

Abstract
Motivation: Easily visualization of complex data features is a necessary step to conduct studies on next-generation sequencing (NGS) data. We developed STAR, an integrated web application that enables online management, visualization and track-based analysis of NGS data.
Results: STAR is a multilayer web service system. On the client side, STAR leverages JavaScript, HTML5 Canvas and asynchronous communications to deliver a smoothly scrolling desktop-like graphical user interface with a suite of in-browser analysis tools that range from providing simple track configuration controls to sophisticated feature detection within datasets. On the server side, STAR supports private session state retention via an account management system and provides data management modules that enable collection, visualization and analysis of third-party sequencing data from the public domain with over thousands of tracks hosted to date. Overall, STAR represents a next-generation data exploration solution to match the requirements of NGS data, enabling both intuitive visualization and dynamic analysis of data.
Availability and implementation: STAR browser system is freely available on the web at http://wanglab.ucsd.edu/star/browser and https://github.com/angell1117/STAR-genome-browser.
Contact: wei-wang@ucsd.edu
1 INTRODUCTION
Using genome-browser software to visualize sequence data is often a productive first step to take when tasked with extracting biological meaning from experimental results. Existing genome-browsers fall mainly into two categories: stand-alone and internet-based. Stand-alone programs, such as seqMINER, CisGenome, MapView, EagleView, NGSView, MagicViewer, integrative genomics viewer and GenomeView (Abeel et al., 2012; Arner et al., 2010; Bao et al., 2009; Fiume et al., 2010; Ge et al., 2011; Hou et al., 2010; Huang and Marth, 2008; Jiang et al., 2010; Robinson et al., 2011; Shannon et al., 2006; Stothard and Wishart, 2005; Ye et al., 2011), are installed locally and allow users to perform analysis and data visualization to the limit of their local machine. Users must ensure software remains up-to-date and are often required to download and reformat datasets for use with the application without a clear channel to share data with others. In contrast, internet-based browsers require no download, installation or update, typically delegate computational load to remote servers, and store data centrally for all users to immediately browse. Some internet-based data exploration platforms such as Cistrome (Liu et al., 2011) and Cistrome Finder (Sun et al., 2013) also provide visualization function. Other popular web-based browsers, such as University of California, Santa Cruz (UCSC) genome browser (Kent et al., 2002), UCSC Cancer Genomics Browser (Zhu et al., 2009) and Ensembl genome browser (Stalker et al., 2004), provide the benefits of web applications but suffer from continued reliance on first-generation web application technology such as postback and server-side drawing, making little use of the computational power of clients.
Several publicly released genome browsers, such as the National Center for Biotechnology Information (NCBI) epigenome viewer (Sayers et al., 2011), Generic Genome Browser (GBrowse2.0) (Stein et al., 2002), Anno-J (Lister et al., 2008), JBrowse (Skinner et al., 2009) and the WashU epigenome browser(Zhou, 2011), offer an enhanced user experience through the use of second-generation (Web 2.0) technologies such as advanced third-party JavaScript libraries, asynchronous communications (AJAX), RESTful architecture, and client-side rendering. These browsers deliver a more desktop-like user experience than their first-generation counterparts and distribute computation more evenly between the server and the client, thereby helping to reduce network loading.
Although second-generation browsers have significantly enhanced the user experience, they have room for improvement on some daily used functions. The NCBI epigenome viewer allows smooth panning and zooming but does not support comparative analysis between data tracks. GBrowse2.0 and JBrowse do not provide registries of public data tracks for ready-to-use visualization and make use of statically generated server-side images that limit client-side interactivity. The WashU epigenome browser displays data tracks from the Human Epigenome Atlas [produced by the National Institutes of Health (NIH) Roadmap Epigenomics project (Bernstein et al., 2010) and the Encyclopedia of DNA Elements (ENCODE) project (ENCODE Project Consortium, 2007)] along with interactive visualization and analysis functions such as correlation and statistics test, but there is no sequencing data analysis tool like peak calling, and it is not fully compatible to Microsoft Internet Explorer and Firefox web browsers. A feature lacking from all second-generation browsers is the inclusion of sophisticated analysis tools as an integrated component.
We present STAR, a next-generation web-based application that aims to provide an integrated solution for both visualization and analysis of deep-sequencing data, along with simple access to public datasets. On the client side, STAR features a next-generation browser that is based on the Anno-J (Lister et al., 2008) genome browser, modifying it extensively to add enhanced visualization options, bug-fixes, improved cross-browser compatibility, dynamic access to an extensive collection of public datasets and a suite of in-browser analysis tools (such as correlation analysis and peak calling). The browser features client-side rendering via HTML5 Canvas, smooth navigation via asynchronous data transfer, dynamic track state changes (zoom, scale, resize) via JavaScript, cross-domain data retrieval and modular track development architecture. On the server side, STAR provides distributed mechanism to collect and process data from public data sources, user access control to protect private data, data track management and data configuration management, portable personal server setup and utilities for storage, parsing and analysis of sequence data.
2 IMPLEMENTATION
The goal of STAR is to provide a scalable system that can process, analyze and visualize private data alongside the large volume of publicly available data (http://wanglab.ucsd.edu/star/browser). The system can be described as three tiered, with a middle layer between the front-end and the back-end (Fig. 1 provides an architectural overview of the system).
Fig. 1.

Architecture and workflow of the STAR system
(1) The back-end (server-side) layer is responsible for managing data and for providing client-accessible hooks via web services. The STAR server periodically downloads data from public databases, such as the NIH gene expression omnibus (GEO) database and the Web sites of the NIH Roadmap Epigenomics and ENCODE consortium. Automated processing modules then parse any downloaded data, build index, deposit them into a distributed database system and create RESTful web services that provide access via the Hypertext Transfer Protocol (HTTP). Where this has been conducted on a remote server, STAR needs only be informed of the URL of the appropriate web service and does not need to mirror data.
(2) The middle layer, implemented as a central Web site, provides a web-based management interface for the system. Through the web interface, user information, user groups, permission controls, track visibility, metadata and visualization options may be edited by an authorized user. For general use, all available (visible) data tracks are searchable, allowing users to select desired tracks and assemble them into a customized list for visualization.
(3) The front-end layer is responsible for data representation and consists of a DHTML (Dynamic HTML) genome browser, built using the ExtJS JavaScript library, which runs in a modern web browser such as Chrome, Safari, Firefox or Internet Explorer. The browser supports extensive client-side view customization, thereby reducing computation load on the server.
2.1 The main web interface
The STAR system provides a central web that allows users to manage and share data from different sources (Fig. 2). In the top-right frame, users can browse and sort all available data tracks by organism, cell type, data type (e.g. ChIP-seq, RNA-seq, MethylC-seq), generation institute, upload date, data accessibility (public, group or private), track type (read or intensity type that does or does not contain read sequences) or type of histone mark. Full text search using key words is also available for quick data retrieval.
Fig. 2.
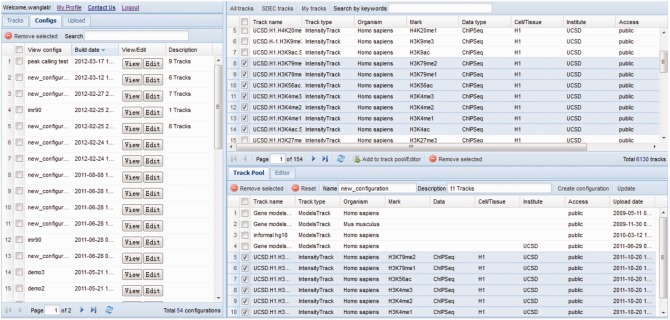
The central web page of the STAR system
Any dataset, selected by the user for visualization, is first added to a track pool in the bottom-right frame. The track pool also includes different gene model tracks, such as human and mouse species for RefSeq gene information. Those selected tracks are then assembled into a ‘view configuration’ that is added to a list of configurations shown in the left frame. Next to each configuration in the list is a ‘view’ button that, when clicked, launches a new browser instance showing only the tracks selected in the configuration (in the order selected). The main interface and graphical features of the STAR browser are shown in Figures 3 and 4 and described in detail in the following sections.
Fig. 3.

Screenshot of the main graphical interface: (A) the information of the data tracks. The tracks can be re-ordered, activated or deactivated, e.g. dragging the track names to re-order; (B) additional information window shows the output of in-browser analysis (e.g. peak found by peak-calling algorithms and correlation coefficient between two tracks) or zoom-in details; (C) global settings and functional analysis buttons to change the global view settings, such as display mode, scaling method, global scaling factor, dragging mode, zooming level, locations and data analysis options; (D) Y-labels and track selector; (E) in-track configuration toolbar to select display mode for Hi-C data; (F) track lane displays data signal and dialog buttons for color changing, scaling factor and lane-height resizing
Fig. 4.

Features of the STAR browser: (A) sequence reads at nucleotide resolution; (B) heatmap display mode; (C) Hi-C interactions in triangle mode; (D) Hi-C interactions in rectangle mode; (E) correlation coefficients between selected track pairs; (F) set operation (e.g. sum/subtraction/merge of two tracks) results
2.2 Browser features
The browser facilitates simple navigation of data visualizations. To change locations, a user may drag the mouse left or right, triggering asynchronous retrieval and rendering of new data as required. Alternatively, users can input a chromosome coordinate in the top toolbar or a gene name in the searching box of gene model track to jump directly to a specific location. Tracking the mouse cursor, an information box shows the current genomic coordinate and the sequencing read count at that position in the track under the cursor. Users have the option to set a baseline level of read counts to only visualize signals stronger than that baseline, and may change the color of data series as desired using a color palette.
2.2.1 Single base pair and chromosome-wide resolution
The STAR browser supports a wide range of zooming levels from single base pair to chromosome-wide resolution. At single base resolution, users can visualize read sequences from each experiment, compare them with a reference genome sequence shown at the top of the panel (Fig. 4A) and readily identify single nucleotide polymorphisms and indels. Chromosome-wide resolution provides a bird’s-eye view of the whole chromosome, facilitating identification and comparison of chromosome-wide patterns across tracks. The zoom level may be adjusted by clicking on the zoom-in/out buttons to zoom by a fixed interval, by typing in a specific zoom ratio in bases per pixel (such as 10:1 or 1:10) or by selecting zoom as a drag mode and highlighting a region to be zoomed in upon.
2.2.2 Flexible data scaling
As signal intensity can vary widely within and between data tracks, it is important to scale data appropriately. The STAR browser supports three scaling modes: global-fixed, individual and uniform. In the global-fixed scaling mode, the signals of all tracks are displayed at the same scale through the entire genome, in contrast to the other two modes in which signals are optimized for display in the current visualization window. The global scaling factor can be set via a y-axis input box. In the uniform scaling mode, all tracks use the same scaling factor within the current view window so that signals in different data tracks can be directly compared. In the individual scaling mode, individual tracks are automatically scaled to optimize the visualization of each individual track in the current window. In addition, users can manually scale signals by either specifying the global (for all tracks) or individual (for individual tracks) scaling factor. Another useful feature is to customize the baseline for visualization, which allows removal of noise.
2.2.3 Flexible display modes
There are two display modes in the browser: histogram and heatmap. Data series are displayed in histogram mode by default, with the height of each bar representing sequence read depth at that location. In heatmap mode, the read count or signal intensity is colored red for high values through to green for low. Heatmap mode permits a more compact display, allowing on-screen viewing of more datasets at once.
2.2.4 Display of chromosomal interaction data
The STAR browser provides a novel function to browse and visualize 2D interaction data such as Hi-C data. In this function, a 2D heat map shows the interaction intensity or correlation between pairs of genomic loci. As the need for visualizing the relationship between different loci is increasing, this unique function makes it possible to interactively visualize such 2D datasets and connect the information with available genomic annotation. There are two display modes for 2D maps: triangle and rectangle (Fig. 3C and 3D). The triangle mode shows the interaction intensity of Hi-C at a certain position and the two interacting loci form a regular triangle (Fig. 3C). In this way, the proximal interactions are displayed in the bottom region and long distance interactions in the upper region. The rectangle mode shows the interaction intensity as a matrix (Fig. 3D) in which the proximal interactions are shown along the diagonal. In both display modes, users can visualize the Hi-C data within the same chromosome or between different chromosomes. Positions of the interacting loci and the interaction intensity are shown for the current position of the mouse cursor.
2.2.5 In-browser analysis
To allow easy interpretation of data, several in-browser analysis tools have been implemented. Users may select tracks for analysis by ticking the checkbox of each track, and the analysis results are displayed in a new track next to the selected track for analysis or in a separate information window. The STAR system currently provides three types of analysis tools that are widely used for NGS data exploration: data statistics, set operations and peak calling.
Statistics functions include ‘intensity’ and correlation analysis. In addition to the function of showing the signal intensity at the mouse cursor's location, users can also measure the average signal intensity for a sample (data track) or for the current view region using the ‘intensity’ operation on the selected tracks. Correlation analysis helps user to evaluate how similar one data track is to other tracks. The first selected one is considered as the reference, and the Pearson correlations with each of other tracks can be obtained by clicking the ‘correlation’ button. The results are shown as histograms in the left bottom information window (Fig. 4E). These ‘statistics functions’ in STAR are region-based, i.e. the result is calculated for the current view range. Such a design provides users the flexibility to explore regions of interest with a fast speed. Users can also zoom out to the entire chromosome to perform these calculations if needed.
Set operations allow comparison of signals between different data tracks, which include merge, intersect, sum and subtract. Those operations provide an easy way to show the difference and common signals between the two selected tracks. For example, ‘merge’ operation takes union of all the signals from two tracks and displays them in a new track with signals colored differently, which allows easy visualization of the common and different signals. ‘Intersect’ displays regions that have signals from all the selected tracks, and it helps to identify common signal peaks/domains and remove some weak signals. ‘Sum’ and ‘subtract’ are also useful when users want to plus or minus reference signals from a data track.
Another in-browser analysis tool is peak calling that is, to our knowledge, not available in any other existing general purpose browser. Peak calling is a computational method widely used to identify peaky signals such as protein binding sites or chromatin modification regions. STAR system has wrapped up Model-based Analysis for ChIP-Seq (MACS) (Zhang et al., 2008) as the peak calling algorithm in back-end. STAR determines whether peak calling can be performed on the selected track, and only enables a check box next to the tracks for which peaks can be called. The peak calling runs in asynchronous mode at the back-end and the results are shown as a new track once available. Users do not have to keep waiting on the page because the peak calling results are stored in the back-end and can be visualized immediately in the user's next login. It is worth noting that the STAR system only provides the mechanism to streamline the whole workflow and handle output data. The analysis results are based on the referenced program itself. These jobs can be easily submitted to a cluster or grid computing environment for using distributed computing resource without too much complexity by modifying job policy.
2.2.6 Data formats and data processing flow
Back-end data processing module processes the data track files, either uploaded by users or downloaded from the public databases. This module recognizes multiple input formats, such as Bowtie (Langmead et al., 2009), Eland and Sequence Alignment/Map (SAM/BAM) formats (Li et al., 2009), for short reads alignment results. Other data formats, including Browser Extensible Data (BED)/bigBed, Wiggle format (WIG)/bigwig, General Feature Format (GFF) and MapView, which usually can be visualized by UCSC genome browser, are also supported. The STAR system automatically determines the input data format and extracts the required information. Although there are many different types of data features for visualization, such as gene model, ChIP-Seq, RNA-Seq and CpG methylation sites, and the browser renders them in different presentation features, they all share essentially the same information such as location, strand, sequence reads and signal intensity values. They can be extracted and abstracted into a uniform internal database format, which is crucial for automating data process (Fig. 5).
Fig. 5.

Data processing workflow of the STAR system
2.2.7 View configuration saving
Users are often interested in specific genomic regions or prefer a specific set of scaling and zooming levels in their visualization. The STAR browser allows users to save their preferences using the ‘save’ button and the user can share the URL with others to display the saved configuration. In addition, ‘snapshot’ function allows the user to save the current view position/configuration that can be loaded (‘load’ button) or removed (‘remove’ button) from the user's account.
2.3 Data management system
A major advantage of web-based browser systems compared with stand-alone browsers is that users can access not only their own datasets but also public datasets without downloading an entire set to a local computer. STAR has a scalable and distributed architecture that meets the requirements of handling large amounts of data. Although data sources may be distributed across many remote sites, metadata and data access services are organized and managed by a central web interface. This promotes better sharing of data tracks between users.
2.3.1 Data access control
Registered users can use the data-access-control module to customize data tracks and their configurations. In addition to visualizing the public data stored in the STAR database, users can also view their own data and select one of the three permission levels to control access to the data: private (only accessible to the user), group (accessible to all users in the defined group) and public (accessible to the public). The group access permission is especially useful for collaborators sharing private data. STAR also allows anonymous login as a guest without a password. Guests gain access to all public data tracks and the full functionality of the STAR system for the current session.
2.3.2 Data track management
Combined with a valid work-around to address cross-domain restrictions in web browsers (such as the use of a properly secured reverse proxy), data may be pulled from multiple remote sites directly into a visualization instance by simply knowing the URL of a data source. This helps balance data loading between multiple servers and provides a simple mechanism for publishing locally stored data. When generating custom data tracks, users can process and store data in a local computer and simply submit the track meta-information to the STAR system by providing access permission, species, data track type and service URL. The STAR system saves this information to provide centralized data management and thus functions as a one-stop shop for users.
2.3.3 Data configuration management
To view data tracks, users need to assemble a configuration that contains a list of desired tracks and display settings. As a user may have thousands of data tracks available to them, it is essential to have an easy way to find and retrieve the tracks of interest. In the STAR system, users can perform a full text search using key words. Alternatively, users can browse all the available data tracks based on information such as species, cell type or experimental type. Retrieved data tracks can then be sorted by generation date, experimenter or some other piece of information. Users can select data tracks to build a new data configuration or can add the selected data tracks to an existing configuration. The data configuration can be viewed using the default settings for factors including chromosome number, start position, display resolution, color and scaling factor. Users can also customize and save visualization settings.
2.4 Back-end database
The STAR system collects diverse sequencing data that are publicly available and makes them ready-to-view for users. STAR gathers data files from different data sources following their data release policy. A data retrieving program can periodically check the Web sites of NCBI gene expression omnibus, ENCODE Consortium and NIH Roadmap Epigenomics Mapping Consortium and downloads any new or updated sequencing data files to the STAR data servers. Automated routines then uncompress the data files and extract needed information, such as location, sequence and signal intensity values. Extracted data can then be deposited into multiple data nodes supported by file system database using a simple internal format (Fig. 5). When a client requests new data, the server retrieves data from the database or from an existing cache and responds with it to the client. Currently, there are >7000 data tracks available in the STAR database that is accessible to the public (Table 1).
Table 1.
Summary of data tracks in STAR database system
| Species | Homo sapiens (>6000), Mus musculus, Arabidopsis thaliana and so forth. |
|---|---|
| Chromatin marks | H2AK5ac; H2BK5ac; H2BK120ac; H2BK12ac; H2BK15ac; H2BK20ac; H3K14ac; H3K18acl H3K23ac H3K27ac; H3K27me3; H3K36me3; H3K4ac; H3K4me1; H3K4me2; H3K4me3; H3K56ac; K3K79me1; H3K79me2; H3K9ac; H3K9me3; H4K20me1; H4K5ac; H4K8ac; H4K91ac; and others. |
| Experimental type | ChIP-Seq; DNaseSeq; Bisufite-Seq; MeDIP-Seq; MRE-Seq; RRBS; RNASeq; and others. |
| Tissue/cell type | Gm12878; H1; H9; Hepg2; Hmec; Hsmm; Huvec; K562; Nhek; Nhlf; IMR90; iPS; and others. |
| Data sources | Epigenomics Roadmap project; ENCODE project; and others. |
To facilitate interpretation of data tracks, the STAR system also provides access to results from computational analysis. Currently, epigenetic states annotated by a hidden Markov model called ChroModule (Won et al., 2013) are available in eight cell types (Gm12878, H1, Hmec, Hsmm, Huvec, K562, Nhek, Nhlf). ChroModule calculates the probability of each 100 bp bin assigned to one of the five annotated categories including promoter, enhancer, intragenic, repressed and background based on the histone modification data. These probabilities are available as data intensity tracks in STAR for visualization.
3 DISCUSSION
The STAR system aims to provide an integrated solution to help researchers easily visualize and analyze sequencing data. To achieve good user experience, fast data access and quick response are crucial aspects. Such features are usually limited by network communications and computational load on servers. For a traditional web-based genome browser, usually the web server or application server retrieves data from a database and renders an HTML page to the client. Almost all the computing load is on the server side and heavy request traffic places heavy load on servers. The STAR system distributes computation between the client and the server(s). Graphical representations of data are constructed purely on the client side and modifications to view state (such as scaling or color) do not require any form of server interaction thanks to the use of HTML Canvas. (Page refreshes are completely avoided through the use of AJAX to provide a smoother browsing experience.) Most modern, W3C-compliant web browsers are in the process of fully adopting the new HTML5 specification, which includes the Canvas element (although most such as Safari, Chrome, Opera and Firefox already supported Canvas many years ago). To provide compatibility with older versions of Internet Explorer, Google’s ExplorerCanvas plug-in provides the required functionality.
When users zoom out to view a larger genomic region, more data must be retrieved from the data server, which takes more time and disk space. To deal with this, JBrowse provides a solution similar to Google Maps by pre-generating small images at different zooming levels. However, because those images are generated on the server side, it is usually difficult to provide a flexible way to interact with the data and change view mode rapidly when compared with the performance of STAR. Instead of generating static images, STAR pre-calculates the coverage of positions at each zooming level. The server retrieves data from a pre-calculated database at each view resolution. This significantly reduces the data amount retrieved and the time for data processing. For the purpose of simplicity, STAR uses the Berkeley DB (BDB) file system, rather than a relational database such as MySQL to deposit sequencing data. This provides key features such as optimized indexing and caching to deposit and retrieve data from files using several customized language-specific APIs. This design has a better performance than most relational databases, and there is no need to run a large system to maintain the sequencing data. To reduce searching space, the input data are deposited into separate file databases in which each file contains all the data features for a specific chromosome. Combined with pre-calculation, file system database and cache technologies, the data preparation is fast and provides almost constant access time regardless of the zooming level. To reduce network transit time, all data are compressed (where supported by the client). Upon receipt and unpacking, data parsing times in the browser are minimal due to the fact that JavaScript Object Notation (JSON) format is natively supported by JavaScript.
If custom data file is too large for network transferring (STAR limits the maximum upload size up to 2 GB), or unpublished data need to be kept private, user would probably prefer to storing data in their own data server and this is supported by STAR. This function is available in the UCSC genome browser using indexed data formats, such as BAM and bigwig to contain data features. Data files are distributed on web accessible servers and queried via HTTP to retrieve the portion that is needed for the chromosomal position that users are currently viewing. In contrast, STAR provides a high efficiency C++ program for data processing and an in-house Common Gateway Interface (CGI) server program for processing data requests. There are several advantages of this design. First, users do not need to install and configure a complicated web server such as Apache. The STAR system handles data request using a simple program that is easy to install and launch. Second, STAR uses a simple data processing program to convert multiple input formats to a uniform internal format, which is easy for data processing. Third, it provides a better solution for data service provision, such as pre-calculation and cache functions to improve system performance. Once the meta-information for each track is generated and submitted through the STAR upload page, data are available for direct viewing and sharing with other users.
We compared the STAR browser system with several other web-based genome browsers (Table 2). Relative to other Web 2.0-enabled browsers, STAR provides a large collection of sequencing data tracks, allowing users to visualize most public datasets. In addition to the features that are available in many recently developed genome browsers, such as rich visualization features and high interactivity, STAR has implemented sophisticated in-browser analysis tools such as peak calling that are not yet available in other browsers. The functionality of in-browser analysis makes data analysis intuitive and straightforward, which is particularly useful for users without much data analysis experience. We are aware that there is still more room to improve the functionality and usability of the STAR system. In the future, we plan to further improve the user-friendly interface, add more graphical features to the browser and include more annotation tracks to help researchers easily extract biological information from sequence data.
Table 2.
Feature comparison between different genome browser systems
| Feature | UCSC genome browser | Gbrowse 2.0 | Anno-J | Jbrowse | The human epigenome browser at Washington University | STAR system |
|---|---|---|---|---|---|---|
| Technology | Traditional web technology | AJAX, Server side render | AJAX, HTML 5.0 Canvas | AJAX, tiled images | AJAX, Google Maps APIs | AJAX, HTML 5.0 Canvas |
| 3D data | N/A | N/A | N/A | N/A | Hi-C | Hi-C |
| Heatmap | Yes | No | No | No | Yes | Yes |
| Web browser | All | All | Chrome, Firefox | All | Chrome, Firefox | Chrome, Internet Explorer, Firefox, Opera |
| Display settings | Predefined | Predefined | Predefined | N/A | Dialog | Toolbar in each track |
| Data Scaling | Server side | N/A | Client Side | Server Side | Both client side and server side | Flexible scaling methods, client side |
| Available tracks | Thousands of data tracks, wide range of data sources | N/A | N/A | N/A | NIH Roadmap Epigenomics; ENCODE | NIH Roadmap Epigenomics; ENCODE; GEO |
| In-browser analysis | Correlation | N/A | N/A | N/A | Correlation; Hypothesis test | Set operation; Correlation; Peak calling |
| Data access control | Public; Custom data URLs | N/A | N/A | N/A | Public; Custom data URLs | Private, group, public permissions on tracks and configurations |
Note: Display settings refer to whether the user can adjust the display properties of the data track such as color and signal shape. Data scaling is how to normalize data to show readable and comparable signals between multiple tracks and locations. N/A, the feature is not implemented or the information is not available.
Funding: NIH (U01 ES017166 to B.R., J.E. and W.W.) (in part).
Conflict of Interest: none declared.
REFERENCES
- Abeel T, et al. GenomeView: a next-generation genome browser. Nucleic Acids Res. 2012;40:e12. doi: 10.1093/nar/gkr995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, et al. NGSView: an extensible open source editor for next-generation sequencing data. Bioinformatics. 2010;26:125–126. doi: 10.1093/bioinformatics/btp611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, et al. MapView: visualization of short reads alignment on a desktop computer. Bioinformatics. 2009;25:1554–1555. doi: 10.1093/bioinformatics/btp255. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiume M, et al. Savant: genome browser for high-throughput sequencing data. Bioinformatics. 2010;26:1938–1944. doi: 10.1093/bioinformatics/btq332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge DL, et al. SVA: software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011;27:1998–2000. doi: 10.1093/bioinformatics/btr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou HB, et al. MagicViewer: integrated solution for next-generation sequencing data visualization and genetic variation detection and annotation. Nucleic Acids Res. 2010;38:W732–W736. doi: 10.1093/nar/gkq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Marth G. EagleView: a genome assembly viewer for next-generation sequencing technologies. Genome Res. 2008;18:1538–1543. doi: 10.1101/gr.076067.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, et al. CisGenome Browser: a flexible tool for genomic data visualization. Bioinformatics. 2010;26:1781–1782. doi: 10.1093/bioinformatics/btq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon PT, et al. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner ME, et al. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalker J, et al. The Ensembl Web site: mechanics of a genome browser. Genome Res. 2004;14:951–955. doi: 10.1101/gr.1863004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, et al. The Generic Genome Browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21:537–539. doi: 10.1093/bioinformatics/bti054. [DOI] [PubMed] [Google Scholar]
- Sun H, et al. CistromeFinder for ChIP-seq and DNase-seq data reuse. Bioinformatics. 2013;29:1352–1354. doi: 10.1093/bioinformatics/btt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won KJ, et al. Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res. 2013;41:4423–4432. doi: 10.1093/nar/gkt143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, et al. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 2011;39:e35. doi: 10.1093/nar/gkq1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X. Human epigenome browser at Washington university. Behav. Genet. 2011;41:944–944. doi: 10.1038/nmeth.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JC, et al. The UCSC Cancer Genomics Browser. Nat. Methods. 2009;6:239–240. doi: 10.1038/nmeth0409-239. [DOI] [PMC free article] [PubMed] [Google Scholar]