

Abstract
Goals
The aim of this report is to delineate the clinical, pathological and enteroendocrine features of PC1/3 deficiency in children
Background
Prohormone convertases play a pivotal role in the activation of biologically inactive hormones. Congenital defects in the enteroendocrine axis such as Prohormone Convertase 1/3 (PC1/3) deficiency have been rarely reported and their pathophysiological mechanisms are largely unknown.
Study
Enteroendocrine function and pathology was evaluated in four males (1, 2, 7 and 10 years old) from 2 families with PC1/3 deficiency at a university children’s hospital. Clinical course, pathology analysis including immunohistochemistry for PC1/3, PC2 and glucagon like peptide 1 (GLP1) and electron microscopy (EM) as well as enteroendocrine function tests (GLP 1, GLP2, oral glucose tolerance test – OGTT) were performed.
Results
All (n=4) suffered from congenital severe diarrhea associated with malabsorption. The diarrhea improved during the first year of life and hyperphagia with excessive weight gain (BMI >97th percentile) became the predominant phenotype at an older age. Analysis of the entero-endocrine axis revealed high pro-insulin levels (57 – 1,116 pmol/l) in all patients, low serum GLP2 levels and impaired insulin and GLP1 secretion following an OGTT at a young age, with improvement in one older child tested. EM showed normal ultrastructure of enterocytes and enteroendocrine cells (EE). Immunohistochemistry revealed normal expression of chromogranin A, a marker of EE cells but markedly reduced immunostaining for PC1/3 and PC2 in all patients.
Conclusions
PC1/3 deficiency is associated with an age dependent, variable clinical phenotype caused by severe abnormalities in intestinal and enteroendocrine functions. Serum level of pro-insulin can be used as an effective screening tool.
Keywords: enteroendocrine cells, insulin, obesity, children, prohormone convertase deficiency, congenital diarrhea, malabsorption
INTRODUCTION
Prohormone convertases are essential enzymes for the cleavage of multiple prohormones into their active hormonal form. Prohormone convertase 1/3 (PC1/3), encoded by PCSK1, is expressed in a variety of neuroendocrine tissues including brain, pituitary gland, adrenal glands, intestinal L-cells and endocrine pancreatic alpha and beta cells. This convertase activates multiple substrates, such as proglucagon, pro-insulin, and propiomelanocortin, to their active form.1
Until recently, three patients had been described with a mutation in PC1/3.2–5 Two of these patients suffered from congenital watery diarrhea while abdominal bloating and loose stools were reported in the third patient during adulthood. Data from one infant, indicated a malabsorption of monosaccharides (fructose and glucose) and fat early in life.3 Whereas impaired growth is a typical finding in the neonatal and infantile period, severe obesity was found in two of the affected patients who survived beyond the neonatal period.4;5 We recently described a comprehensive analysis of the genotype-phenotype characteristics of 13 children with PC1/3 deficiency, including four described in this current manuscript.6 However, the evolution of the clinical phenotype over time in patients with PC1/3 deficiency, the impact of PCSK1 mutation on enteroendocrine function, pathological characteristics and the underlying mechanisms of disease are largely unknown.
Multiple peptide hormones are produced in the gastrointestinal and pancreatic system to aid in the regulation of energy homeostasis, metabolism and gut development. PC1/3 is required for the processing of some of these hormones, including insulin 7 and glucagon-like peptides 1 and 2 (GLP-1 and GLP-2). GLP-1 and GLP-2 are secreted locally by intestinal L-cells following nutrient ingestion, and have a wide range of activity, including stimulation of insulin synthesis and secretion, regulation of intestinal absorption, motility and proliferation and appetite control.8–12 GLP-1 and 2 are absent in intestinal tissues from PCSK1 knockout mice,13 but it is not known if GLP-1 and GLP-2 production is affected in children with PC1/3 deficiency.
The aims of this study were to describe the clinical course over time in PC 1/3 deficient patients and investigate the impact of PC1/3 deficiency on intestine, enteroendocrine (EE) and endocrine pancreas beta cell function in four pediatric patients with a mutation in the PC1/3 gene. To the best of our knowledge, this is the first detailed report on intestine pathology and on EE and pancreatic beta cell function in children with PC1/3 deficiency.
MATERIALS AND METHODS
Subjects
A retrospective chart review was performed to gather data on family history and progression of clinical course over time including symptoms, development of new pathologies, trends of weight and height, stool consistency and frequency, total parenteral nutrition requirements and enteral food intake characteristics. Where appropriate, z-scores were calculated.14 Results of previously undertaken diagnostic tests were collected and analyzed. Informed consent was obtained from each of the primary caretakers to undertake the diagnostic investigations and for publication of the data.
Histology
Per-endoscopic biopsies of duodenum (patients 1, 2, 3) and terminal ileum (patients 1, 2) were fixed in 10% neutral-buffered formalin, embedded in paraffin, sectioned and stained with haematoxylin and eosin using routine procedures. Age-matched controls with normal intestinal biopsies and unremarkable past medical history were selected.
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded sections, according to established protocols. The tissue sections were immunostained with antibodies against PC1 (Chemicon International, Temecula, CA, 1:200 dilution in DAKO antibody diluent), PC2 (Chemicon International, Temecula, CA, 1:200) and GLP1 (kindly provided by Dr. Daniel Drucker, University of Toronto, Canada, 1:1000), using Ventana automated immunostainer with Ultraview Ventana Kit. Microscopic images were visualized and captured using a Nikon Eclipse E600 microscope. Sections from each patient were compared to sections from the healthy age- matched controls. Quantification of immunopositive cells for each antibody was performed in a blinded fashion by an experienced pathologist (RC). The number of immunopositive cells per 20 well-oriented crypts was counted and the mean values for the patients group and controls were calculated.
Transmission electron microscopic (EM) assessment of intestinal EE cells was performed on samples fixed in universal fixative and embedded in Epon, according to standard protocol. Epon blocks cut at one micron in thickness were stained with toluidine blue to select representative areas of intestinal mucosa. Ultra-thin sections were contrasted with uranyl acetate and lead citrate and examined under a JEOL 1011 electron microscope at an accelerating voltage of 80 kV.
Oral glucose tolerance test
Subjects were admitted to the Endocrinology Testing Unit at the Hospital for Sick Children. After an overnight fast, a dextrose solution was given orally at a dose of 1.75 g/kg body weight to a maximum of 75 g. Blood samples were then taken at t=0, 15, 30, 60, 90 and 120 min., plasma was separated by centrifugation, and then stored at −80° C until analysis. Samples were analyzed for glucose, insulin, C-peptide and GLP-1 levels. The homeostatic model assessment (HOMA), a measure of the degree of insulin resistance, was calculated as fasting glucose × fasting insulin, divided by 22.5. The insulinogenic index, a measure of the insulin secretory response, was calculated as insulin (milliunits per liter) at 30 min- fasting insulin (milliunits per liter)/glucose (millimoles per liter) at 30 min – fasting glucose (millimoles per liter). Using values derived from the oral glucose tolerance test, whole body insulin sensitivity (WBISI) was calculated using the Matsuda index: 10 000/√ [(fasting glucose × fasting insulin) × (mean glucose × mean insulin)] where glucose is measured in mg/dl and insulin in lU/ml.15 Test result of the patients were compared to published values in children and with those obtained in two healthy subjects, age 29 and 37, BMI 19 and 20, respectively.
Biochemical analyses
Plasma levels of bioactive GLP-1 (7–36NH2), insulin, glucagon and leptin were determined using a multiplex sandwich-immunoassay (Meso Scale Discovery, Gaithersburg, MD). C-peptide and pro-insulin concentrations were determined using a commercial radio-immunoassay (Millipore, Billerica, MA). Pro-insulin does not cross-react with insulin in this assay. Active GLP-2 levels were measured following a standard meal, according to Hartmann et al.16
Statistical analyses
Most analyses are descriptive considering the small sample size. A t-test was performed to compare the immunohistochemistry results in duodenal biopsies between patients and controls.
RESULTS
Genotype and clinical phenotype
Two families were identified, each with two affected male siblings (family A: patient 1 - 1 year old and patient 2 - 10.8 year old; family B: patient 3 - 2.2 year old and patient 4 - 7.8 year old). The two youngest patients were referred to the Hospital for Sick Children as neonates. Family A is of Arab origin and the parents are second cousins; Family B is of African origin and the parents are not known to be consanguineous. Genetic testing revealed homozygote true null mutations in all four patients; patient 1 and 2: p.M1X, c.2T>C; patient 3 and 4: p.R405X, c.1213C>T.6 As we reported earlier, the p.M1X mutation alters the PCSK1’s first amino acid, or the initiation of translation codon, and the subsequent in-frame substitute codon is located within exon 3 at M125. The p.R405X mutation results in an entire deletion of the protein’s P and C-terminal tail domains. We did not assess the functional consequence of the p.M1X mutation since it does not result in a protein, and the recombinant PC1/3 protein that contained the p.R405X mutation was secreted by HEK cells, but lacked any enzymatic activity. These data that were recently published demonstrates that the four affected subjects have a true PC1/3 null genotype. All four patients were born at term with normal birth weight (mean 3.3 kg, range 2.95 – 3.7) without dysmorphic features (Table 1). Although there was no history of polyhydramnios indicative of diarrhea in utero, from day one of life each newborn had osmotic diarrhea leading to hospital admissions between days 3 and 48 of life due to weight loss and dehydration accompanied by systemic metabolic acidosis and hypoglycemia. Three patients (1, 2, 3) required total parenteral nutrition during the first year of life to ensure adequate hydration and normal linear growth velocity. In the fourth patient, despite the diarrhea the mother was able to replace all fluid and caloric losses orally.
TABLE 1.
Clinical characteristics at different age groups of 4 patients with PC1/3 deficiency
| Characteristic | Unit | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|---|
| Current age | years | 1 | 10.8 | 2.2 | 7.8 |
| Ethnicity | Arab | Arab | African | African | |
| Gender | male | male | male | male | |
| Birth weight | kg | 2.95 | 3.13 | 3.55 | 3.7 |
| Duration of pregnancy | weeks | 37 | 42 | 40 | 40 |
| Age at presentation | days | 9 | 13 | 3 | 48 |
| Stool consistency at presentation | watery | watery | watery | watery | |
| Duration of TPN | months | 12 | 11 | 20 | Not required |
| Current stool frequency | x/day | 1–2 | 1 | 3 | 2–3 |
| Current stool consistency | loose-formed | loose | formed | formed | |
| Current weight (z-score) | kg | 9.1 (−1.3) | 66.1 (2.4) | 14.1 (0.6) | 38 (2.0) |
| Current height (z-score) | cm | 73 (−1.1) | 139 (−0.5) | 83.4 (−1.6) | 126.5 (−0.1) |
| Current BMI (z-score) | 17.(1.2) | 35.7 (>3) | 20.3 (2.8) | 24.3 (>3) | |
| Proinsulin (norm 6.4–9.4 pmol/l) | pmol/l:age in years | 71 : 0.33 550 : 0.83 | 1116 : 10 | 681 : 1.75 | 539 : 7.2 |
Elemental formula was introduced in all 3 patients, who were on TPN, as diarrhea improved with age. Stool consistency and frequency improved in all children after the first year of life. Poor weight gain and impaired linear growth have improved over the first year of life, leading to a gradual taper and then discontinuation of TPN (Table 1). Features of significant weight gain despite tapering of TPN were evident in all patients (Fig. 1). Patients 3 and 4 had crossed above the 50th percentile for weight by months of age (Fig. 1A). The two older patients (2, 4) had crossed the 97th percentile for weight by 5 yrs of age (Fig. 1B). Hyperphagia was evident in all children after the first year of life.
Figure 1.

Representative growth charts for weight of patients with PC1/3 deficiency at different ages (Fig 1A; 0– 30 months, Fig 1B; 2–11 years). Patient 1 (▲), patient 2 (■), patient 3 (●) and patient 4 (⋆).
Dysmorphic facial features were not present in any of the patients; however, micropenis was detected in patient 4 and genu varum developed in two patients (2, 3) since they began walking at age 15 and 13 months respectively. Involvement of hypothalamic-pituitary endocrine pathways was manifested to a variable extent at different ages. The two older patients (2, 4) had diabetes insipidus-like symptoms, with polyuria and nocturnal enuresis at age 9.5 and 4.5 years. However, both were able to concentrate their urine in a water deprivation test (serum sodium 142 mmol/l, serum osmolarity 294 mmol/l, urine osmolarity 595 mmol/l in patient 2 and serum sodium 144 mmol/l, serum osmolarity 313 mmol/l, urine osmolarity 543 mmol/l in patient 4) and patient 4 had a normal vasopressin concentration of 2.7 ng/l (norm <6.8 ng/l). Patient 2 was treated with desmopressin which improved his clinical symptoms. Growth hormone deficiency was confirmed in one patient (2) at 9 years of age by a provocative stimulation test (maximal growth hormone level 5.1 ug/l during clonidine/arginine stimulation test) and suspected in two others with appropriate nutritional status (1, 4) due to very low IGF-1 levels for age (<25 ug/l and 51 ug/l at age 1 year and 7.5 years, respectively).
Intestinal function
Fat malabsorption was tested in two patients (1, 4) at 2 months and 6 years of age, respectively. Both had elevated fecal fat content (18% and 19% of dietary intake, respectively; normal < 10%). Fecal elastase was measured in two patients (2, 3) at age 1 and 3 months, respectively and was within the normal range (355 ug/g and 288 ug/g, respectively, normal >200). Stool electrolytes were normal in all four patients at age 3 weeks- 5 months (sodium 28 – 90 mmol/l, potassium 14 – 45 mmol/l and chloride 15 – 31 mmol/l). Alpha -1-antitrypsin clearance was determined in one patient (4) with a normal result (< 1 ml/d, normal < 22) at age 2 months.
Brush border disaccharidases were measured in two patients (2, 3) at age 9 years and 1 month, respectively. Patient 2 had low lactase and sucrase activity on duodenal biopsy, whereas patient 3 low lactase, sucrase and maltase levels. Serum citrulline, measured as a marker of absorptive enterocyte mass were around the 25th percentile in three patients in whom it was measured, compared to published control levels (1 2–12 umol/l, 3 10–21 umol/l, 4 14 umol/l).17
Enteroendocrine and endocrine pancreas function
The two youngest patients (1 and 3) suffered from multiple hypoglycemic episodes in the first year of life. However, episodes of hypoglycemia were not reported after the first year of life in none of the patients. Proinsulin levels in serum and plasma were elevated in all four patients (range 57–1116 pmol/l; laboratory reference 6.4–9.4 pmol/l) at different ages (Table 1).
In three of 4 patients a glucose tolerance test was performed (Fig. 2). Venous access for repeated blood sampling was not obtained in the fourth child due to poor venous access (patient 3). Fasting C-peptide responses, prior to the glucose tolerance test, were undetectable in the youngest patient with a mild hypoglycemia (1). There were no clear abnormalities of glucose excursion throughout the testing. There was, however, a markedly low insulin and C-peptide response in the affected infant (1) and patient 4. By contrast, the oldest patient (2) had a response similar to the two control subjects. The calculated insulinogenic index was low in all patients, albeit lowest in patients 1 and 4, compared to controls and lower than previously described in non-obese children,18;19 findings that are consistent with impaired insulin secretion. Calculation of insulin sensitivity, by HOMA and WBISI, revealed age dependent results, with the younger child (1), demonstrating high insulin sensitivity and the older boy (2) demonstrating relatively low insulin sensitivity. The values in patient 4 were comparable to children of similar age and BMI without PC1/3 deficiency,20 and similar to adult control subjects depicted in Table 2.
Figure 2.

Glucose, glucagon, GLP-1, insulin and C-peptide response to an oral glucose tolerance test in three patients (patient 1, 2 and 4) with PC1/3 deficiency and two control subjects (C1, C2).
TABLE 2.
Parameters for insulin secretion and sensitivity during an oral glucose tolerance test in PC1/3 deficient patients and controls
| Glucose tolerance status |
Reference values |
Patient 1 | Patient 2 | Patient 4 | Control 1 | Control 2 |
|---|---|---|---|---|---|---|
| Fasting glucose (mmol/l) | 4.4–5.5 | 3.6 | 4.5 | 4.0 | 5.4 | 5.1 |
| Fasting C-peptide (nmol/l) | <0.90 | 0.00 | 0.67 | 0.10 | 0.21 | 0.39 |
| Fasting insulin (pg/ml) | <860 | 58 | 930 | 190 | 292 | 242 |
| HOMA | <3 | 0.3 | 5.4 | 1.0 | 2.0 | 1.6 |
| WBISI | 10.3 | 0.5 | 2.1 | 1.1 | 1.6 | |
| IGI | 0.88 | 5.78 | 2.33 | 26.68 | 14.08 |
HOMA, homeostatic model assessment; WBISI, whole body insulin sensitivity; IGI, insulinogenic index.
GLP-1 response was also profoundly impaired in the two young patients (1, 4) and low in the third, older patient (2). Serum glucagon concentrations were adequate for the levels of fasting glucose, indicating a normal glucagon response. GLP-2 levels were below the normal range 21 in all patients both in the fasting state and 45 minutes after ingestion of a standardized mixed meal: patient 1: 4 and 4 pmol/l; 2: 2 and 8 pmol/l; 3: 4 pmol/l (fasting); 4: 6 and 12 pmol/l. Leptin concentrations were 29.6 µg/l in patient 2, 9.1 µg/l in patient 4 and 1.3 µg/l in patient 1, in the normal range for healthy children based on age and BMI, as provided in Table 1.22
Pathology findings
Histopathology
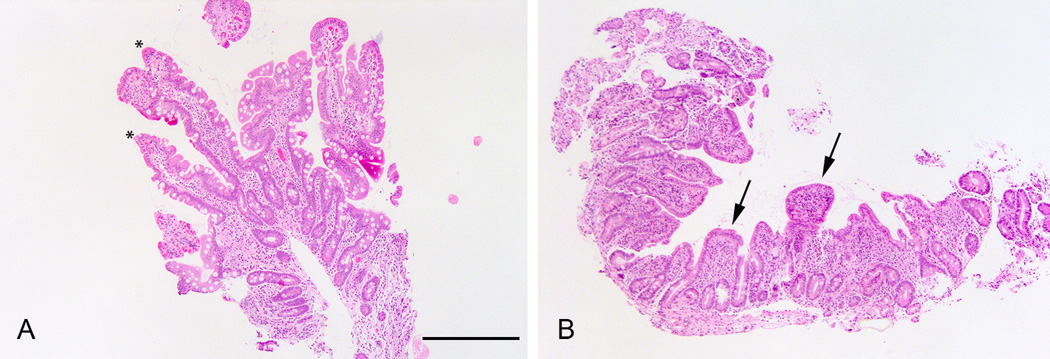
H&E stained sections from both duodenal and ileal biopsies of patients 1 and 2 showed an intestinal mucosa with well preserved villi and intact lining surface epithelium without inflammation in the underlying lamina propria. The duodenal biopsy from patient 3 showed mild to moderate villous atrophy with well preserved enterocytes without mucosal inflammation (Fig. 3).
Figure 3.

Duodenal histology (Hematoxylin & Eosin, magnification bar for A&B, 500 microns) of patient 2 (A) showing normal length villi (asterixes) and patient 3 showing blunted villi (B, arrows).
Immunohistochemistry
Sections of duodenum immunostained for chromogranin A (CGA) and serotonin (5-HT, not shown), used as general markers for EE cells showed numerous scattered immunopositive cells in biopsies from all patients in a distribution and number that is comparable to the age-matched controls (Figs. 4A&B). Positively immunostained cells for CGA and 5-HT were localized mostly in the crypts and mid-villus regions. Immunostaining for PC1 showed numerous positive cells within the epithelium of crypts and villi of controls (Fig. 4C), while only very occasional PC1-positive cells were observed (Fig 4 D) in all three patients (1, 2, 3). Statistical analysis confirmed significant difference (P<0.01) compared to age-matched controls (Table 3). Similarly, immunostaining for PC2 showed scattered positive cells in biopsies from controls (Fig 4E) while in biopsies from the patients (Fig 4F) only very occasional positive cells were present (P<0.01). Immunostaining for GLP-1 was also reduced in duodenal biopsies of patients compared to controls (Figs 4G&H, P<0.05).
Figure 4.

Immunohistochemistry for enteroendocrine cell (EE) markers in duodenal biopsies from patients with PC1/3 deficiency compared to age matched controls.
(A) Immunostaining for chromogranin A (CGA) in control showing immunoreactive EE cell (dark brown) scattered within epithelium of crypts and villi;(B) the same immunostain as in A in patient with PC 1/3 deficiency shows similar number and distribution of CGA immunoreactive cells (immunostaning for CGA ,magnification bar for A&B, 100 microns).
(C) Immunostaning for PC1 in control showing numerous PC1 immunoreactive cells (dark brown) distributed within crypt and villous epithelium; (D) in patient with PC1/3 deficiency only very occasional PC1 immunoreactive cells (arrow) are present. (Immunostaning for PC 1, magnification bar for C&D, 50 microns).
(E) Immunostaning for PC2 in control biopsy shows scattered positive cells (arrows) within villous epithelium. (F) only a single PC2 positive cell (arrow) is seen in duodenal mucosa of PC1/3 deficient patient. (Immunostaning for PC2, magnification bar same as in Fig C–D).
(G) Immunostaning for GLP1 in control with several immunoreactive cells within villous epithelium (arrows);(H) very occasional GLP1 immunoreactive cells (arrows) are noted in patient with PC1/3 deficiency. (Immunostaning for GLP 1, magnification bar same as Fig C–D).
TABLE 3.
Immunohistochemistry for chromogranin A, PC1, PC2 and GLP-1 in duodenal and ileal tissue from PC1/3 deficiency patients and controls.
| Chromogranin | PC1 | PC2 | GLP-1 | |
|---|---|---|---|---|
| duodenum | ||||
| Patients (n=3) | 3.5 (0.3) | 0.7 (0.2)* | 0.2 (0.1)* | 0.3 (0.1)† |
| Controls (n=6) | 3.9 (0.4) | 3.5 (0.6) | 1.2 (0.4) | 0.8 (0.4) |
| ileum | ||||
| Patients (n=2) | 3.0 (0.3) | 1.0 (0.4) | 0 | 0.8 (0.3) |
| Controls (n=5) | 3.0 (0.7) | 2.5 (0.3) | 0 | 1.0 (0.2) |
Values are mean number of positive cells per crypt (Standard Deviation).
P<0.01
P<0.05 compared to controls.
The overall number and distribution of CGA and 5-HT positive cells in ileal biopsies from patients (1, 2) and controls were similar (not shown). The mean number of PC1 positive cells in patient-derived biopsies was lower compared to controls. Immunostaining for PC2 was negative in ileal biopsies from both patients and controls, whereas the mean number of GLP-1 immunopositive cells was comparable between patients and controls. No statistical analysis was possible due to small sample size.
Transmission electron microscopy
Ultrastructural examination of duodenal biopsies confirmed well preserved enterocytes with an intact microvillous brush border. The ultrastructural features of goblet and Paneth cells were within normal limits. Scattered enteroendocrine (EE) cells were identified at the base of the crypts or mid-villous region. Based on different size and ultrastructural appearance of secretory granules (dense-core vesicles), the storage site of amine and peptides, several types of enteroendocrine cells - in agreement with previous descriptions - were observed (Figs 5A&B).23 The most common subtype of EE cells contained numerous spherical, electron dense granules with a mean size of 200 nm (range 106–308 nm) concentrated in the basal cytoplasm (Fig. 5B), which was comparable to controls.23 The other common subtype was enterochromaffin (EC) type cells with pleiomorphic electron-dense granules with variable size ranging from 97 nm to 295 nm (Fig 5A) and was similar to EC cells observed in duodenal biopsy of a control patient (Fig 5C). There were no specific ultrastructural alterations in the cytoplasmic organelles of any of the EE cells examined.
Figure 5.
Electron microscopy features of endocrine cells in duodenal biopsy from patients with PC1/3 deficiency and control.
(A) Classical enterochromaffin-type cell in duodenum of patient with PC1/3 deficiency showing well preserved ultrastructure including pleiomorphic electron-dense secretory granules (arrow), mitochondria (mi)and nucleus (Nu). (B) Different subtype of enteroendocrine cell in same biopsy as Fig A with spherical electron-dense granules (arrow) and well preserved cytoplasmic organelles including mitochondria (mi) and nucleus (Nu). (C) Enterochromaffin-type cell in duodenum of control showing similar ultrastructural features as in Fig A. (Magnification bar for A, B&C, 2 microns).
DISCUSSION
There is limited clinical knowledge on the function of prohormone convertases and the consequences of deficiencies of these proteins in humans. More than a decade ago, the first patient with a deficiency in PC1/3 was described but information on the clinical course over time, pathology findings and the endocrine and enteroendocrine function in this rare disease is still very limited.2–5 The results from the four patients presented here, therefore, provide meaningful new insights into the effects of loss of PC 1/3 function in humans.
One of the hallmarks of PC1/3 deficiency is the changing and diverse clinical phenotype over time as demonstrated by the 4 patients described in this report. Infants present with severe neonatal diarrhea and are usually TPN dependent. Unlike any other congenital diarrhea, the diarrhea in children with PC1/3 deficiency improves over time and the patients become obese, TPN independent with loose-formed stools. The second hallmark of the disease is a diverse clinical presentation with varying penetrance of a variety of endocrinopathies that develop as the children age. The endocrinopathies reflect the impact of PC1/3 on the processing of prohormones and include GH deficiency, diabetes insipidus like phenomena and hypogonadism with micropenis. Congenital enteropathies are often difficult to diagnose and the final diagnosis relies on a combination of clinical, laboratory and pathology findings and, if the defected gene is known, on genetic analysis. Neonates with PC1/3 deficiency present with non-specific clinical and laboratory findings of watery osmotic diarrhea, failure to thrive and metabolic acidosis. Intestinal biopsies are also non specific with normal or mildly distorted intestinal architecture with mild villous blunting. EM findings, reported here for the first time, are normal and non-specific as well. However the use of EM in our cases was useful in ruling out other congenital enteropathies as well as demonstrating normal ultra structure of EE cells indicating that PC1/3 deficiency does not affect subcellular organization of these cells. The diagnosis in PC 1/3 deficiency can be made retrospectively after the neonatal period based on the changing clinical phenotype overtime, following an improvement in the diarrhea and the development of obesity and endocrinopathies. For the neonatal period and beyond, pro-insulin can serve as a valuable screening test as pro-insulin levels were markedly elevated in our patients. Immunohistochemistry of intestinal biopsies for expression of PC1 can be used as an adjunct confirmatory diagnostic test based on our findings of very low PC1 expression in the duodenum. The residual PC 1 expression is probably the result of background staining or antibody cross reactivity and not direct binding to residual PC1 protein based on the genetic analysis in our patients with null mutations in all of them.6
Normal intestinal function appears to require functional PC1/3 activity in the neonatal period and infancy. Functional investigations in both the previously published and the current patients indicate malabsorption of fat and carbohydrates.3;4 The importance of PC1/3 and the enteroendocrine system in normal gut function and development is clearly demonstrated by the unique clinical phenotype of PC1/3 deficient children. Lack of PC1/3 associated enteric hormones and trophic factors could explain the diarrhea in all patients and the mild villous atrophy described in patient 3. The PC1/3 dependent enteric hormones GLP-1 and GLP-2 are potential candidates in the pathophysiology of PC1/3 deficiency and indeed both were low in our patients. It is likely that impaired processing of GLP-2 contributes to the malabsorption and diarrhea of PC 1/3 deficiency, since GLP-2 is involved in intestinal lipid and carbohydrate uptake 24;25 and enterocyte proliferation.26;27
GLP-1 secretion and immunostaining in duodenal biopsies were also severely disturbed in young affected subjects. However, GLP-1 does not appear to have a function in intestinal proliferation and regeneration or nutrient absorption; rather it is responsible for decreasing gastric emptying and intestinal motility.8;28 Absence of GLP-1, therefore, might influence absorption indirectly by increasing intestinal transit time. The reason for the spontaneous improvement in diarrhea after the first year of life is unclear, although compensatory and adaptive mechanisms are likely. Based on immunohistochemistry results, it is unlikely that PC2 is able to compensate for the intestinal functions of PC1/3. The virtual absence of PC2 staining in duodenal samples is consistent with previous findings where PC2 mRNA was not detected in PCSK1 deficient mice.29
Insulin and C-peptide responses after the oral glucose tolerance test were impaired in the two youngest patients, but were in the normal range in the older patient as compared to the adult controls and published data. Ideally, comparison with age-matched control subjects would have been performed. However, it proved to be unfeasible to recruit healthy children for this study. Nevertheless, it appears that normal insulin secretion is disturbed in young patients, but has the possibility to improve with age despite the presence of the same mutation in patients 1 and 2, probably following an unknown adaptive process. In the previously described infant and in an affected 6-year-old child a relatively normal fasting insulin concentration was reported, but stimulation tests were not done.3;4 The adult patient described by O’Rahilly et al.5 was found to have a very low insulin concentrations 2 hours post glucose ingestion. Previous animal data indicates that PC1/3 is required for normal pro-insulin processing.7
The glucose tolerance test results in the patients did not demonstrate clear glucose intolerance despite the absence of a normal insulin and GLP-1 response. GLP-1 secretion, which appears to be affected in young patients, is a well-known enhancer of insulin secretion. Whereas mice in one mouse model of PC1/3 deficiency had normal glucose tolerance,13 mice in a more recently developed model develop massive obesity and glucose intolerance related to impaired insulin secretion.29 Potential explanations for the normal glucose tolerance in humans might be related to increased sensitivity to insulin in young age as was shown in our patients and the fact that pro-insulin also has biological activity 30 thereby reducing the impact of a lack of insulin. The biological activity of pro-insulin and its prolonged half life of 20–30 minutes in combination with increase insulin sensitivity and glucose malabsorption might explain the hypoglycemic episodes over the first year found in the patients reported herein and in another affected patient described previously. The resolution of these episodes overtime is probably related to the reduced insulin sensitivity with aging and other compensatory mechanism leading to an improvement in insulin secretion.
The cause of the hyperphagia and obesity at an older age in patients with PC1/3 deficiency is not clear but is probably related to abnormal processing of appetite controlling hormones. Signs of impaired hypothalamic pro-opiomelanocortin (POMC) processing have been described in previous patients with PC1/3 deficiency.3 Impaired processing of POMC is considered responsible for the reduction of melanocyte-stimulating hormones, which plays a direct role in appetite control.31 Of interest, data have indicated that intestinally-derived GLP-1 reduces food intake,32 and the reduced GLP-1 response observed in our patients could play a role in the observed hyperphagia. In addition, the anorexigenic hormone cholesystokinin and the orexigenic hormone ghrelin, are also activated by PC1/3 and might play a role in the observed hyperphagia.33–34 Leptin concentrations in our patients in relation to BMI were within a similar range to healthy subjects,22 and unlikely to explain the hyperphagia in our patients. Interestingly an association between single nucleotide polymorphism in PCSK1 (rs6232, rs6234–rs6235) and obesity in children and adults has been described.35 More recently, carriers of mutations causing partial PC1/3 deficiency were found to have an 8.7-fold higher risk for obesity.36 Further studies are, therefore, warranted to provide additional insights into the role of these appetite controlling hormones and PCSK1 mutations in patients with PC1/3 deficiency.
In conclusion, the results arising from this study indicate that PC1/3 deficiency is associated with significant abnormalities in intestinal and enteroendocrine function in humans. The results underscore the diversity of the clinical phenotype and its evolution with advancing chronologic age. Measuring serum levels of pro-insulin concentration is recommended as a tool to screen for PC1/3 deficiency when the diagnosis is suspected in the relevant clinical setting.
Acknowledgments
Source of Funding: This work was supported in part by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (#DK083762).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors have no conflict of interest to disclose in relation to this study.
REFERENCES
- 1.Seidah NG. The proprotein convertases, 20 years later. Methods Mol Biol. 2011;768:23–57. doi: 10.1007/978-1-61779-204-5_3. [DOI] [PubMed] [Google Scholar]
- 2.Jackson RS, Creemers JW, Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 3.Jackson RS, Creemers JW, Farooqi IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112:1550–1560. doi: 10.1172/JCI18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farooqi IS, Volders K, Stanhope R, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab. 2007;92:3369–3373. doi: 10.1210/jc.2007-0687. [DOI] [PubMed] [Google Scholar]
- 5.O'Rahilly S, Gray H, Humphreys PJ, et al. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995;333:1386–1390. doi: 10.1056/NEJM199511233332104. [DOI] [PubMed] [Google Scholar]
- 6.Martin MG, Lindberg I, Solorzano-Vargas RS, et al. Congenital Proprotein Convertase 1/3 Deficiency Causes Malabsorptive Diarrhea and other Endocrinopathies in a Pediatric Cohort. Gastroenterology. 2013 doi: 10.1053/j.gastro.2013.03.048. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X, Orci L, Carroll R, et al. Severe block in processing of proinsulin to insulin accompanied by elevation of des-64,65 proinsulin intermediates in islets of mice lacking prohormone convertase 1/3. Proc Natl Acsad Sci U S A. 2002;99:10299–10304. doi: 10.1073/pnas.162352799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 9.Burrin DG, Stoll B, Jiang R, et al. GLP-2 stimulates intestinal growth in premature TPN-fed pigs by suppressing proteolysis and apoptosis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G1249–G1256. doi: 10.1152/ajpgi.2000.279.6.G1249. [DOI] [PubMed] [Google Scholar]
- 10.Drucker DJ, Erlich P, Asa SL, et al. Induction of intestinal epithelial proliferation by glucagon-like peptide 2. Proc Natl Acad Sci U S A. 1996;93:7911–7916. doi: 10.1073/pnas.93.15.7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Estall JL, Drucker DJ. Glucagon-like Peptide-2. Annu Rev Nutr. 2006;26:391–411. doi: 10.1146/annurev.nutr.26.061505.111223. [DOI] [PubMed] [Google Scholar]
- 12.Adeli K, Lewis GF. Intestinal lipoprotein overproduction in insulin-resistant states. Curr Opin Lipido.l. 2008;19:221–228. doi: 10.1097/MOL.0b013e3282ffaf82. [DOI] [PubMed] [Google Scholar]
- 13.Zhu X, Zhou A, Dey A, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc Natl Acad Sci U S A. 2002;99:10293–10298. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.CDC Growth Charts. 2012 Avaiable at: http://www.cdc.gov/growthcharts.
- 15.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 16.Hartmann B, Johnsen AH, Orskov C, et al. Structure, measurement, and secretion of human glucagon-like peptide-2. Peptides. 2000;21:73–80. doi: 10.1016/s0196-9781(99)00176-x. [DOI] [PubMed] [Google Scholar]
- 17.Goossens L, Bouvry M, Vanhaesebrouck P, et al. Citrulline levels in a paediatric age group: Does measurement on dried blood spots have additional value? Clin Chim Acta. 2011;412:661–664. doi: 10.1016/j.cca.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 18.Ong KK, Petry CJ, Emmett PM, et al. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia. 2004;47:1064–1070. doi: 10.1007/s00125-004-1405-8. [DOI] [PubMed] [Google Scholar]
- 19.Salgin B, Ong KK, Thankamony A, et al. Higher Fasting Plasma Free Fatty Acid Levels Are Associated with Lower Insulin Secretion in Children and Adults and a Higher Incidence of Type-2 Diabetes. J Clin Endocrinol Metab. 2012;97:3302–3309. doi: 10.1210/jc.2012-1428. [DOI] [PubMed] [Google Scholar]
- 20.Yeckel CW, Weiss R, Dziura J, et al. Validation of insulin sensitivity indices from oral glucose tolerance test parameters in obese children and adolescents. J Clin Endocrinol Metab. 2004;89:1096–1101. doi: 10.1210/jc.2003-031503. [DOI] [PubMed] [Google Scholar]
- 21.Amin H, Holst JJ, Hartmann B, et al. Functional ontogeny of the proglucagon-derived peptide axis in the premature human neonate. Pediatrics. 2008;121:e180–e186. doi: 10.1542/peds.2007-1461. [DOI] [PubMed] [Google Scholar]
- 22.Lahlou N, Landais P, De BD, et al. Circulating leptin in normal children and during the dynamic phase of juvenile obesity: relation to body fatness, energy metabolism, caloric intake, and sexual dimorphism. Diabetes. 1997;46:989–993. doi: 10.2337/diab.46.6.989. [DOI] [PubMed] [Google Scholar]
- 23.Lechago J. Endocrine Pathology. 3rd edition ed. Baltimore: Williams and Wilkins; 1997. Endocrine cells of the gastrointestinal tract; pp. 1463–1493. [Google Scholar]
- 24.Hsieh J, Longuet C, Maida A, et al. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology. 2009;137:997–1005. doi: 10.1053/j.gastro.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 25.Cheeseman CI. Upregulation of SGLT-1 transport activity in rat jejunum induced by GLP-2 infusion in vivo. Am J Physiol. 1997;273:R1965–R1971. doi: 10.1152/ajpregu.1997.273.6.R1965. [DOI] [PubMed] [Google Scholar]
- 26.Martin GR, Wallace LE, Sigalet DL. Glucagon-like peptide-2 induces intestinal adaptation in parenterally fed rats with short bowel syndrome. Am J Physiol Gastrointest Liver Physiol. 2004;286:G964–G972. doi: 10.1152/ajpgi.00509.2003. [DOI] [PubMed] [Google Scholar]
- 27.Bjerknes M, Cheng H. Modulation of specific intestinal epithelial progenitors by enteric neurons. Proc Natl Acad Sci U S A. 2001;98:12497–12502. doi: 10.1073/pnas.211278098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tolessa T, Gutniak M, Holst JJ, et al. Inhibitory effect of glucagon-like peptide-1 on small bowel motility. Fasting but not fed motility inhibited via nitric oxide independently of insulin and somatostatin. J Clin Invest. 1998;102:764–774. doi: 10.1172/JCI942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lloyd DJ, Bohan S, Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Hum Mol Genet. 2006;15:1884–1893. doi: 10.1093/hmg/ddl111. [DOI] [PubMed] [Google Scholar]
- 30.Revers RR, Henry R, Schmeiser L, et al. The effects of biosynthetic human proinsulin on carbohydrate metabolism. Diabetes. 1984;33:762–770. doi: 10.2337/diab.33.8.762. [DOI] [PubMed] [Google Scholar]
- 31.Gao Q, Horvath TL. Neurobiology of feeding and energy expenditure. Annu Rev Neurosci. 2007;30:367–398. doi: 10.1146/annurev.neuro.30.051606.094324. [DOI] [PubMed] [Google Scholar]
- 32.De SA, Salem V, Long CJ, et al. The gut hormones PYY 3–36 and GLP-1 7–36 amide reduce food intake and modulate brain activity in appetite centers in humans. Cell Metab. 2011;14:700–706. doi: 10.1016/j.cmet.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehfeld JF, Bundgaard JR, Hannibal J, et al. The cell-specific pattern of cholecystokinin peptides in endocrine cells versus neurons is governed by the expression of prohormone convertases 1/3, 2, and 5/6. Endocrinology. 2008;149:1600–1608. doi: 10.1210/en.2007-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Cao Y, Voogd K, et al. On the processing of proghrelin to ghrelin. J Biol Chem. 2006;281:38867–38870. doi: 10.1074/jbc.M607955200. [DOI] [PubMed] [Google Scholar]
- 35.Benzinou M, Creemers JW, Choquet H, et al. Common nonsynonymous variants in PCSK1 confer risk of obesity. Nat Genet. 2008;40:943–945. doi: 10.1038/ng.177. [DOI] [PubMed] [Google Scholar]
- 36.Creemers JW, Choquet H, Stijnen P, et al. Heterozygous mutations causing partial prohormone convertase 1 deficiency contribute to human obesity. Diabetes. 2012;61:383–390. doi: 10.2337/db11-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]