

Abstract
Background
To engineer Saccharomyces cerevisiae for efficient xylose utilization, a fungal pathway consisting of xylose reductase, xylitol dehydrogenase, and xylulose kinase is often introduced to the host strain. Despite extensive in vitro studies on the xylose pathway, the intracellular metabolism rewiring in response to the heterologous xylose pathway remains largely unknown. In this study, we applied 13C metabolic flux analysis and stoichiometric modeling to systemically investigate the flux distributions in a series of xylose utilizing S. cerevisiae strains.
Results
As revealed by 13C metabolic flux analysis, the oxidative pentose phosphate pathway was actively used for producing NADPH required by the fungal xylose pathway during xylose utilization of recombinant S. cerevisiae strains. The TCA cycle activity was found to be tightly correlated with the requirements of maintenance energy and biomass yield. Based on in silico simulations of metabolic fluxes, reducing the cell maintenance energy was found crucial to achieve the optimal xylose-based ethanol production. The stoichiometric modeling also suggested that both the cofactor-imbalanced and cofactor-balanced pathways could lead to optimal ethanol production, by flexibly adjusting the metabolic fluxes in futile cycle. However, compared to the cofactor-imbalanced pathway, the cofactor-balanced xylose pathway can lead to optimal ethanol production in a wider range of fermentation conditions.
Conclusions
By applying 13C-MFA and in silico flux balance analysis to a series of recombinant xylose-utilizing S. cerevisiae strains, this work brings new knowledge about xylose utilization in two aspects. First, the interplays between the fungal xylose pathway and the native host metabolism were uncovered. Specifically, we found that the high cell maintenance energy was one of the key factors involved in xylose utilization. Potential strategies to reduce the cell maintenance energy, such as adding exogenous nutrients and evolutionary adaptation, were suggested based on the in vivo and in silico flux analysis in this study. In addition, the impacts of cofactor balance issues on xylose utilization were systemically investigated. The futile pathways were identified as the key factor to adapt to different degrees of cofactor imbalances and suggested as the targets for further engineering to tackle cofactor-balance issues.
Keywords: 13C-MFA, Fungal xylose pathway, Maintenance, Cofactor engineering
Introduction
Biofuel, especially cellulosic biofuel, plays an increasingly important role in sustainable energy supply and greenhouse gas emissions reduction [1]. Thanks to a number of breakthroughs in metabolic engineering and synthetic biology, a series of industrial microorganisms such as Escherichia coli[2] and Saccharomyces cerevisiae[3,4] have been engineered to use non-food feedstock to produce a variety of biofuels. Among all these endeavors, one of the most promising strategies is to engineer S. cerevisiae to utilize xylose for bioethanol production. To this end, a heterologous xylose pathway identified from fungal species such as Scheffersomyces stipitis and Candida tenuis is often introduced into S. cerevisiae by functionally expressing xylose reductase (XR), xylitol dehydrogenase (XDH), and xylulose kinase (XKS). When engineering the fungal xylose pathway in recombinant S. cerevisiae strains, the unbalanced utilization of various cofactors by XR and XDH [5] is often believed to be an issue that may affect the performance of xylose fermentation. Basically, NADPH is preferred to be consumed by XR with the production of NADP+, while NAD+ is required by XDH. Such cofactor imbalance accounts for xylitol accumulation due to the insufficient regeneration of NAD+ for the XDH reaction from the heterologous xylose pathway and the endogenous central carbon metabolism, which leads to low ethanol yield [4,6,7].
Numerous efforts have been made in the past three decades to alter the cofactor specificity of XR and XDH. For example, protein engineering was used to increase the specificity of XR to NADH [8-10], and improve the specificity of XDH to NADP+[11]. In the meantime, a series of metabolic engineering strategies were used to optimize the cofactor utilizations in xylose metabolism. In general, three independent approaches have been developed, including: 1) altering the preference of XR to use NADH as the cofactor [12-17]; 2) altering the preference of XDH to use NADP+ as the cofactor [10,18-21]; and 3) engineering cofactor dependent metabolic pathways in the native S. cerevisiae such as 6-phosphogluconate dehydrogenase in the oxidative pentose phosphate pathway [22] and glyceraldehyde-3-phosphate dehydrogenase in the glycolysis pathway [23]. As expected, the decreased xylitol production and enhanced ethanol yield could be achieved by using any of these strategies, which raises an interesting question: which strategy or strategies should be pursued to push the xylose utilization to the theoretical limit?
To address this question, detailed understanding of the interplays between the heterologous xylose pathway and the native host metabolism is required. In the past decade, the in vivo metabolic behaviors of recombinant S. cerevisiae strains when metabolizing xylose have been analyzed by stoichiometric flux analysis [24,25] and metabolomics analysis [26,27]. In this study, we attempted to rigorously investigate the global effect of the heterologous xylose pathways by selecting six representative engineered xylose utilizing S. cerevisiae strains from our previous studies [28] and applying 13C-metabolic flux analysis (13C-MFA) to systemically characterize the in vivo carbon flux distributions. We found that the oxidative pentose phosphate pathway was actively used in the recombinant S. cerevisiae strains in order to supply enough NADPH for the fungal xylose pathway. Carbon fluxes into the TCA cycle were regulated in response to the metabolic burdens of expressing a heterologous xylose pathway in nutrient limited medium. Based on the discovery from 13C-MFA, an in silico study was carried out to evaluate the impact of cofactor engineering strategies and cell maintenance energy on xylose-based ethanol production under different fermentation conditions. To augment previous efforts on engineering S. cerevisiae for efficient xylose utilization, this study provides quantitative readouts of xylose metabolism and uncovers new insights about metabolic regulations of a heterologous xylose pathway.
Material and methods
Strains, media, and culture conditions
The S. cerevisiae strains used in this study are summarized in Table 1. All S. cerevisiae strains were stored in 25% glycerol at −80°C. To culture S. cerevisiae strains, seed cultures were grown in YPAD media (1% yeast extract, 2% peptone, 0.01% adenine hemisulfate, 20 g/L glucose) at 30°C for overnight. The seed cultures were then inoculated (0.1%, v/v) into the defined minimal medium as described elsewhere [29,30], with 5 g/L xylose as the sole carbon source. The yeast strains were grown with triplicates at 30°C, 250 rpm in capped culture tubes (14 mL) containing 6 mL defined minimal medium to achieve the oxygen limited condition. Cell growth was monitored at OD600. In the exponential phase, two samples were taken for growth rate calculation and extracellular metabolites measurement.
Table 1.
S. cerevisiae strains used in this study
| S. cerevisiae strains | Origins of XR | Origins of XDH | Origins of XKS |
|---|---|---|---|
| CF1 |
Aspergillus flavus |
Candida dubliniensis |
Nectria haematococca |
| CF3 |
Aspergillus nidulans |
Aspergillus niger |
Pencillium chrysogenum |
| CF9 |
Aspergillus flavus |
Pichia guilliermondii |
Candida dubliniensis |
| CS1 |
Candida dubliensis |
Nectria haematococca |
Aspergillus niger |
| CS5 |
Zygosaccharomyces rouxii |
Aspergillus niger |
Nectria haematococca |
| CS10 | Candida dubliensis | Scheffersomyces stipitis | Podospora anserina |
XR: xylose reductase; XDH: xylitol dehydrogenase; XKS: xylulose kinase.
For 13C tracer experiments in defined minimal medium, the same culture conditions were used to culture S. cerevisiae strains, except that the carbon source was replaced with 5 g/L [1-13C] xylose (Cambridge Isotope Laboratories). For 13C tracer experiments in YPAX medium (1% yeast extract, 2% peptone, 0.01% adenine hemisulfate, 40 g/L xylose), 0.1% inoculum was transferred from the seed culture to YPAX medium with 40 g/L [1-13C] xylose as the carbon source. For all 13C tracer experiments, S. cerevisiae strains were grown in duplicates, and the cells (i.e. biomass) were harvested at the mid-log phase based on the growth curve previously determined using non-labeled xylose in defined minimal medium and YPAX medium [28].
The concentrations of extracellular metabolites, including xylose, xylitol, and ethanol were analyzed by HPLC equipped with a refractive index detector (Shimadzu Scientific Instruments, Columbia, MD) and an HPX-87H column (BioRad, Hercules, CA). The flow rate was 0.6 mL/min at 60°C using 5 mM sulfuric acid as the mobile phase. No byproducts but xylose was detected in the supernatant.
Isotopic analysis
For GC-MS measurement of isotopomer labeling patterns in proteinogenic amino acids, the biomass was harvested by centrifugation and hydrolyzed using 6 M HCl (24 h at 100°C). The amino acids were derivatized in 50 μl of tetrahydrofuran and 50 μl of N-(tert-butyl dimethylsilyl)-N-methyl-trifluoroacetamide (Sigma-Aldrich). A gas chromatograph (GC2010, Shimadzu) equipped with a DB5-MS column (J&W Scientific, Folsom, CA) and a mass spectrometer (QP2010, Shimadzu) were used for analyzing metabolite labeling profiles. Three types of charged fragments were detected by GC-MS for Ala, Gly, Ser, Asp, Glu, Phe, and Leu: the [M-57]+ group (containing unfragmented amino acids); and the [M-159]+ or [M-85]+ group (containing amino acids that had lost an α-carboxyl group). For each type of fragments, the labeling patterns were represented by M0, M1, M2, etc., which were fractions of unlabeled, singly labeled, and doubly labeled amino acids (Additional file 1: Text S1). The effects of natural isotopes on isotopomer labeling patterns were corrected by previously reported algorithms [31]. The 13C enrichment of proteinogenic amino acids was calculated as , where M i is the GC-MS isotopomer fraction for a given amino acid; C is the total number of carbon atoms in the amino acid molecule.
13C metabolic flux analysis
The central metabolic pathway map of recombinant S. cerevisiae strains was generated based on genome annotation from the KEGG database (Kyoto Encyclopedia of Genes and Genomes) and previous fluxomics studies [24,25,29,32]. The simplified pathway map includes the fungal xylose pathway, oxidative and reductive pentose phosphate pathway, glycolysis, futile pathways, transport pathways between cytosol and mitochondria, and TCA cycle (Additional file 1: Text S2). As discussed before [33], to develop a pseudo-steady-state flux model, the xylose uptake rates were measured at the mid-log phase and normalized to 100 units. The biomass composition of S. cerevisiae strains was referred to a previous study [29], and used as loose constraints for the fluxes into building block synthesis (i.e., allowed to have variations of up to 20%). To determine the remaining unknown fluxes of the metabolic network, a nonlinear optimization problem was formulated to iteratively search a set of fluxes that can satisfy the reaction stoichiometry and minimize the objective function [33]: , where v n are the unknown fluxes to be optimized in the program, M i are the measured isotopomer labeling patterns of proteinogenic amino acids, N i are the model-simulated isotopomer labeling patterns of proteinogenic amino acids using isotopomer mapping matrices (IMM), δ i are the standard deviations of the GC-MS data from two biological replicates. The nonlinear optimization was finished by using “fmincon” command in MATLAB (MathWorks, USA). The optimization was run for 100 times with different initial guesses to search for a likely global solution for the unknown fluxes. The observed and simulated isotopomer labeling patterns of proteinogenic amino acids were compared in Additional file 1: Figure S1. The confidence intervals of the calculated fluxes were generated via a Monte Carlo approach as described in our previous research [33]. In short, the isotopomer labeling patterns of the proteinogenic amino acids were randomly perturbed for 100 times within the standard derivation. For each perturbed dataset, the 13C-MFA was applied and the flux distributions were calculated. Then, the standard derivations of each flux were derived from such 100 simulations.
Since the relative activities of isoenzymes cannot be decided, the cofactor balancing for NADPH was not included in the mass balancing of 13C-MFA. For the NADH production, only the glycolysis pathway and the TCA cycle (without considering isocitrate dehydroganase) was taken into calculation. Considering that no fermentation products (e.g., ethanol) can be detected, it was hence assumed that all of the NADH was used for ATP production via oxidative phosphorylation.
In silico simulations
A stoichiometric model developed by Christen and Sauer [29] was modified and used in this study to simulate the ideal ethanol production under different xylose utilization conditions. Such stoichiometric model includes the same metabolic pathways as those in the 13C-MFA. A parameter “f” was introduced to describe the degree of cofactor imbalance in the heterologous xylose pathway: Xylose + f ∙NADPH - f ∙NADH + ATP → Xylulose 5-phosphate + f ∙NADP+ - f ∙NAD+ + ADP. To simplify the model simulation, the cofactors for aldehyde dehydrogenase (EC 1.2.1.5), malic enzyme (EC 1.1.1.38), and the isocitrate dehydrogenase (EC 1.1.1.42) were set as NADPH. Similar to the 13C-MFA, NADH was only considered to be produced from the glycolysis pathway and the TCA cycle and was consumed for either ATP production through oxidative phosphorylation or ethanol production through the fermentation pathway.
To find the optimal ethanol production, we normalized the xylose uptake rate into 100 units and sampled multiple biomass yields, growth associated maintenance requirements, and oxygen uptake conditions as the model inputs. For each of the model input, the metabolic fluxes were constrained by reaction stoichiometry [34] to find the maximal ethanol yield (equation 1). The mathematical optimization was then solved by linear optimization solver “linprog” in MATLAB (MathWorks, USA):
| (1) |
where S is the stoichiometric matrix and is correlated with the degree of cofactor imbalance in the xylose pathway (i.e., f) and the growth associated maintenance requirement (i.e., GAM in mmol ATP/g DCW), v is the vector of metabolic fluxes, v Xylose is the xylose uptake rate normalized as 100 units, v Oxygen is the normalized oxygen uptake flux, v Biomass is the normalized biomass yield, lb and ub are the lower and upper boundary of metabolic fluxes, respectively, ub O2 is the upper boundary of the oxygen uptake level set in the model.
Enzymatic activity assay
The enzyme activities of XR and XDH were analyzed in a 96-well plate using a Biotek Synergy2 microplate reader (Winooski, VT) by monitoring the absorbance at 340 nm for depletion or generation of NAD(P)H as described previously [28]. The specific activities were reported as units (i.e. micromoles of NAD(P)H reduced or oxidized per minute) per milligram of protein. Cultures in the defined minimal medium or YPAX medium were harvested at mid-log phase. Cell mass equivalent to an OD of 20 was collected for lysis with YPER Extraction Reagent (Pierce, Rockford, IL). BCA Protein Assay Reagent (Pierce) was used to determine the protein concentrations and Albumin Standard (Pierce) was used for the protein concentration standard curve. The XR reaction mixture was made as 50 mM KH2PO4 buffer (pH 7), 0.2 mM NADPH or 0.2 mM NADH, and 200 mM xylose. The XDH reaction mixture was made 50 mM KH2PO4 buffer (pH 7), 50 mM MgCl2, 1 mM NAD+, and 200 mM xylitol.
In order to measure the specific activity of XKS, a modified procedure with a glycerol kit manufactured by R-Biopharm (Darmstadt, Germany) was used. Since the XKS assay was coupled to pyruvate kinase and lactate dehydrogenase [35], the NADH depletion can be monitored and linearly related to XKS activity. The standard protocol of the glycerol kit was followed, with the substitution of galactose kinase with crude enzyme XKS and using 5 mM D-xylulose rather than glycerol as the substrate [28].
Results
Metabolic behaviors of selected S. cerevisiae strains
To investigate the effects of heterologous xylose pathways on native carbon metabolism of S. cerevisiae, six recombinant strains with different origins of XR, XDH and XKS in the xylose pathway (Table 1) were selected from our previous study [28]. In that previous study, the XR, XDH and XKS enzymes cloned from different microorganisms were assembled via a high-throughput method (i.e. DNA assembler [36]) to construct a library of xylose pathways. The resulting library of xylose pathways was then transformed into S. cerevisiae, from which some of the recombinant strains were successfully selected based on their significantly improved metabolic behaviors in terms of growth rates, xylose consumption rates, and ethanol yields when culturing in YPAX medium.
Among the six strains, three of them (i.e. CF1, CF3, and CF9) were originally identified as the “fast-growers” with higher growth rates (ranging from 0.10 ~ 0.14 h-1, Table 2) and ethanol yields (ranging from 0.18 ~ 0.25 g/g) during library screening in YPAX medium, while the other three strains (i.e. CS1, CS5, and CS10) were found to be “slow-growers” with lower growth rates (ranging from 0.05 ~ 0.07 h-1) and ethanol yields (ranging from to 0.07 ~ 0.15 g/g). When growing the selected strains in minimal medium with xylose as the sole carbon source under oxygen limited conditions, the production of ethanol and other products (e.g. acetate, glycerol, and xylitol) was not detected, indicating the consumed xylose was almost exclusively used for biomass production. Such fermentation behaviors were also reported by a recent research on evolutionary engineering of S. cerevisiae for efficient xylose utilization [37]. Among the selected strains in this study, diverse metabolic behaviors were identified, with growth rates ranging from 0.02 ~ 0.05 h-1, xylose uptake rates ranging from 0.54 ~ 3.08 mmol/g DCW/h, and biomass yield ranging from 0.08 ~ 0.47 g/g. The “fast-growers” and “slow-growers” were hence re-grouped based on the performances in the minimal medium. In general, when culturing in minimal medium, one of the “fast-growers” (i.e. CF3) had lower growth rates than two of the “slow-growers” (i.e. CS1 and CS10) identified in YPAX culturing. The diverged fermentation profiles of the same recombinant strains in the minimal medium and the YPAX medium suggested that the metabolic responses to the xylose utilization were different under diverse culturing conditions.
Table 2.
Fermentation profiles of S. cerevisiae strains
|
S. cerevisiae
strains |
Minimal medium |
YPAX medium |
|||||
|---|---|---|---|---|---|---|---|
| Xylose uptake (mmol/g/h) | Growth rate (h -1 ) | Biomass yield (g/g) | Xylose uptake (mmol/g/h) | Growth rate (h -1 ) | Biomass yield (g/g) | Ethanol yield (g/g) | |
| CF1 |
0.69 ± 0.21 |
0.046 ± 0.005 |
0.469 ± 0.149 |
2.07 ± 0.21 |
0.14 ± 0.01 |
0.45 ± 0.02 |
0.25 ± 0.03 |
| CF3 |
0.83 ± 0.30 |
0.026 ± 0.002 |
0.184 ± 0.064 |
1.83 ± 0.09 |
0.13 ± 0.01 |
0.47 ± 0.02 |
0.20 ± 0.03 |
| CF9 |
1.07 ± 0.17 |
0.048 ± 0.004 |
0.299 ± 0.024 |
1.44 ± 0.07 |
0.10 ± 0.01 |
0.46 ± 0.02 |
0.18 ± 0.01 |
| CS1 |
3.08 ± 0.21 |
0.034 ± 0.009 |
0.078 ± 0.033 |
1.40 ± 0.28 |
0.05 ± 0.00 |
0.24 ± 0.01 |
0.07 ± 0.00 |
| CS5 |
0.57 ± 0.03 |
0.018 ± 0.003 |
0.234 ± 0.032 |
1.42 ± 0.28 |
0.06 ± 0.00 |
0.28 ± 0.01 |
0.11 ± 0.02 |
| CS10 | 0.54 ± 0.14 | 0.030 ± 0.011 | 0.369 ± 0.125 | 1.43 ± 0.29 | 0.07 ± 0.00 | 0.33 ± 0.02 | 0.15 ± 0.01 |
The One-step ANOVA for the biomass yields in minimal medium returns p value as low as 0.0102, concluding that there is strong evidence that the biomass yields in the all the groups (i.e., CF1, CF3, CF9, CS1, CS5, and CS10) differ. The fermentation profiles of S. cerevisiae strains in YPAX medium were referred to previous report [28].
Carbon metabolism of recombinant S. cerevisiae strains
By feeding [1-13C] xylose into the minimal medium and analyzing the corresponding isotopomer enrichment patterns in proteinogenic amino acids, the carbon fluxes through the central metabolic networks were calculated for the selected S. cerevisiae strains (Figure 1). The xylose uptake fluxes of each recombinant S. cerevisiae strain were normalized to relative flux value as 100. Based on 13C-MFA, in all of the S. cerevisiae strains, the oxidative pentose phosphate pathway had high fluxes (relative flux value as 17.2 ~ 47.4), which is consistent with transcriptional analysis showing the up-regulation of the oxidative pentose phosphate pathway in xylose growth [8]. The metabolic fluxes split at the node of F6P to enter either oxidative pentose phosphate pathway or the glycolysis pathway, at the ratio close to 1:1. The flux into reductive pentose phosphate pathway merged with the glycolytic flux at the node of GAP with a ratio close to 1:1. The futile cycle such as pyruvate carboxylase and phosphoenolpyruvate carboxykinase was found to be active and served as the major pathway for net production of oxaloacetate to refuel the TCA cycle in mitochondria.
Figure 1.

Heatmap of metabolic flux distributions of S. cerevisiae strains. The xylose uptake flux in each of the S. cerevisiae strains was normalized as 100 units.
While the similar flux distribution patterns were followed by all the selected strains, differences among various xylose metabolisms were still observed. Particularly, the metabolic fluxes through the TCA cycle differed from strain to strain (ranging from 102.3 ~ 142.3 for relative flux value), and were found to be inversely correlated with the biomass yields (Figure 2). For example, the CF1 strain had nearly the lowest TCA cycle activity (relative flux value as 102.5) but the highest biomass yield (0.469 ± 0.149 g/g), while the CS1 strain had the highest TCA cycle activity (relative flux value as 142.3) but the lowest biomass yield (0.078 ± 0.033 g/g). Since two carbons were lost as CO2 in one TCA cycle, the strong activity of TCA cycle will make poor use of the carbon substrates, which may explain the inverse correlations between biomass yield and TCA cycle activity. The fluxes into oxidative pentose phosphate pathway could be correlated with biomass yields in two ways. On one hand, the NADPH produced from the oxidative pentose phosphate pathway were the major source for synthesis of biomass building blocks. On the other hand, the CO2 released from the oxidative pentose phosphate pathway can lead to the decrease of biomass yields. The trade-off between such two effects could explain that neither positive or negative correlation was observed between fluxes into oxidative pentose phosphate pathway and biomass yields (Figure 2).
Figure 2.

Correlation between biomass yields (g DCW/g xylose) and the fluxes in TCA cycle or oxidative pentose phosphate pathway.
Energy metabolism of recombinant S. cerevisiae strains
The balancing of cofactors including NADPH, NADH and ATP was not included when calculating the carbon flux by 13C-MFA, due to the lack of information about the relative activities of isoenzymes. For instance, the isoenzymes of isocitrate dehydrogenase can use either NAD+ (EC 1.1.1.41) or NADP+ (EC 1.1.1.42) in the TCA cycle. Therefore, it becomes difficult to quantify exactly how much NADH or NADPH was produced by isocitrate dehydrogenase without knowing the relative activities of different isoenzymes. On the other hand, it has been suggested in many 13C-MFA studies [38,39] that the flux distributions can be calculated independently without the cofactor balancing, followed by using the carbon flux distributions to uncover the in vivo utilization of cofactors. In this study, the active oxidative pentose phosphate pathway contributes to 11 ~ 74% of NADPH required by the fungal xylose pathway and the building block synthesis in biomass production. The aldehyde dehydrogenase (EC 1.2.1.5), malic enzyme (EC 1.1.1.38), and the isocitrate dehydrogenase (EC 1.1.1.42) may also contribute to the NADPH production in xylose metabolism.
For NADH and ATP metabolism, the TCA cycle in recombinant S. cerevisiae strains showed strong activities, generating huge amount of NADH as the redox for oxidative phosphorylation. As a result, significant amount of ATP was produced, which leads to high requirements for maintenance energy in xylose metabolism of the recombinant S. cerevisiae strains (Figure 3). Interestingly, since the TCA cycle was mainly used for energetic instead of biosynthetic purpose, the requirements for cell maintenance energy were found to be inversely correlated with the biomass yields. That is, the higher TCA cycle activity is, the more ATP was generated through oxidative phosphorylation as maintenance energy, while more carbons were lost as CO2, resulting in lower biomass yields.
Figure 3.
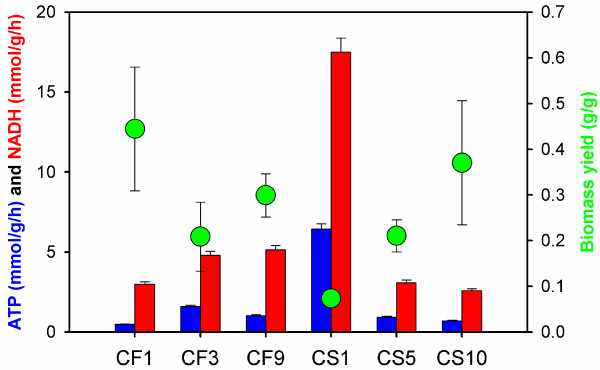
Correlation between biomass yields (g DCW/g xylose) and the net produced ATP and NADH as required for cell maintenance (mmol ATP/g DCW).
In silico simulation of xylose metabolism in recombinant S. cerevisiae strains
To evaluate the impact of cofactor imbalance issue on xylose metabolism of recombinant S. cerevisiae strains, we applied a stoichiometric model for in silico simulation of the maximal ethanol yields (Figure 4). Since the XR enzymes are not strictly NADH or NADPH dependent, using the different XR enzymes in the heterologous xylose pathway could lead to various degrees of cofactor imbalance in S. cerevisiae strains. In general, we introduced “f” (ranging from 0 to 1) as a parameter to quantify the degree of cofactor imbalance in the heterologous xylose pathway (i.e., Xylose + f ∙NADPH - f ∙NADH + ATP → Xylulose 5-phosphate + f ∙NADP+ - f ∙NAD+ + ADP). For each value of “f”, we sampled different biomass yields, growth associated maintenance requirements (i.e., GAMs), and oxygen uptake conditions as the model inputs and searched for the maximal ethanol yields under the given growth condition and with the designated xylose pathway.
Figure 4.

In silico simulations of maximal ethanol yield (g ethanol/g xylose) by using different cofactor engineering strategies (i.e., “f” as in the pathway of Xylose + f ·NADPH - f ·NADH + ATP → Xylulose 5-phosphate + f ·NADP+ - f ·NAD+ + ADP) under various fermentation conditions with combinations of diverse biomass yields, GAM, and aerobic conditions (represented by YO2 as the oxygen uptake in mol O2/mol xylose).
Based on the in silico simulation, the ideal ethanol yield (i.e., over 95% of the theoretical yield) can be only achieved within a narrow range of biomass yields and GAMs. When biomass yield and/or GAM are high, the requirement for ATP becomes so strong that the NADH can no longer be used for ethanol production, but instead fueled into oxidative phosphorylation for ATP production. Therefore, no ethanol can be produced under these fermentation conditions. The cofactor-balanced xylose pathway (i.e., f = 0 as in bacterial isomerase pathway) could indeed be more beneficial to the xylose-based ethanol production than its counterparts (i.e., f = 0.33, 0.37, 1.00), by providing a wider range of fermentation conditions for optimal fermentation conditions. The advantages of using cofactor-balanced pathway are more significant when oxygen uptake level is lower. However, the ideal ethanol production can be possibly achieved in all of the oxygen conditions regardless of cofactor imbalance issue. The metabolic flux distributions that led to optimal ethanol production have similar patterns, including a negligible fluxes into oxidative pentose phosphate pathway, small fluxes (relative flux value less than 35) into TCA cycle, and strong fluxes towards ethanol production (relative flux value higher than 150). However, the fluxes into futile pathways were found to be very flexible corresponding to different degrees of cofactor-imbalance issues (Figure 5). For example, the fluxes of conversing oxaloacetate to malate with the consumption of one NADH were found to be positively correlated with the degrees of cofactor-imbalance issues. That is, the greater the cofactor-imbalance issue was, the more fluxes were diverted into this futile pathway to consume excessive NADH.
Figure 5.
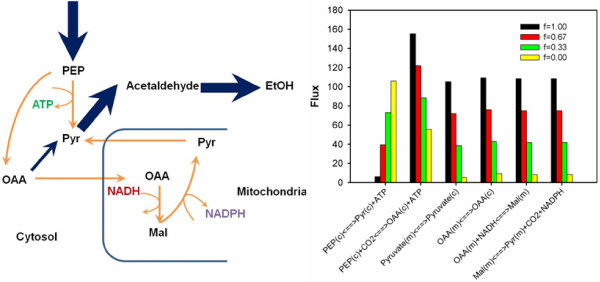
The futile pathways flexibly used to achieve the optimal ethanol production under different cofactor engineering strategies, under aerobic conditions (i.e., 0 < YO2 < ∞), with GAM set as 300 mmol ATP/g DCW. The orange arrows indicate the futile pathways. The thickness of the blue arrows is proportional to the relative flux value.
13C tracer experiments of S. cerevisiae strains in nutrient rich medium
In this study, we cultured strains in the oxygen limited conditions with defined minimal medium based on several considerations. First, as reported by many groups, few S. cerevisiae strains can grow in defined minimal medium under anaerobic conditions with xylose as the sole carbon source, since the oxygen is needed to resolve the cofactor imbalance [40]. On the other hand, the oxygen-limited condition has been suggested as optimal for ethanol production by an in silico simulation [4,40]. In addition, the application of 13C-MFA strictly requires the using of defined minimal medium for culturing recombinant S. cerevisiae strains. However, many labs, including ours, often choose nutrient-rich medium (e.g. YPAX) to create yeast mutants and characterize the xylose fermentation. Compared to the previous characterization of the same strains in nutrient-rich medium (i.e. YPAX), the metabolic behaviors are significantly different in defined minimal medium in terms of growth rates and ethanol production. In order to confirm that the discoveries from the 13C-MFA in minimal medium is also applicable to the studies of xylose utilization in rich medium, we have designed another set of isotopic tracing experiments by providing 4% [1-13C] xylose into the YPAX medium. The 13C enrichments of all the amino acids detected were only 20 ~ 50% of their counterparts from the minimal medium (Figure 6), indicating that the nutrient transport rather than de novo synthesis was the major pathway for building block production. As revealed by 13C-MFA in this study, the GAM plays a pivotal role in xylose utilization, especially in the stressful cultivation conditions. With the building blocks, such as amino acids, provided by the direct uptake from the medium instead of de novo synthesis, nutrient-rich medium is less stressful than the minimal medium, leading to a lower maintenance requirement and better fermentation performance. In addition, compared to those in the nutrient-rich medium, the in vitro enzyme activities of XR and XDH activities in the minimal medium (Figure 7) were within the same level [28]. However, the activities of XKS were about two magnitudes lower (0.001 ~ 0.003 U/mg compared to 0.1 ~ 0.4 U/mg), which could be another reason for the inconsistent metabolic behaviors of the same strain in different media.
Figure 6.

13C enrichment of proteinogenic amino acids in S. cerevisiae strains cultured in A) defined minimal medium and B) YPAX medium. Note: the mass spectrum of Asp, Glu and Phe in CF3 strain cultured in YPAX medium had too much noise to derive the reliable readouts of 13C enrichment.
Figure 7.

Enzyme activities of xylose reductase (XR), xylitol dehydrogenase (XDH), and xylulose kinase (XKS) in S. cerevisiae strains. The activities of XRs with both NADPH and NADH as the cofactors were measured.
Discussion
In spite of thorough in vitro investigation of the heterologous xylose pathway, the in vivo metabolic behaviors of recombinant S. cerevisiae strains in xylose metabolism still remain largely as a black box. To make it worse, the conclusions drawn from the in vitro analysis may not be extended into the in vivo studies, considering the difference between intracellular and extracellular environments. In this case, 13C-MFA becomes one of the valuable tools to provide rigorous and quantitative readouts of in vivo network-scale metabolic behaviors. As discovered by 13C-MFA in this study, the requirements for maintenance energy [41] of the recombinant S. cerevisiae strains for xylose utilizations were so high that no ethanol can be produced. The metabolic burdens of expressing the heterologous enzymes in nutrient limited medium could account for the high requirements for maintenance energy during xylose utilization. Our previous 13C-MFA studies have compared the flux distributions in wild-type S. cerevisiae strain without expressing heterologous proteins and the engineered S. cerevisiae strains expressing heterologous XR, XDH, and XKS in glucose cultures [42] and found that the metabolic burdens of expressing the heterologous enzymes in nutrient limited medium led to 30 ~ 38% increase of TCA cycle fluxes in the recombinant strains so that more ATP was produced through oxidative phosphorylation to satisfy the higher requirement for the maintenance energy. The similar metabolic responses (i.e., more flux into TCA cycle) to metabolic burdens of heterologous protein expression in carbon-limited medium were also uncovered recently in several other 13C-MFA study of recombinant Pichia pastoris[43-45]. In these studies, the up-regulated TCA cycle was correlated to the decreased biomass yield since more carbons got lost as CO2 instead of being used for building block synthesis in TCA cycle. In this study, we found the similar linkage between the decreased biomass yield and the increased flux in TCA cycle, confirming that the metabolic burdens of expressing heterologous proteins could also play pivotal roles in xylose utilization.
It is believed that the cofactor-balanced pathway (e.g., bacterial isomerase pathway) is more advantageous for ethanol production. As suggested by the in silico simulation in this study, adopting the cofactor-balanced pathway does not show significant advantages to the cofactor-imbalanced pathways in terms of the maximal ethanol yield that can be achieved. However, it indeed provides a wider range of fermentation conditions for optimal ethanol production, especially under the oxygen limited conditions. Compared to the cofactor-imbalanced pathway, the cofactor-balanced pathway does not generate NADH, which can be more beneficial for cell growth when the oxygen uptake level is low, as the capacity to convert NADH to ATP via oxidative phosphorylation is limited. In addition, the futile pathways were found to be the key pathway for the optimal ethanol production in both cofactor-imbalanced and cofactor-balanced pathway, since they could be flexibly adjusted to adapt to different cofactor usages. It is hence possible that the futile pathways could be engineered in the future to tackle the cofactor imbalance issue in xylose utilization. Beside the cofactor imbalance issue, another issue that may need the attention is the metabolic burden caused by heterologous protein expression in stress conditions. As illustrated from both the in silico simulation and the 13C-MFA in this study, no ethanol can be produced when metabolic burden becomes too large to be neglected. In addition to the effects on ethanol yields, expressing the heterologous fungal pathway in S. cerevisiae strains also had impacts on other physiological parameters, such as growth rate and xylose consumption rate. As shown in Table 2, the growth rate of CS5 was poor while the xylose consumption rates of CS5 and CS10 were much lower than those of the other yeast strains. Deciphering the mechanisms behind the aforementioned effects could be challenging for 13C-MFA in this study since not only the intracellular metabolism rewiring but also the protein properties such as protein solubility, and catalytic capacities could be involved in affecting the growth rate and xylose consumption rate. Therefore, the comprehensive characterization of enzyme properties needs to be finished in the future.
In summary, by systemically applying 13C-MFA to a series of recombinant xylose-utilizing S. cerevisiae strains, the interplays between the fungal xylose pathway and the native host metabolism were uncovered. The oxidative pentose phosphate pathway of S. cerevisiae strains was activated to provide NADPH for the fungal xylose pathway. The TCA cycle was found to be tightly regulated in response to the requirements of maintenance energy caused by heterologous protein expression. Based on in silico simulations, reducing the maintenance energy can be a crucial strategy to improve ethanol production. The addition of exogenous nutrients such as amino acids to reduce the de novo synthesis of biomass building blocks can be an efficient strategy to reduce the maintenance energy. Also, metabolic engineering strategies, such as directed evolution, can be potentially helpful to reduce the maintenance requirement and improve the xylose based ethanol production.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XF and HZ conceived this study. XF performed the experiments; XF and HZ interpreted and analyzed data. XF and HZ wrote the manuscript and prepared a revised version. All authors approved the final version.
Supplementary Material
Isotopomer labeling patterns of proteinogenic amino acids. The variations between the biological replicates were less than 2%. Text S2. Metabolic flux calculation by 13C-MFA. Figure S1. Measured and simulated isotopomer labeling patterns of proteinogenic amino acids in enzyme-based-library of S. cerevisiae strains.
Contributor Information
Xueyang Feng, Email: xfeng@igb.illinois.edu.
Huimin Zhao, Email: zhao5@illinois.edu.
Acknowledgements
This work was supported by Energy Biosciences Institute. XF thanks for the Shen postdoc fellowship from the Department of Chemical and Biomolecular Engineering at the University of Illinois at Urbana-Champaign.
References
- van Maris A, Abbott D, Bellissimi E, van den Brink J, Kuyper M, Luttik M, Wisselink H, Scheffers W, van Dijken J, Pronk J. Alcoholic fermentation of carbon sources in biomass hydrolysates by Saccharomyces cerevisiae: current status. Antonie Van Leeuwenhoek. 2006;90:391–418. doi: 10.1007/s10482-006-9085-7. [DOI] [PubMed] [Google Scholar]
- Bokinsky G, Peralta-Yahya P, George A, Holmes B, Steen E, Dietrich J, Soon Lee T, Tullman-Ercek D, Voigt C, Simmons B, Keasling J. Synthesis of three advanced biofuels from ionic liquid-pretreated switchgrass using engineered Escherichia coli. Proc Natl Acad Sci U S A. 2011;108:19949–19954. doi: 10.1073/pnas.1106958108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Cheng J-s, Wang B, Fink GR, Stephanopoulos G. Xylose isomerase overexpression along with engineering of the pentose phosphate pathway and evolutionary engineering enable rapid xylose utilization and ethanol production by Saccharomyces cerevisiae. Metab Eng. 2012. In press. [DOI] [PubMed]
- Jeffries T, Jin Y. Metabolic engineering for improved fermentation of pentoses by yeasts. Appl Microbiol Biotechnol. 2004;63:495–509. doi: 10.1007/s00253-003-1450-0. [DOI] [PubMed] [Google Scholar]
- Jeffries TW. Engineering yeasts for xylose metabolism. Curr Opin Biotechnol. 2006;17:320–326. doi: 10.1016/j.copbio.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Bruinenberg PM, Bot PHM, Dijken JP, Scheffers WA. The role of redox balances in the anaerobic fermentation of xylose by yeasts. Eur J Appl Microbiol Biotechnol. 1983;18:287–292. doi: 10.1007/BF00500493. [DOI] [Google Scholar]
- Van Vleet JH, Jeffries TW. Yeast metabolic engineering for hemicellulosic ethanol production. Curr Opin Biotechnol. 2009;20:300–306. doi: 10.1016/j.copbio.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Runquist D, Hahn-Hägerdal B, Bettiga M. Increased expression of the oxidative pentose phosphate pathway and gluconeogenesis in anaerobically growing xylose-utilizing Saccharomyces cerevisiae. Microb Cell Fact. 2009;8:49. doi: 10.1186/1475-2859-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi M, Erlemann P, Bui-Thanh N-A, Dellweg H. Xylose fermentation by yeasts 4. Purification and kinetic studies of xylose reductase from Pichia stipitis. Appl Microbiol Biotechnol. 1988;29:148–154. doi: 10.1007/BF00939299. [DOI] [Google Scholar]
- Watanabe S, Saleh A, Pack S, Annaluru N, Kodaki T, Makino K. Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein engineered NADP+ −dependent xylitol dehydrogenase. J Biotechnol. 2007;130:316–319. doi: 10.1016/j.jbiotec.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Kodaki T, Makino K. Complete reversal of coenzyme specificity of xylitol dehydrogenase and increase of thermostability by the introduction of structural zinc. J Biol Chem. 2005;280:10340–10349. doi: 10.1074/jbc.M409443200. [DOI] [PubMed] [Google Scholar]
- Jeppsson M, Bengtsson O, Franke K, Lee H, Hahn-Hägerdal B, Gorwa-Grauslund M. The expression of a Pichia stipitis xylose reductase mutant with higher KM for NADPH increases ethanol production from xylose in recombinant Saccharomyces cerevisiae. Biotechnol Bioeng. 2006;93:665–673. doi: 10.1002/bit.20737. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Abu SA, Pack S, Annaluru N, Kodaki T, Makino K. Ethanol production from xylose by recombinant Saccharomyces cerevisiae expressing protein-engineered NADH-preferring xylose reductase from Pichia stipitis. Microbiology. 2007;153:3044–3054. doi: 10.1099/mic.0.2007/007856-0. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Pack S, Saleh A, Annaluru N, Kodaki T, Makino K. The positive effect of the decreased NADPH-preferring activity of xylose reductase from Pichia stipitis on ethanol production using xylose-fermenting recombinant Saccharomyces cerevisiae. Biosci Biotechnol Biochem. 2007;71:1365–1369. doi: 10.1271/bbb.70104. [DOI] [PubMed] [Google Scholar]
- Petschacher B, Nidetzky B. Altering the coenzyme preference of xylose reductase to favor utilization of NADH enhances ethanol yield from xylose in a metabolically engineered strain of Saccharomyces cerevisiae. Microb Cell Fact. 2008;7:9. doi: 10.1186/1475-2859-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson O, Hahn-Hägerdal B, Gorwa-Grauslund M. Xylose reductase from Pichia stipitis with altered coenzyme preference improves ethanolic xylose fermentation by recombinant Saccharomyces cerevisiae. Biotechnol Biofuels. 2009;2:9. doi: 10.1186/1754-6834-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runquist D, Hahn-Hägerdal B, Bettiga M. Increased ethanol productivity in xylose-utilizing Saccharomyces cerevisiae via a randomly mutagenized xylose reductase. Appl Environ Microbiol. 2010;76:7796–7802. doi: 10.1128/AEM.01505-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Shen Y, Li X, Bao X. Effect of the reversal of coenzyme specificity by expression of mutated Pichia stipitis xylitol dehydrogenase in recombinant Saccharomyces cerevisiae. Lett Appl Microbiol. 2007;45:184–189. doi: 10.1111/j.1472-765X.2007.02165.x. [DOI] [PubMed] [Google Scholar]
- Matsushika A, Watanabe S, Kodaki T, Makino K, Inoue H, Murakami K, Takimura O, Sawayama S. Expression of protein engineered NADP+−dependent xylitol dehydrogenase increases ethanol production from xylose in recombinant Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2008;81:243–255. doi: 10.1007/s00253-008-1649-1. [DOI] [PubMed] [Google Scholar]
- Matsushika A, Inoue H, Watanabe S, Kodaki T, Makino K, Sawayama S. Efficient bioethanol production by a recombinant flocculent Saccharomyces cerevisiae strain with a genome-integrated NADP+ −dependent xylitol dehydrogenase gene. Appl Environ Microbiol. 2009;75:3818–3822. doi: 10.1128/AEM.02636-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krahulec S, Klimacek M, Nidetzky B. Engineering of a matched pair of xylose reductase and xylitol dehydrogenase for xylose fermentation by Saccharomyces cerevisiae. Biotechnol J. 2009;4:684–694. doi: 10.1002/biot.200800334. [DOI] [PubMed] [Google Scholar]
- Jeppsson M, Johansson B, Hahn-Hägerdal B, Gorwa-Grauslund MF. Reduced oxidative pentose phosphate pathway flux in recombinant xylose-utilizing Saccharomyces cerevisiae strains improves the ethanol yield from xylose. Appl Environ Microbiol. 2002;68:1604–1609. doi: 10.1128/AEM.68.4.1604-1609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verho R, Londesborough J, Penttila M, Richard P. Engineering redox cofactor regeneration for improved pentose fermentation in Saccharomyces cerevisiae. Appl Environ Microbiol. 2003;69:5892–5897. doi: 10.1128/AEM.69.10.5892-5897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen J, Aristidou A, Salusjärvi L, Ruohonen L, Penttilä M. Metabolic flux analysis of xylose metabolism in recombinant Saccharomyces cerevisiae using continuous culture. Metab Eng. 2003;5:16–31. doi: 10.1016/S1096-7176(02)00012-5. [DOI] [PubMed] [Google Scholar]
- Sonderegger M, Jeppsson M, Hahn-Hägerdal B, Sauer U. Molecular basis for anaerobic growth of Saccharomyces cerevisiae on xylose, investigated by global gene expression and metabolic flux analysis. Appl Environ Microbiol. 2004;70:2307–2317. doi: 10.1128/AEM.70.4.2307-2317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergdahl B, Heer D, Sauer U, Hahn-Hägerdal B, van Niel E. Dynamic metabolomics differentiates between carbon and energy starvation in recombinant Saccharomyces cerevisiae fermenting xylose. Biotechnol Biofuels. 2012;5:34. doi: 10.1186/1754-6834-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jin M, Balan V, Jones AD, Li X, Li B-Z, Dale BE, Yuan Y-J. Comparative metabolic profiling revealed limitations in xylose-fermenting yeast during co-fermentation of glucose and xylose in the presence of inhibitors. Biotechnol Bioeng. 2013. In press. [DOI] [PubMed]
- Kim B, Du J, Eriksen D, Zhao H. Combinatorial design of a highly efficient xylose utilizing pathway for cellulosic biofuels production in Saccharomyces cerevisiae. Appl Environ Microbiol. 2012;79:931–941. doi: 10.1128/AEM.02736-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen S, Sauer U. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res. 2011;11:263–272. doi: 10.1111/j.1567-1364.2010.00713.x. [DOI] [PubMed] [Google Scholar]
- Blank LM, Sauer U. TCA cycle activity in Saccharomyces cerevisiae is a function of the environmentally determined specific growth and glucose uptake rates. Microbiology. 2004;150:1085–1093. doi: 10.1099/mic.0.26845-0. [DOI] [PubMed] [Google Scholar]
- Wahl SA, Dauner M, Wiechert W. New tools for mass isotopomer data evaluation in 13C flux analysis: mass isotope correction, data consistency checking, and precursor relationships. Biotechnol Bioeng. 2004;85:259–268. doi: 10.1002/bit.10909. [DOI] [PubMed] [Google Scholar]
- Maaheimo H, Fiaux J, Cakar Z, Bailey J, Sauer U, Szyperski T. Central carbon metabolism of Saccharomyces cerevisiae explored by biosynthetic fractional 13C labeling of common amino acids. Eur J Biochem. 2001;268:2464–2479. doi: 10.1046/j.1432-1327.2001.02126.x. [DOI] [PubMed] [Google Scholar]
- Feng X, Tang K-H, Blankenship RE, Tang YJ. Metabolic flux analysis of the mixotrophic metabolisms in the green sulfur bacterium Chlorobaculum tepidum. J Biol Chem. 2010;285:35104–35112. doi: 10.1074/jbc.M110.163303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Thiele I, Palsson BØ. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson FJ. D-Xylulokinase. Methods Enzymol. 1966;9:454–458. [Google Scholar]
- Shao Z, Zhao H, Zhao H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009;27:e16. doi: 10.1093/nar/gkn991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalcinati G, Otero J, Van Vleet J, Jeffries T, Olsson L, Nielsen J. Evolutionary engineering of Saccharomyces cerevisiae for efficient aerobic xylose consumption. FEMS Yeast Res. 2012;12:582–597. doi: 10.1111/j.1567-1364.2012.00808.x. [DOI] [PubMed] [Google Scholar]
- Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E. The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem. 2004;279:6613–6619. doi: 10.1074/jbc.M311657200. [DOI] [PubMed] [Google Scholar]
- Rühl M, Le Coq D, Aymerich S, Sauer U. 13C-flux analysis reveals NADPH-balancing transhydrogenation cycles in stationary phase of nitrogen-starving Bacillus subtilis. J Biol Chem. 2012;287:27959–27970. doi: 10.1074/jbc.M112.366492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y-S, Jeffries TW. Stoichiometric network constraints on xylose metabolism by recombinant Saccharomyces cerevisiae. Metab Eng. 2004;6:229–238. doi: 10.1016/j.ymben.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Förster J, Famili I, Fu P, Palsson B, Nielsen J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 2003;13:244–253. doi: 10.1101/gr.234503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Zhao H. 13C metabolic flux analysis of glucose and xylose metabolism in Saccharomyces cerevisiae and Scheffersomyces stipitis. AIChE J. 2013;59:3195–3202. doi: 10.1002/aic.14182. [DOI] [Google Scholar]
- Jordà J, Jouhten P, Cámara E, Maaheimo H, Albiol J, Ferrer P. Metabolic flux profiling of recombinant protein secreting Pichia pastoris growing on glucose:methanol mixtures. Microb Cell Fact. 2012;11:57. doi: 10.1186/1475-2859-11-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyland J, Fu J, Blank L, Schmid A. Quantitative physiology of Pichia pastoris during glucose-limited high-cell density fed-batch cultivation for recombinant protein production. Biotechnol Bioeng. 2010;107:357–368. doi: 10.1002/bit.22836. [DOI] [PubMed] [Google Scholar]
- Heyland J, Fu J, Blank L, Schmid A. Carbon metabolism limits recombinant protein production in Pichia pastoris. Biotechnol Bioeng. 2011;108:1942–1953. doi: 10.1002/bit.23114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Isotopomer labeling patterns of proteinogenic amino acids. The variations between the biological replicates were less than 2%. Text S2. Metabolic flux calculation by 13C-MFA. Figure S1. Measured and simulated isotopomer labeling patterns of proteinogenic amino acids in enzyme-based-library of S. cerevisiae strains.