot analyses were done with anti-HER2, anti-CXCR4, and anti-Giα-3 antibodies.
We next used the detergent solubilization method to isolate lipid rafts and cell surface proteins from biotinylated prostate cancer cells to study CXCL12-induced signaling (Fig. 2A). In both PC-3 and C4-2B cells, CXCL12 induced phosphorylation of lipid raft-associated HER2 without significantly changing the HER2 phosphorylation in the cell surface nonraft proteins. Similarly, CXCL12-induced HER2 phosphorylation was detectable in lipid rafts isolated from the nonbiotinylated PC-3 and C42B cells without significantly changing the HER2 phosphorylation in cytosol and membrane fractions (Fig. 2B). CXCL12-induced HER2 phosphorylation identified as early as 5 minutes and peaked 15 minutes in both PC-3 and C4-2B cells (Fig. 2C), demonstrating a time-dependent activation of HER2. AMD3100 is a small-molecule bicyclam reagent, which has been developed as an antagonist for CXCR4. Treatment of AMD3100 with PC-3 cells abrogated CXCL12-mediated phosphorylation of HER2 in lipid rafts without significantly changing HER2 phosphorylation of cytosol and membrane fractions in PC3 cells (Fig. 2D). Our group as well as others (2, 23) have shown that Ser473 phosphorylation predominantly localized to the cytosol and membrane fraction and that CXCL12 can enhance Ser473 phosphorylation of Akt in PC-3 cells. Furthermore, AMD3100 treatment abrogated the CXCL12-induced Ser473 phosphorylation of Akt in PC-3 cells (Fig. 2E). Together, these data showed that CXCL12-induced HER2 transactivation occurs in lipid rafts and subsequent signaling through Akt phosphorylation occurs in cytosol and membrane fractions in prostate cancer cells.
FIGURE 2.

CXCL12 and CXCR4 transactivation of HER2 in lipid rafts of prostate cancer cells. A. Serum-starved PC-3 and C4-2B cells were treated with CXCL12 and biotinylated, and lipid rafts and cell surface proteins were captured with anti-avidin agarose beads. Western blot was done with anti-pHER21248 antibodies. B. Serum-starved PC-3 and C4-2B cells were treated with or without 200 μg/mL CXCL12. Lipid rafts and cytosol + membrane fractions were isolated from PC-3 cells using successive detergent solubilization method. Equal amounts of proteins were immunoblotted with anti-HER2Tyr1248, anti-HER2, and anti-CXCR4 antibodies. C. Serum-starved PC-3 and C42-B cells were treated with 200 ng/mL CXCL12 for 5, 15, and 30 min. Lipid rafts and cytosol + membrane fractions were isolated from untreated and CXCL12-treated prostate cancer cells. Equal amounts of proteins from both fractions were immunoblotted with anti-HER2Tyr1248 antibodies. D. Serum-starved PC-3 cells were either treated or not treated with 5 μmol/L concentration of AMD3100. Both groups were treated with 200 ng/mL CXCL12 for 15 min or medium alone without CXCL12. Equal amounts of proteins from lipid rafts and cytosol + membrane fractions were immunoblotted with anti-HER2Tyr1248 antibodies. Relative quantitation of HER2Tyr1248 band intensities were shown at the top of the gel. E. The cytosol and membrane proteins from the experiment mentioned in D were immunoblotted with anti-AktSer473 antibodies. Relative quantitation of AktSer473 band intensities were shown at the top of the gel.
Src Kinase Signaling Mediates CXCL12/CXCR4 –Induced HER2 Transactivation in Prostate Cancer Cells
To further understand the Src kinase role in CXCL12/CXCR4–induced HER2 transactivation, we measured the Src kinase expression and activation in the lipid rafts versus cytosol and membrane fractions in prostate cancer cells. Src kinase is expressed in both lipid rafts and nonlipid rafts of prostate cancer cells (Fig. 3A). Interestingly, Tyr416 phosphorylation of Src kinase is highly associated with lipid rafts compared with cytosol and membrane fractions of prostate cancer cells (Fig. 3A). In PC-3 cells, Src phosphorylation is higher than C4-2B cells under serum-starved conditions in lipid rafts. CXCL12 treatment enhanced Src phosphorylation in lipid rafts of C4-2B cells without significantly changing the phosphorylation of cytosol and membrane fraction–associated Src kinase. However, in PC-3 cells, CXCL12 treatment marginally enhanced lipid raft–associated Src phosphorylation compared with C4-2B cells (Fig. 3A).
FIGURE 3.

CXCL12 activates Src kinase in lipid rafts of prostate cancer cells. A. Serum-starved PC-3 and C42-B cells were treated with 200 ng/mL CXCL12 for 15 min or medium alone. Lipid rafts and cytosol + membrane fractions were isolated using successive detergent solubilization method. Equal amounts of both fractions were immunoblotted with anti-SrcTyr416 and anti-Src antibodies. Relative quantitation of SrcTyr416 band intensities of lipid raft fractions were shown at the top of the gel. B. Serum-starved PC-3 and C4-2B cells were either treated or not treated with PP2 (2.5 μmol/L). Both groups were treated with 200 μg/mL CXCL12 for 15 min or medium alone without CXCL12. Equal amounts of proteins from lipid raft fractions were immunoblotted with anti-SrcTyr416 and anti-Src antibodies. C. Serum-starved PC-3 cells were treated with CXCL12 and with PP2 compound as described in B. Lipid rafts and cytosol + membrane fractions were isolated using successive detergent solubilization method and equal amounts of proteins were immunoblotted with anti-pHER2Tyr1248 antibodies. D. Cytosol and membrane fractions described in C were immunoblotted with anti-AktSer473 antibodies. All the blots are representative of two independent experiments.
As expected, pretreatment of PC-3 and C4-2B cells with PP2 compound inhibited Src phosphorylation in the lipid rafts (Fig. 3B). The basal and CXCL12-induced phosphorylations of Src were inhibited by the PP2 compound in C4-2B cells. Interestingly, the PP2 compound also inhibited CXCL12-induced HER2 phosphorylation in lipid rafts. In addition, the basal HER2 phosphorylation observed in cytosol and membrane fractions of PC-3 cells was also sensitive to Src kinase inhibition with PP2 compound, which suggests that constitutive HER2 phosphorylation in prostate cancer cells seems to be mediated by the Src kinase (Fig. 3C). The PP2 compound also inhibited basal and CXCL12-induced Ser473 Akt phosphorylations in cytosol and membrane fractions in PC-3 cells (Fig. 3D). These findings suggest that Src kinase activation in the lipid rafts is required for CXCL12/CXCR4–mediated HER2 phosphorylation in prostate cancer cells. Temporally, Src kinase act in between CXCL12/CXCR4 and HER2 in lipid rafts of prostate cancer cells. In addition, Src is an upstream kinase for HER2 in nonraft domains in prostate cancer cells.
PC-3 Cells Use Growth Factor Receptor Signaling for CXCL12-Mediated Chemoinvasion
We used CI-1033, which is a pan-HER inhibitor shown to inhibit members of HER family receptors in cell culture models (24). Treatment of PC-3 cells with different concentrations of CI-1033 showed that cell proliferation was not altered at 1 μmol/L concentration for 24 hours (Fig. 4A). Western blot analysis of prostate cancer cell lysates showed that CI-1033 treatment resulted in down-regulation of total and phosphorylated HER2 (Fig. 4B) and EGFR (data not shown). At the same time, total Akt protein was unchanged whereas phosphorylated Akt was reduced (Fig. 4B) in PC-3 cells. This is consistent with CI-1033 sensitivity toward Akt activation in breast cancer cells (25). These data suggested that Akt activation depends on HER family receptor signaling in prostate cancer cells. We previously showed that CXCR4-mediated Akt kinase activation is required for migration, invasion, and MMP-9 secretion by PC-3 cells (2). Based on the fact that CXCR4 transactivation of HER2 was localized to lipid rafts and pharmacologic inhibition of HER family receptor function inhibited Akt activation, we hypothesized that CXCL12-induced Akt activation and subsequent MMP-9 expression is mediated by HER family receptors in PC-3 cells. We treated PC-3 cells with CI-1033 (Fig. 4C). We found that both basal and CXCL12-stimulated levels of phosphorylated HER2 were inhibited by this compound in lipid rafts. Phosphorylated HER2, which is associated with the cytosol and the membrane fraction, was also sensitive to this compound irrespective of CXCL12 treatment. Moreover, CI-1033 also inhibited CXCL12-stimulated phosphorylation of Akt at Ser473 (Fig. 4C) in the cytosol and membrane fractions of PC-3 cells. In addition, basal and CXCL12-stimulated pro–MMP-9 secretion (Fig. 5A and B) and chemoinvasion (Fig. 5C and D) were also inhibited by CI-1033. These data show that HER family receptors in prostate cancer cells function as downstream targets of CXCL12 and CXCR4 signaling, and mediate Akt activation, MMP-9 secretion, and cellular invasion.
FIGURE 4.
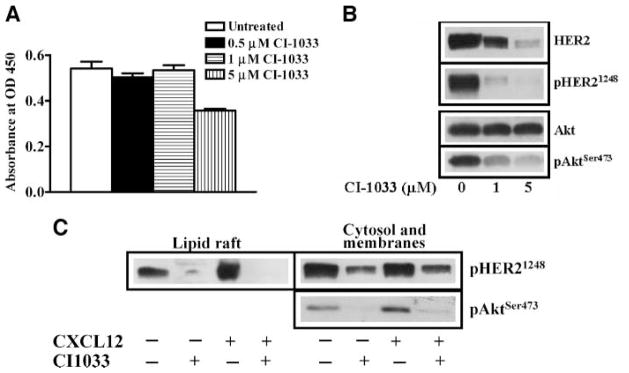
CI-1033 inhibits HER2 expression and signaling in PC-3 cell. A. PC-3 cells were treated with different concentrations of CI-1033 for 24 h and cell proliferation was assessed with the WST assay. B. Total cellular proteins were isolated from PC-3 cells, which were either treated with 1 and 5 μmol/L of CI-1033 or not treated with CI-1033. The cellular proteins were immunoblotted with anti-HER2, anti-pHER21248, anti-Akt, and anti-AktSer473. C. Lipid raft and cytosol + membrane fractions were isolated from PC-3 cells, which were untreated, treated alone with CI-1033 (1 μmol/L for 24 h), CXCL12 (200 ng/mL for 15 min), or a combination of CI-1033 (1 μmol/L for 24 h) followed by CXCL12 (200 ng/mL for 15 min). The equal amounts of protein fractions were immunoblotted with anti-pHER21248 antibodies. Cytosol and membrane fractions were immunoblotted with anti-AktSer473 antibodies.
FIGURE 5.

CI-1033 inhibits MMP-9 expression and invasion in prostate cancer cells. A. The conditioned medium was prepared from PC-3 cells that were untreated (lane 2), treated with CI-1033 (1 μmol/L) for 24 h (lane 3), and treated with the combination of CI-1033 (1 μmol/L) for 24 h followed by CXCL12 (200 ng/mL; lane 4). The conditioned medium was analyzed on gelatin zymography. Recombinant pro-MMP9 was also run as a positive control in the zymographic analysis (lane 1). B. The conditioned medium was obtained from PC-3 cells, which was untreated (lane 1) or treated with CXCL12 (200 ng/mL). The conditioned medium was analyzed on gelatin zymography. C. PC-3 cell invasion was done using Matrigel-coated filters. Before chemoinvasion, PC-3 cells were either untreated or treated with 1 μmol/L CI-1033 for 24 h. During chemoinvasion, CI-1033 (1 μmol/L) was included in the top well and CXCL12 (200 ng/mL) was included in bottom well. Two independent experiments were done in triplicate, and the data were analyzed using ANOVA. The Tukey posttest was done for pair-wise comparisons. *, P < 0.05. D. A representative field of PC-3 cells that adhered to the bottom of the Matrigel-coated filters.
Lipid Raft–Destabilizing Agents Alter the CXCL12/CXCR4 Signaling and Chemoinvasion of PC-3 Cells
Methyl β-cyclodextrin (MBCD) is a cholesterol-binding agent known to disrupt lipid raft signaling in cancer cells. MBCD treatment (Fig. 6A) resulted in inhibition of basal and CXCL12-induced HER2 phosphorylation in lipid rafts. CXCR4 levels are slightly decreased in lipid rafts and are increased in the cytosol and membrane fractions upon MBCD treatment of PC-3 cells. The basal and CXCL12-induced cellular chemoinvasions were also inhibited in MBCD-treated cells (Fig. 6B). Similarly, amphotericin B, which is a fungal antibiotic known to disrupt lipid rafts by cholesterol binding, also inhibited basal and CXCL12-induced cellular chemoinvasions in PC-3 cells (Fig. 6B). These results show that intact lipid raft architecture is required for CXCL12/CXCR4 transactivation of HER2 and PC-3 cell invasion, and disruption of lipid rafts with destabilizing agents inhibits both CXCL12/CXCR4 transactivation of HER2 in lipid rafts and cellular chemoinvasion.
FIGURE 6.

Lipid raft – dispersing agents inhibit CXCL12 signaling and chemoinvasion of PC-3 cells. A. PC-3 cells were serum starved and either untreated or pretreated with MBCD (5 mmol/L) for 1 h. Following MBCD treatment, cells were stimulated with CXCL12. Lipid rafts and cytosol + membrane fractions were isolated using successive detergent solubilization method. Western blot analysis was done with anti-HER2Tyr1248 and anti-CXCR4 antibodies. B. Serum-starved PC-3 cells were treated with MBCD and amphotericin B for 1 h, and Matrigel chemoinvasion assay was done as indicated in Materials and Methods. CXCL12 (200 ng/mL) is placed in the lower chamber as a chemoattractant. The data shown were representative of two independent experiments that were done in triplicate.
CXCR4 Overexpression Enhances HER2 Phosphorylation and Invasion of PC-3 Cells
Western blot and fluorescence-activated cell sorting analyses of PC-3-Neo, PC-3-CXCR4-2.1, and PC-3-CXCR4-2.3 cells show that transfection of PC-3 cells yielded stable clones. Western blot analysis of total CXCR4 levels in transfected cells shows that CXCR4 protein levels increased by 3-fold in CXCR4-2.1 and CXCR42.3 compared with neo-expressing cells (Fig. 7A). Similarly, cell surface expression of CXCR4 also enhanced in CXCR4-2.1 and CXCR4-2.3 cells compared with neo-expressing cells (Fig. 7B). Western blot analysis for phosphorylated HER2 in lipid raft fractions of neo- and CXCR4-transfected cells resulted in an enhancement of basal pHER2 phosphorylation in CXCR4-transfected clones compared with neo clones, and all the clones retained the capability of CXCL12-induced HER2 transactivation (Fig. 8A). These cells were tested for chemoinvasiveness in Matrigel-coated filters. Both transfected cells showed enhanced chemoinvasion compared with neo-transfected cells in Matrigel-coated filters (Fig. 8B). The CXCL12-induced chemoinvasion of PC-3 transfectants were abrogated in the presence of neutralizing antibodies to the CXCR4 (Fig. 8B).
FIGURE 7.

Characterization of CXCR4-overexpressing stable PC-3 cell clones. A. Western blot analysis of cell lysates prepared from PC-3 Neo, PC-3 CXCR4-2.1, and PC-3 CXCR4-2.3 cells with anti-CXCR4 antibodyandanti–glyceraldehyde-3-phosphate dehydrogenase antibody. Bottom, quantitation of pixel intensities of immunoreactive CXCR4 and glyceraldehyde-3-phosphate dehydrogenase. B. Fluorescence-activated cell sorting analysis of PC-3 Neo, PC-3 CXCR4-2.1, and PC-3 CXCR4-2.3 cells with phyco-erythrin (PE) – conjugated anti-CXCR4 antibody. Isotype antibody staining of cells was subtracted from the CXCR4-stained cells. The number of CXCR4-positive cells was shown as a percentage of total gated cells.
FIGURE 8.
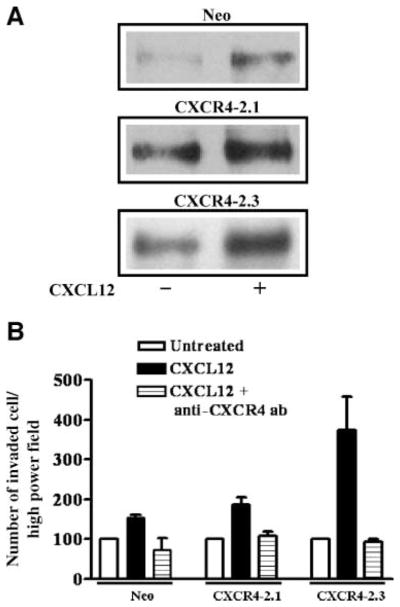
CXCR4 overexpression enhances HER2 phosphorylation and chemoinvasion of PC-3 cell clones. A. PC-3 Neo, PC-3 CXCR4-2.1, and PC-3 CXCR4-2.3 cells were serum starved and either untreated or treated with CXCL12 for 15 min. Lipid rafts were extracted using successive detergent solubilization and were immunoblotted with anti-HER2Tyr1248 antibodies. B. PC-3 Neo, PC-3, CXCR4-2.1, and PC-3 CXCR4-2.3 cells were serum starved, and chemoinvasion was done using Matrigel-coated filters. CXCL12 was included in the bottom chamber as a chemoattractant. Anti-CXCR4 – neutralizing antibodies were added to the upper chamber.
CXCR4 Overexpression Enhances Intraosseous Tumor Growth of Prostate Cancer Cells
We then wished to determine whether CXCR4 expression would enhance growth of prostate cancer cells in the bone microenvironment. As a model of expansion to the metastatic deposit, we injected the modified prostate cancer cells into previously implanted human bone fragments (26). Three weeks after injection, the developing bone tumors were harvested. Ex vivo radiographs (Fig. 9A) showed obvious enhancement of trabecular destruction in bone tumors containing CXCR4-transfected cells. H&E and cytokeratin staining of PC-3 bone tumor tissue sections shows less number of tumor cells in PC-3 neo bone tumors compared with PC-3 CXCR4 bone tumors (Fig. 9B). Quantitative histomorphometric analysis confirmed a reduction in trabecular area in bones injected with CXCR4-transfected cells (Fig. 9C). The reduction in the bone area was accompanied by an increase in the tumor area (Fig. 9D). Further, proliferation marker Ki-67 staining of tumors shows more number of Ki-67–positive cells in PC-3 CXCR4 bone tumors than in PC-3 neo bone tumors (Fig. 10). This study showed that prostate cancer cell expression of CXCR4 promotes rapid intraosseous bone tumor growth and bone destruction.
FIGURE 9.

CXCR4 overexpression in PC-3 cells enhances bone tumor growth in severe combined immunodeficient – human model. PC-3 Neo, PC-3 CXCR4-2.1, and PC-3 CXCR4-2.3 cells were injected into human fetal bone fragments implanted in severe combined immunodeficient mice. Each arm of the animal experiment has n = 6. A. X-ray analysis of representative bone tumor from each group. B. H&E-stained sections of bone tumors were shown at ×5 and ×20 magnifications. Bottom, the anti-cytokeratin antibody staining of PC-3 bone tumor tissues at ×20 magnification. Histomorphometric analysis of PC-3 Neo, PC-3 CXCR4-2.1, and PC-3 CXCR4-2.3 cell bone tumors, showing the trabecular bone area in C and tumor area in D. The data were analyzed using ANOVA. The Tukey posttest was done for pair-wise comparisons. *, P < 0.05.
FIGURE 10.
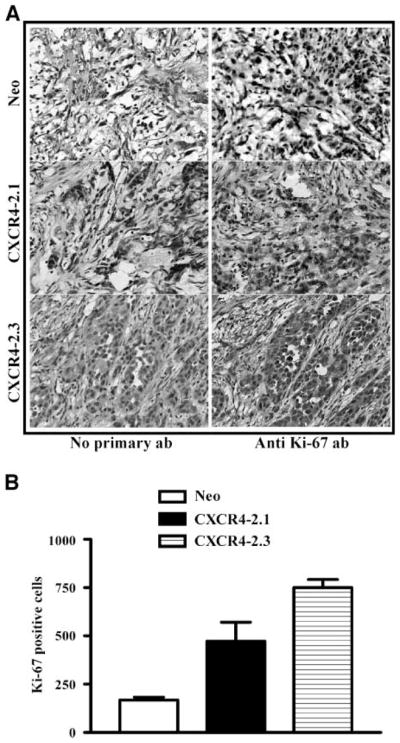
CXCR4 overexpression enhances proliferation index of PC-3 bone tumors A. A representative section of immunohistochemical analysis of PC-3 bone tumor sections with anti-Ki67 antibody. Left panels, negative control, immunohistochemistry was done without the primary antibody (ab). B. The number of Ki-67 – positive nuclei per 1,000 tumor cells in each of the bone tumor. Two bone tumors from each of the group were analyzed for Ki-67 immunohistochemistry analysis.
Discussion
In this study, we have shown that CXCL12/CXCR4 transactivates HER2 in lipid raft microdomains in prostate cancer cells. Based on the results of our study, we conclude that (a) CXCR4 was highly localized to the lipid rafts, whereas a relatively small pool of HER2 was localized to lipid rafts in prostate cancer cells; (b) CXCL12, which is the only known ligand for CXCR4, can transactivate HER2 in lipid rafts of prostate cancer cells without significantly altering the phosphorylation status of the HER2 in nonlipid raft locations in prostate cancer cells; (c) an intracellular nonreceptor tyrosine kinase, Src, mediates CXCL12/CXCR4 transactivation of HER2 in prostate cancer cells and, moreover, Src kinase seems to be responsible for constitutive HER2 phosphorylation in nonlipid raft domains of prostate cancer cells; (d) HER receptor inhibition abrogated the CXCL12/CXCR4 transactivation of HER2 and chemoinvasive process of prostate cancer cells; (e) CXCR4/Src/HER2 signaling module in lipid rafts is the critical player of CXCL12-induced prostate cancer cell chemoinvasive process; (f) enhanced exogenous CXCR4 expression contributed to downstream HER2 receptor activation and chemoinvasiveness of prostate cancer cells; and (g) CXCR4 overexpression enhanced in vivo bone tumor proliferative growth and osteolysis. These findings suggest that lipid rafts are key sites for CXCL12/CXCR4 signaling in prostate cancer cells and CXCL12/CXCR4 transactivation of HER2 contributes to chemoinvasive and growth function of intraosseous prostate cancer tumors (Fig. 11).
FIGURE 11.

A proposed model showing CXCL12/CXCR4 signaling in lipid rafts of prostate cancer cells.
Three independent analyses of lipid raft isolation were done: (a) cell surface biotinylation followed by lipid raft isolation; (b) sucrose density centrifugation of cell lysates; and (c) successive detergent solubilization method, which shows that CXCR4 was highly localized in prostate cancer cells under the culture conditions. In addition, lipid raft–destabilizing agents abrogated the CXCL12-mediated chemoinvasion of prostate cancer cells. These data suggest that lipid raft association of CXCR4 is a prerequisite for CXCL12-induced chemoinvasion of prostate cancer cells. Lipid raft association of CXCR4 in prostate cancer cells seems to be constitutive, and CXCL12 activation does not further enhance CXCR4 association with lipid rafts. Consistent with our finding, CXCR4 association with lipid rafts in CD34-positive stem cells primes these cells for CXCL12 responsiveness (27). In contrast to constitutive association of CXCR4 in lipid rafts of prostate cancer cells, CXCR4 lateral movement into lipid rafts in response to extracellular matrix proteins (27) and CXCL12 (28) actively takes place in stem cells. This difference can be explained by the findings that prostate cancer cells have been shown to express the CXCL12 gene (2, 29) and prostate cancer cell–expressed CXCL12 could constitutively localize the CXCR4 in the lipid raft microdomains. Together, these observations suggest that prostate cancer cells behave differently than stem cells in CXCR4 localization in response to CXCL12, and this constitutive lipid raft association of CXCR4 could contribute to the growth advantage of prostate cancer cells in CXCL12-rich metastatic environment.
A pool of HER2 was also associated with lipid rafts in prostate cancer cells although a majority of HER2 was present elsewhere in the cells. A similar association of HER2 with lipid rafts has been shown in breast tumor cells (30) and fibroblasts (31). The lipid raft–associated HER2 seems to be non-phosphorylated compared with HER2 present in other sites in the cell, and our data suggest that this fraction of HER2 is a target for nontraditional growth factor ligands such as CXCL12 in prostate cancer cells. Our data further suggest that the CXCL12/CXCR4 axis, which is highly localized in lipid rafts, is an upstream activator of HER2. In accordance with our findings, Cabliogu et al. (7) showed that CXCL12 induced HER2 phosphorylation in breast cancer cells; similarly, Porcile et al. (8) found that CXCL12 induced EGFR phosphorylation in ovarian cancer cells. However, to our knowledge, our findings are the first to identify lipid raft microdomains as the site for this transactivation. Our attempts to show a direct interaction between CXCR4 and HER2 by immunoprecipitation were unsuccessful (not shown); therefore, the transactivation process is likely to be indirect, using other lipid raft constituent signaling components in prostate cancer cells. Similar attempts to isolate CXCR4/HER2 complex in breast cancer cells also failed (7), suggesting an indirect transactivation process in other types of cancer cells. Other members of G-protein–coupled receptors have been shown to transactivate growth factor receptor systems in several cell types, leading to mitogenic signaling in cells. One mechanism involves the release of membrane-bound growth factors by proteolysis ectodomain shedding (5). We have observed that HER2 and EGFR are present as a complex in prostate cancer cells (not shown); the presence of a dimeric complex supports the idea that proteolytic shedding of a membrane-bound growth factor ligand may lead to HER2 signaling in lipid rafts (32). This idea is further supported by the findings from ovarian cancer cells, where AG1478, an EGFR kinase inhibitor, blocked CXCL12-dependent cell proliferation and downstream signaling pathways involving Erk1/2 and Akt proteins. Ligands for HER family receptors were also expressed in androgen-independent cancer cell lines, which suggests the existence of an autocrine loop operating in these cells, and resulting in signaling events contributing to the enhanced proliferation in these cells (33). This observation further fit into the model of CXCL12/CXCR4–mediated transactivation of HER family receptor activation by processing membrane-bound ligands and contributing to subsequent proliferation of prostate cancer cells. In support of this model, Sun et al. (29) recently showed that prostate cancer cell–expressed CXCL12 could act as mitogenic factor in cell culture models.
The other mechanism involving G-protein–coupled receptor–mediated transactivation process includes Src kinase, which has been shown to be associated with the carboxyl-terminal region of the HER2 receptor through its SH2 domain. Src kinase was present in lipid rafts as well as in nonrafts in prostate cancer cells (Fig. 3). However, CXCL12 treatment enhanced lipid raft–associated Src phosphorylation at Tyr416 without significantly enhancing nonraft-associated Src in C4-2B cells. Pharmacologic inhibition of Src kinase activation by PP2 compound abrogated the basal as well as CXCL12-induced HER2 phosphorylation in lipid rafts of prostate cancer cells. Furthermore, PP2 compound inhibited the constitutive Tyr1248 phosphorylation of HER2 in nonraft fractions in prostate cancer cells, suggesting the crucial role of Src in HER2 receptor activity. Based on the pharmacologic inhibition of Src data, we conclude that Src is a mediator of CXCL12-induced HER2 activation in lipid rafts of prostate cancer cells. This Src interaction with HER2 seems to be required for HER2 receptor – mediated invasive and migratory properties of epithelial cells (34). In breast cancer cells, Src involvement has been documented in the CXCR4 and HER2 transactivation, and in these cells HER2 abundance dictates the transactivation process (7). We have consistently observed the presence of higher levels of phosphorylated HER2 in non–lipid raft fractions (Fig. 2). The Src kinase inhibitor PP2 abrogated the phosphorylation of HER2 in these fractions, which are lacking lipid rafts (Fig. 3). Activated Src has been shown to be physically associated with EGFR family members, and this association further activates the receptor leading to phosphorylation of downstream targets. Src also actively participates in sorting of activated growth factor receptors into internalizing endosomes and further degradation (35). Our data with Src inhibitors further suggest that the presence of phosphorylated HER2 in the cytosol and membrane fractions is the consequence of known downstream processes of growth factor receptors. Higher phosphorylated HER2 in cell surface fractions (Fig. 2A), which are devoid of lipid rafts, could potentially result in the presence of autocrine loop involving growth factor ligands that are shown to be expressed in prostate cancer cells (33). Src protein has been shown to be associated with lipid rafts so its involvement in CXCR4-mediated HER2 transactivation in prostate cancer cells seems to be relevant to prostate cancer cell invasion and metastasis.
Pharmacologic inhibition of expression and activation of HER family members leads to complete abrogation of MMP-9 secretion from PC-3 cells (Fig. 5A). These data are inline with previously published reports showing the CXCL12/CXCR4–mediated expression of MMP-9 in cancer cells (2, 36–40). Furthermore, these data suggest that CXCL12/CXCR4 transactivation of HER2 is an upstream event of Akt activation in prostate cancer cells. Several studies further suggest that MMP-9 plays a key role in tumor cell migration and invasion by remodeling the matrix; this fact is further supported by MMP-9 knockdown studies (41–43). In prostate cancer bone metastasis, MMP-9 plays a key role in the initial establishment of metastatic formation by fostering the tumor cell–induced pathophysiologic matrix remodeling and subsequent expansion of tumor deposits in bone tissue (42). Thus, targeting the CXCL12/CXCR4/HER2 pathway as an upstream activator of MMP-9 expression is an attractive avenue for the clinical management of prostate cancer bone metastasis.
Lipid raft–destabilizing agents, such as MBCD and filipin, are known to phosphorylate EGFR in cells (44–46). Interestingly, MBCD treatment with PC-3 cells inhibited basal and CXCL12-induced HER2 phosphorylation in lipid rafts. CXCR4 levels were slightly decreased in lipid rafts and concomitantly enhanced in cytosol and membrane fractions upon MBCD treatment (Fig. 6A). These data suggest that lipid rafts are key sites for CXCL12/CXCR4 transactivation of HER2 in prostate cancer cells. As a consequence of lipid raft disruption, EGFR downstream signaling pathways were inhibited in prostate cancer cells (47). Our data further show that cellular consequences of lipid raft disruption resulted in an inhibition of basal and CXCL12-mediated chemoinvasion of PC-3 cells (Fig. 6B), suggesting the key role of lipid raft signaling in prostate cancer metastasis to bone microenvironment.
A recent report further supports the role of CXCL12/CXCR4 in prostate cancer bone metastasis using the intratibial model in which inhibition of CXCR4 function reduced bone tumor growth (4). Experimental manipulation of CXCR4 also affects secondary tumor growth in lymph nodes. CXCR4 overexpression enhanced lymphatic metastasis of oral squamous cancer cells by activating Akt and Erk kinase pathways; inhibition of both pathways by pharmacologic inhibitors reduced metastasis (48). Similarly, neutralization of CXCR4 function by antibody or small interfering RNA–mediated approaches reduced metastasis formation in several model systems (4, 49–51). Collectively, these studies suggest that the CXCL12/CXCR4 axis not only affects “homing” but also tumor growth in organ microenvironments rich in CXCL12. Our data further suggest that CXCL12/CXCR4–mediated transactivation of growth factor signaling promotes expansion of the metastatic deposit. Although our in vitro data strongly support the CXCL12/CXCR4 transactivation of HER2 enhanced in CXCR4 transfectants, the in vivo data can be viewed as this enhanced transactivation contributing to intraosseous tumor growth. In the absence of neutralization strategy of CXCL12/CXCR4/HER2/pro-MMP9 axis and lipid rafts in the in vivo model system, we cannot rule out the existence of alternate mechanisms downstream of CXCL12/CXCR4/HER2, contributing to enhanced intraosseous tumor growth in prostate cancer bone metastasis.
Based on our results, a mechanistic pathway of CXCR4-mediated metastatic tumor growth begins with CXCR4 and HER2 localization in lipid rafts of prostate cancer cells. In high CXCL12 microenvironments (e.g., bone and lymph nodes), binding of CXCL12 to CXCR4 leads to transactivation of HER2 receptor via Src kinase activation, Akt signaling, MMP-9 expression, cancer cell invasion, and ultimately expansion of the metastatic tumor. The lipid raft–destabilizing agent (BMCD), EGFR family inhibitor, and Src inhibitor could disrupt lipid raft–associated CXCR4 signaling events in prostate cancer cells (Fig. 11). In the bone microenvironment, this pathway is associated with rapid expansion of the metastatic deposit and matrix degradation. Our observations help provide a mechanistic link between CXCL12/CXCR4 signaling and transactivation of HER2 in prostate cancer bone metastasis.
Materials and Methods
Cell Culture
PC-3 cells were obtained from American Type Culture Collection, and C4-2B cells were obtained from Dr. Leland Chung (Emory University of School of Medicine, Atlanta, GA; ref. 52). PC-3 cells were cultured in RPMI 1640 and C4-2B cells were cultured in T medium (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin.
Cell Surface Biotinylation, Lipid Raft Fractionation, and Western Blot Analysis
PC-3 and C4-2B cells were washed with PBS and biotinylated with 0.5 mg/mL of EZ-Link Sulfo-NHS-SS-Biotin (Pierce Biotechnology, Inc.) for 30 min at 4°C. Cells were washed with ice-cold PBS and quenched with 50 mmol/L NH4Cl for 10 min at 4°C. The lipid raft fractionation was done as previously described (2). Triton X-100– soluble fractions containing cytosolic and membrane proteins that are devoid of lipid rafts were termed as the “cytosol and membrane” fraction, whereas Triton X-100–insoluble but β-octylglucoside–soluble fractions were termed as the “lipid raft” fraction. The soluble fractions containing lipid rafts, and the cytosol and membrane fractions, were diluted with NP40 lysis buffer containing 25 mmol/L Tris (pH 7.5), 1% NP40, 100 mmol/L NaCl, and 1× protease inhibitor cocktail. Biotinylated proteins were incubated with Avidin beads (Pierce Biotechnology) overnight, and bead complexes were washed seven times with buffer containing 0.5% SDS, 60 mmol/L Tris (pH 7.5), 2 mmol/L EDTA, and 2.5% Triton X-100. The pellet containing biotinylated lipid raft and nonraft cell surface proteins were subjected to immunoblot analysis with antibodies against CXCR4 (Chemicon International, Inc.), HER2, and pHER21248 (Upstate USA, Inc.). After secondary antibody incubation, chemiluminescence reaction was done with SuperSignal Western femto or pico substrate (Pierce Biotechnology). The band intensities were determined by quantitation of pixel intensities using Un-Scan-It software (version 5.1; Orem).
Sucrose Density Centrifugation
PC-3 and C4-2B cells were washed with PBS and cell pellets were resuspended in 1 mL of cell lysis buffer [1% Triton X-100, 25 mmol/L MES (pH 6.5), 150 mmol/L NaCl, and protease and phosphatase inhibitors]. Cells were homogenized with the Dounce homogenizer followed by passing the cell lysates through a 22-gauge needle 10 times. The cell lysates were mixed with equal volume of 70% sucrose, placed in a centrifuge tube, and layered with 4 mL of 30% sucrose and 3 mL of 5% sucrose. The tubes were centrifuged at 100,000 × g for 16 h. One-milliliter fractions were collected from the top of the tube and Western blot analyses were done with antibodies against HER2, CXCR4, and Gi-α3 proteins (Calbiochem).
Cell Proliferation Assay
PC-3 cells (1 × 104) were seeded in a 96-well plate. The following day, cells were exposed to different concentrations of CI-1033 (Pfizer), which was dissolved in sterile water. After 24 h, cells were washed with PBS and exposed to tetrazolium salt WST-1. WST-1 was cleaved to form soluble yellow formazan product by viable cells and the formazan product was measured in a 96-well plate reader per manufacturer’s recommendations (Roche Diagnostics).
Transfection of PC-3 Cells and Characterization of CXCR4-Expressing Clones
PC-3 cells were transfected with pCDNA3 (Invitrogen Life Technologies) and pCDNA3-CXCR4 (53) expression vectors using the Lipofectamine reagent. Forty-eight hours after transfection, cells were exposed to neomycin at a concentration of 500 μg/mL. Distinct clones developed from a single transfected cell were picked and expanded. The clone isolated from the pCDNA3 was termed PC-3 Neo and pCDNA3-CXCR4 was termed PC-3 CXCR4-2.1 and PC-3 CXCR4-2.3. Fluorescence-activated cell sorting analysis and chemoinvasion studies were done on clones as previously described (2).
Gelatin Zymography
PC-3 cells were serum starved for 16 h and were exposed to CXCL12 and CI-1033 compound. Equal volumes and/or equal amounts of protein from particulate-free conditioned medium were analyzed for gelatin zymography as previously described (2).
Immunohistochemistry of Severe Combined Immunodeficient–Human PC-3 Bone Tumors
PC-3 transfectants (1 × 106) were injected into human fetal femur fragments previously implanted into severe combined immunodeficient mice according to previously described methods (26). Each cell line tested in the experiment was injected in six animals, which were previously implanted with bone fragments. Three weeks later, the bones were excised, fixed, decalcified, sectioned, and stained with H&E, and antibodies against cytokeratin and Ki-67 antigen using the Vecta Stain ABC kit (Vector Laboratories, Inc.).
Histomorphometry
Digital photomicrographs of the entire histologic section were captured at multiple fields at ×5 magnification using AxioVision software (Carl Zeiss). All the fields were merged to reconstruct a digital picture of the entire histologic section using Adobe Photoshop 7.0 (Adobe Systems). Tumor tissue (cytokeratin positive areas) and trabecular bone were isolated into separate layers and their areas were quantitated. The whole tissue cross-sectional area (considered 100%) was then highlighted and the area occupied by either tumor or bone was calculated using Adobe Photoshop 7.0 software.
Statistical Analysis
Statistical significance was determined by the nonparametric ANOVA test followed by Tukey posttest to compare all pairs of a column using GraphPad Prism software version 3.0 (GraphPad). P < 0.05 was considered to be statistically significant.
Acknowledgments
Grant support: New Investigator award from the Department of Defense DAMD17-03-1-0120 (S.R. Chinni) and RO1 DK 067687 from the NIH (M.L. Cher).
We thank Dr. Stephen C. Peiper (Medical College of Georgia, Augusta, GA) for providing pCDNA3-CXCR4 expression vector; Drs. Stephen Ethier (Wayne State University, Detroit, MI) and Richard Leopold (Pfizer, Ann Arbor, MI) for providing the CI-1033 compound; Sivasakthy Sivalogan, Juan Cai, and Allen Saliganan for expert technical assistance; and Pridvi Kandagatla for Ki-67 analysis of PC-3 bone tumor tissues.
References
- 1.Zlotnik A. Chemokines in neoplastic progression. Semin Cancer Biol. 2004;14:181– 5. doi: 10.1016/j.semcancer.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 2.Chinni SR, Sivalogan S, Dong Z, et al. CXCL12/CXCR4 signaling activates Akt-1 and MMP-9 expression in prostate cancer cells: the role of bone microenvironment-associated CXCL12. Prostate. 2006;66:32– 48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]
- 3.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832– 7. [PubMed] [Google Scholar]
- 4.Sun YX, Schneider A, Jung Y, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res. 2005;20:318– 29. doi: 10.1359/JBMR.041109. [DOI] [PubMed] [Google Scholar]
- 5.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203– 8. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 6.Luttrell DK, Luttrell LM. Not so strange bedfellows: G-protein-coupled receptors and Src family kinases. Oncogene. 2004;23:7969– 78. doi: 10.1038/sj.onc.1208162. [DOI] [PubMed] [Google Scholar]
- 7.Cabioglu N, Summy J, Miller C, et al. CXCL-12/stromal cell-derived factor-1α transactivates HER2-neu in breast cancer cells by a novel pathway involving Src kinase activation. Cancer Res. 2005;65:6493– 7. doi: 10.1158/0008-5472.CAN-04-1303. [DOI] [PubMed] [Google Scholar]
- 8.Porcile C, Bajetto A, Barbieri F, et al. Stromal cell-derived factor-1α (SDF-1α/CXCL12) stimulates ovarian cancer cell growth through the EGF receptor transactivation. Exp Cell Res. 2005;308:241– 53. doi: 10.1016/j.yexcr.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 9.Spencer KS, Graus-Porta D, Leng J, Hynes NE, Klemke RL. ErbB2 is necessary for induction of carcinoma cell invasion by ErbB family receptor tyrosine kinases. J Cell Biol. 2000;148:385– 97. doi: 10.1083/jcb.148.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhan L, Xiang B, Muthuswamy SK. Controlled activation of ErbB1/ErbB2 heterodimers promote invasion of three-dimensional organized epithelia in an ErbB1-dependent manner: implications for progression of ErbB2-overexpressing tumors. Cancer Res. 2006;66:5201– 8. doi: 10.1158/0008-5472.CAN-05-4081. [DOI] [PubMed] [Google Scholar]
- 11.Woods Ignatoski KM, Livant DL, Markwart S, Grewal NK, Ethier SP. The role of phosphatidylinositol 3′-kinase and its downstream signals in erbB-2-mediated transformation. Mol Cancer Res. 2003;1:551– 60. [PubMed] [Google Scholar]
- 12.Meric-Bernstam F, Hung MC. Advances in targeting human epidermal growth factor receptor-2 signaling for cancer therapy. Clin Cancer Res. 2006;12:6326– 30. doi: 10.1158/1078-0432.CCR-06-1732. [DOI] [PubMed] [Google Scholar]
- 13.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517– 27. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 14.Gregory CW, Whang YE, McCall W, et al. Heregulin-induced activation of HER2 and HER3 increases androgen receptor transactivation and CWR-R1 human recurrent prostate cancer cell growth. Clin Cancer Res. 2005;11:1704– 12. doi: 10.1158/1078-0432.CCR-04-1158. [DOI] [PubMed] [Google Scholar]
- 15.Edwards J, Traynor P, Munro AF, Pirret CF, Dunne B, Bartlett JM. The role of HER1-4 and EGFRvIII in hormone-refractory prostate cancer. Clin Cancer Res. 2006;12:123– 30. doi: 10.1158/1078-0432.CCR-05-1445. [DOI] [PubMed] [Google Scholar]
- 16.Carles J, Lloreta J, Salido M, et al. Her-2/neu expression in prostate cancer: a dynamic process? Clin Cancer Res. 2004;10:4742– 5. doi: 10.1158/1078-0432.CCR-04-0115. [DOI] [PubMed] [Google Scholar]
- 17.Solit DB, Rosen N. Targeting HER2 in prostate cancer: where to next? J Clin Oncol. 2007;25:241– 3. doi: 10.1200/JCO.2006.08.8187. [DOI] [PubMed] [Google Scholar]
- 18.Agus DB, Sweeney CJ, Morris MJ, et al. Efficacy and safety of single-agent pertuzumab (rhuMAb 2C4), a human epidermal growth factor receptor dimerization inhibitor, in castration-resistant prostate cancer after progression from taxane-based therapy. J Clin Oncol. 2007;25:675– 81. doi: 10.1200/JCO.2006.07.0649. [DOI] [PubMed] [Google Scholar]
- 19.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31– 9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 20.Couet J, Sargiacomo M, Lisanti MP. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem. 1997;272:30429– 38. doi: 10.1074/jbc.272.48.30429. [DOI] [PubMed] [Google Scholar]
- 21.Waugh MG, Lawson D, Hsuan JJ. Epidermal growth factor receptor activation is localized within low-buoyant density, non-caveolar membrane domains. Biochem J. 1999;337:591– 7. [PMC free article] [PubMed] [Google Scholar]
- 22.Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res. 2002;62:2227– 31. [PubMed] [Google Scholar]
- 23.Adam RM, Mukhopadhyay NK, Kim J, et al. Cholesterol sensitivity of endogenous and myristoylated Akt. Cancer Res. 2007;67:6238– 46. doi: 10.1158/0008-5472.CAN-07-0288. [DOI] [PubMed] [Google Scholar]
- 24.Smaill JB, Rewcastle GW, Loo JA, et al. Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenyl-amino)quinazoline- and 4-(phenylamino)pyrido[3,2-d]pyrimidine-6-acrylamides bearing additional solubilizing functions. J Med Chem. 2000;43:1380– 97. doi: 10.1021/jm990482t. [DOI] [PubMed] [Google Scholar]
- 25.Nelson JM, Fry DW. Akt, MAPK (Erk1/2), and p38 act in concert to promote apoptosis in response to ErbB receptor family inhibition. J Biol Chem. 2001;276:14842– 7. doi: 10.1074/jbc.M008786200. [DOI] [PubMed] [Google Scholar]
- 26.Nemeth JA, Harb JF, Barroso U, Jr, He Z, Grignon DJ, Cher ML. Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 1999;59:1987– 93. [PubMed] [Google Scholar]
- 27.Wysoczynski M, Reca R, Ratajczak J, et al. Incorporation of CXCR4 into membrane lipid rafts primes homing-related responses of hematopoietic stem/progenitor cells to an SDF-1 gradient. Blood. 2005;105:40– 8. doi: 10.1182/blood-2004-04-1430. [DOI] [PubMed] [Google Scholar]
- 28.van Buul JD, Voermans C, van Gelderen J, Anthony EC, van der Schoot CE, Hordijk PL. Leukocyte-endothelium interaction promotes SDF-1-dependent polarization of CXCR4. J Biol Chem. 2003;278:30302– 10. doi: 10.1074/jbc.M304764200. [DOI] [PubMed] [Google Scholar]
- 29.Sun YX, Wang J, Shelburne CE, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89:462– 73. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- 30.Nagy P, Vereb G, Sebestyen Z, et al. Lipid rafts and the local density of ErbB proteins influence the biological role of homo- and heteroassociations of ErbB2. J Cell Sci. 2002;115:4251– 62. doi: 10.1242/jcs.00118. [DOI] [PubMed] [Google Scholar]
- 31.Mineo C, Gill GN, Anderson RG. Regulated migration of epidermal growth factor receptor from caveolae. J Biol Chem. 1999;274:30636– 43. doi: 10.1074/jbc.274.43.30636. [DOI] [PubMed] [Google Scholar]
- 32.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127– 37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 33.Torring N, Jorgensen PE, Sorensen BS, Nexo E. Increased expression of heparin binding EGF (HB-EGF), amphiregulin, TGF α and epiregulin in androgen-independent prostate cancer cell lines. Anticancer Res. 2000;20:91– 5. [PubMed] [Google Scholar]
- 34.Kim H, Chan R, Dankort DL, et al. The c-Src tyrosine kinase associates with the catalytic domain of ErbB-2: implications for ErbB-2 mediated signaling and transformation. Oncogene. 2005;24:7599– 607. doi: 10.1038/sj.onc.1208898. [DOI] [PubMed] [Google Scholar]
- 35.Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004;6:209– 14. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Chu H, Zhou H, Liu Y, Liu X, Hu Y, Zhang J. Functional expression of CXC chemokine recepter-4 mediates the secretion of matrix metalloproteinases from mouse hepatocarcinoma cell lines with different lymphatic metastasis ability. Int J Biochem Cell Biol. 2007;39:197– 205. doi: 10.1016/j.biocel.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Redondo-Munoz J, Escobar-Diaz E, Samaniego R, Terol MJ, Garcia-Marco JA, Garcia-Pardo A. MMP-9 in B-cell chronic lymphocytic leukemia is up-regulated by α4β1 integrin or CXCR4 engagement via distinct signaling pathways, localizes to podosomes, and is involved in cell invasion and migration. Blood. 2006;108:3143– 51. doi: 10.1182/blood-2006-03-007294. [DOI] [PubMed] [Google Scholar]
- 38.Brand S, Dambacher J, Beigel F, et al. CXCR4 and CXCL12 are inversely expressed in colorectal cancer cells and modulate cancer cell migration, invasion and MMP-9 activation. Exp Cell Res. 2005;310:117– 30. doi: 10.1016/j.yexcr.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Singh S, Singh UP, Grizzle WE, Lillard JW., Jr CXCL12-4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84:1666– 76. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- 40.Samara GJ, Lawrence DM, Chiarelli CJ, et al. CXCR4-mediated adhesion and MMP-9 secretion in head and neck squamous cell carcinoma. Cancer Lett. 2004;214:231– 41. doi: 10.1016/j.canlet.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 41.Lakka SS, Gondi CS, Yanamandra N, et al. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene. 2004;23:4681– 9. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- 42.Dong Z, Bonfil RD, Chinni S, et al. Matrix metalloproteinase activity and osteoclasts in experimental prostate cancer bone metastasis tissue. Am J Pathol. 2005;166:1173– 86. doi: 10.1016/S0002-9440(10)62337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanceau J, Truchet S, Bauvois B. Matrix metalloproteinase-9 silencing by RNA interference triggers the migratory-adhesive switch in Ewing’s sarcoma cells. J Biol Chem. 2003;278:36537– 46. doi: 10.1074/jbc.M304300200. [DOI] [PubMed] [Google Scholar]
- 44.Westover EJ, Covey DF, Brockman HL, Brown RE, Pike LJ. Cholesterol depletion results in site-specific increases in epidermal growth factor receptor phosphorylation due to membrane level effects. Studies with cholesterol enantiomers. J Biol Chem. 2003;278:51125– 33. doi: 10.1074/jbc.M304332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X, Resh MD. Cholesterol depletion from the plasma membrane triggers ligand-independent activation of the epidermal growth factor receptor. J Biol Chem. 2002;277:49631– 7. doi: 10.1074/jbc.M208327200. [DOI] [PubMed] [Google Scholar]
- 46.Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006;168:1107– 18. doi: 10.2353/ajpath.2006.050959. quiz 1404 – 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oh HY, Lee EJ, Yoon S, Chung BH, Cho KS, Hong SJ. Cholesterol level of lipid raft microdomains regulates apoptotic cell death in prostate cancer cells through EGFR-mediated Akt and ERK signal transduction. Prostate. 2007;67:1061– 9. doi: 10.1002/pros.20593. [DOI] [PubMed] [Google Scholar]
- 48.Uchida D, Begum NM, Tomizuka Y, et al. Acquisition of lymph node, but not distant metastatic potentials, by the overexpression of CXCR4 in human oral squamous cell carcinoma. Lab Invest. 2004;84:1538– 46. doi: 10.1038/labinvest.3700190. [DOI] [PubMed] [Google Scholar]
- 49.Muller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50– 6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 50.Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65:967– 71. [PMC free article] [PubMed] [Google Scholar]
- 51.Lapteva N, Yang AG, Sanders DE, Strube RW, Chen SY. CXCR4 knockdown by small interfering RNA abrogates breast tumor growth in vivo. Cancer Gene Ther. 2005;12:84– 9. doi: 10.1038/sj.cgt.7700770. [DOI] [PubMed] [Google Scholar]
- 52.Thalmann GN, Anezinis PE, Chang SM, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577– 81. [PubMed] [Google Scholar]
- 53.Lu Z, Berson JF, Chen Y, et al. Evolution of HIV-1 coreceptor usage through interactions with distinct CCR5 and CXCR4 domains. Proc Natl Acad Sci U S A. 1997;94:6426– 31. doi: 10.1073/pnas.94.12.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]