

Abstract
Large scale analysis of proteins by mass spectrometry is becoming increasingly routine; however, the presence of peptide isomers remains a significant challenge for both identification and quantitation in proteomics. Classes of isomers include sequence inversions, structural isomers, and localization variants. In many cases, liquid chromatography is inadequate for separation of peptide isomers. The resulting tandem mass spectra are composite, containing fragments from multiple precursor ions. The benefits of high-field asymmetric waveform ion mobility spectrometry (FAIMS) for proteomics have been demonstrated by a number of groups, but previously work has focused on extending proteome coverage generally. Here, we present a systematic study of the benefits of FAIMS for a key challenge in proteomics, that of peptide isomers. We have applied FAIMS to the analysis of a phosphopeptide library comprising the sequences GPSGXVpSXAQLX(K/R) and SXPFKXpSPLXFG(K/R), where X = ADEFGLSTVY. The library has defined limits enabling us to make valid conclusions regarding FAIMS performance. The library contains numerous sequence inversions and structural isomers. In addition, there are large numbers of theoretical localization variants, allowing false localization rates to be determined. The FAIMS approach is compared with reversed-phase liquid chromatography and strong cation exchange chromatography. The FAIMS approach identified 35% of the peptide library, whereas LC–MS/MS alone identified 8% and LC–MS/MS with strong cation exchange chromatography prefractionation identified 17.3% of the library.
A challenge facing mass spectrometry-based proteomics is the presence of peptide isomers, which cannot be distinguished on the basis of m/z. In addition to enantiomers and diastereoisomers, isomers arise through localization variants of post-translational modifications (that is, peptides with identical sequences but differing sites of modification), peptides containing isomeric amino acid residues (e.g., leucine/isoleucine), and sequence inversions in which the order of amino acid residues are altered. In addition, structural isomers may arise through different combinations of amino acid residues. For example, a peptide containing the residues serine and alanine has identical mass to one in which those residues are substituted for threonine and glycine. Sequence variants (sequence inversions and structural isomers) are typically straightforward to distinguish following tandem mass spectrometry (MS/MS), either by collision induced dissociation or electron transfer (or capture) dissociation, provided that a single peptide isomer is isolated for fragmentation. Difficulties arise when multiple precursors are selected resulting in compound MS/MS spectra. The presence of fragments from a variety of precursors both confounds the protein database search and leads to incorrect quantitation.1,2 Sequence variants can be separated to some extent by liquid chromatography prior to MS analysis (LC–MS). Nevertheless, Clemmer and co-workers3 showed that this approach lacks the peak capacity to separate large numbers of isomers. For a 4000 member peptide library, ∼50% of the isobars could be separated by LC.3
Ion mobility spectrometry can be applied for the separation of peptide sequence isomers. Hudgins et al. showed the position of lysine within polyalanine peptides had a strong influence on drift times as measured by conventional ion mobility.4 Clemmer and co-workers applied conventional ion mobility to the analysis of large peptide libraries containing numerous sequence variants.3,5 More recently, we have shown that sequence inversions of peptides containing nitrotyrosine can be separated by use of high-field asymmetric waveform ion mobility spectrometry (FAIMS, also known as differential ion mobility).6
FAIMS was introduced by Buryakov et al.7,8 and is based on the differences in mobility of ions in high and low electric fields. Ions are infused between two parallel electrodes across which a voltage is applied via an asymmetric waveform. As a result of their differential ion mobility in high and low electric fields, ions travel a greater distance toward one electrode than the other, eventually colliding with the electrode. To avoid this occurrence, a compensation voltage (CV) is applied to one electrode. By changing the compensation voltage, it is possible to selectively transmit ions through the FAIMS device. FAIMS has successfully been coupled to online nano-LC–MS/MS for proteomic analyses. Saba et al. showed a 55% increase in the number of assigned spectra and saw a 10-fold increase in detection limits when using FAIMS compared to a standard LC–MS/MS approach.9 Swearingen et al.10 demonstrated the improved proteome coverage observed when comparing LC–MS/MS with multiple LC–FAIMS-MS/MS runs at different compensation voltages. Improved proteome coverage by use of FAIMS was also demonstrated in our work on human SUM52 breast carcinoma cells.11 FAIMS is also able to separate localization variants of post-translationally modified peptides, and Bridon et al. have demonstrated the advantages of FAIMS for phosphoproteomics, identifying several isobaric phosphopeptides.12 Typically, large scale phosphoproteomics is carried out using two-dimensional liquid chromatography, often strong cation exchange chromatography coupled with reversed-phase chromatography.13 As for standard proteomics, phosphoproteomics is complicated by the presence of sequence and positional isomers and coeluting peptides. The problem was highlighted by a recent study where 3–6% of phosphopeptides identified from TiO2 enrichment of mouse, fly, and rat protein extracts were isomers.14 An in silico analysis of the human proteome revealed that approximately one-fifth of all tryptic human phosphopeptides are potentially multiply phosphorylated,4 suggesting the problem is greater than previously thought. Unlike electron transfer (or capture) dissociation (ETD, ECD),15,16 analysis of phosphopeptides by CID generally results in the neutral loss of phosphoric acid17 (particularly for serine and threonine phosphorylation), often at the expense of sequence fragments, further complicating identification and localization of the peptide/phosphate. Methods have been introduced, which combine CID with ETD or ECD, such as neutral loss triggered ECD18 or decision tree analysis,19 to address this limitation. Bridon et al. used the decision tree approach in their FAIMS analysis of Drosophila melanogaster.12
Here, we describe the reversed phase (RP) LC–FAIMS-ETD-MS/MS analysis of a phosphopeptide library comprising 4000 members of equal abundance. The library peptides have the sequences GPSGXVpSXAQLX(K/R) and SXPFKXpSPLXFG(K/R), where X = ADEFGLSTVY. The library has numerous sequence inversions and structural isomers. The use of a defined library enables us to assess fully the performance of FAIMS in terms of ability to separate sequence isomers. The results are compared with RPLC–ETD-MS/MS in order to assess the benefits of gas-phase fractionation in FAIMS and with the more widely used two-dimensional approach which combines prefractionation by strong cation exchange chromatography with RPLC–ETD-MS/MS. ETD was employed because of its advantages for phosphopeptide analysis. Again, as a result of using a library of known constraints, we are able to calculate false localization rates for the sites of phosphorylation.
Experimental Section
Synthetic Peptides
Two peptide libraries were synthesized (Alta Biosciences, Birmingham, U.K.) with the basic sequences GPSGXVpSXAQLX(K/R) and SXPFKXpSPLXFG(K/R), where X = ADEFGLSTVY, resulting in a total of 4000 peptides. The libraries were combined and resuspended in water to a concentration of 1 mg/mL. The stock solution was diluted to a final concentration of ∼35 fmol/μL for RPLC–MS/MS analyses.
Strong Cation Exchange (SCX) Liquid Chromatography
The combined synthetic libraries (1 mg) were resuspended in 100 μL of mobile phase A (10 mM KH3PO4, 25% acetonitrile, pH 3, Sigma Aldrich) and loaded onto a 100 × 2.1 mm polysulfethyl A column (5 μm particle size, 20 nm pore size, PolyLC, Columbia, MD) at a flow rate of 200 μL/min. Peptides were separated with a gradient from 0 to 50% mobile phase B (10 mM KH3PO4, 25% acetonitrile, 500 mM KCl, pH 3, Sigma Aldrich) over 40 min, increasing to 70% B over 5 min before returning to 100% A. A total of 15 fractions were collected over 54 min. Fractions were combined as follows: 1, 2, 3, 4, 5, 6, 7 + 8, 9 + 10 + 11, and 12 + 13 + 14 + 15. The combined fractions were dried and resuspended in trifluoroacetic acid (Sigma Aldrich, 0.5%). Prior to analysis, the peptides were desalted (C8 cartridge, Michrom) as follows: Samples were acidified with trifluoroacetic acid (0.5%). The trap was washed with acetonitrile (200 μL) followed by acetonitrile and water (50/50 + 0.1% trifluoroacetic acid, 200 μL). The trap was equilibrated with trifluoroacetic acid (0.1%, 300 μL), and the sample was loaded and washed with trifluoroacetic acid (0.1%, 300 μL). The sample was eluted in acetonitrile and water (70/30 + 0.1% trifluoroacetic acid, 100 μL). Each desalted sample was resuspended in formic acid (0.1%, 110 μL) and diluted by a factor of 5 to give a final concentration of ∼35 fmol/μL. (Peptide concentrations are approximate due to the uneven distribution of peptides between the SCXLC fractions).
RPLC–MS/MS Analysis
Peptides (approximately 173 fmol of each peptide) were loaded onto a 150 mm Acclaim PepMap100 C18 column (LC Packings, Sunnyvale, CA) in mobile phase A (0.1% formic acid, JT Baker, Holland). Peptides were separated over a 30 min linear gradient from 3.2% to 44% mobile phase B (acetonitrile +0.1% formic acid, (JT Baker, Sigma Aldrich) with a flow rate of 350 nL/min. The column was then washed with 90% mobile phase B (10 min) before re-equilibrating at 3.2% mobile phase B (15 min). The column oven was heated to 35 °C. For standard (non-FAIMS) RPLC–MS/MS, the LC system was coupled to an Advion Triversa Nanomate (Advion, Ithaca, NY) which infused the peptides with a spray voltage of 1.7 kV. For the FAIMS analyses, the LC system was coupled to an ADPC-IMS PicoFrit nano-ESI probe (New Objective, Woburn, MA). The spray voltage was 2.6 kV. In both cases, ionized peptides were sampled (introduced) into the Velos Orbitrap ETD mass spectrometer (Thermo Fisher Scientific, Bremen, Germany).
ETD-MS/MS Analysis
The mass spectrometer performed a full FT-MS scan (m/z 380–1600) and subsequent ETD MS/MS scans of the seven most abundant ions above a threshold of 5 000. Survey scans were acquired in the Orbitrap with a resolution of 30 000 at m/z 400. Precursor ions were subjected to ETD with supplemental activation (saETD) in the linear ion trap. The width of the precursor isolation window was 2 Th, and only multiply charged precursor ions were subjected to saETD. saETD was performed with fluoranthene ions. Automatic gain control (AGC) was used to accumulate a sufficient number of charges (fluoranthene, target 1 × 105, maximum fill time 50 ms). Precursor ions (AGC target 1 × 104, maximum fill time 100 ms) were activated for 100 ms (charge dependent activation time was enabled). The dynamic exclusion repeat count was set to 1 with a duration of 60 s. Data acquisition was controlled by Xcalibur 2.1 (Thermo Fisher Scientific).
RPLC–FAIMS-MS/MS Analysis
The mass spectrometer parameters in RPLC–FAIMS-MS/MS were the same as those described above (ETD-MS/MS). The FAIMS device (Thermo Fisher Scientific, San Jose, CA) was operated under the following conditions: gas flow rate of 3.5 L/min and a composition of 50/50 He:N2, the dispersion voltage (DV) was set to −5000 V, and the inner and outer electrodes temperatures were 70 and 90 °C, respectively. The dwell time was set to 50 ms. Nine separate RPLC–FAIMS-MS/MS analyses were performed at compensation voltages (CV) of −25, −27.5, −30, −32.5, −35, −37.5, −40, −42.5, and −45 V. (The compensation voltage range used here was determined as described in the Supporting Information and in Supplemental Figures 1 and 2).
Database Searching
A fasta format database was constructed containing the 4000 synthetic peptides. Raw data were converted to dta files in Proteome Discoverer 1.0 (minimum peak count 1, signal/noise 3, and maximum precursor mass 5000 Da) and searched against the database using Mascot 2.3 and Mascot Daemon 2.2.2. Data from the FAIMS and non-FAIMS analyses were searched with the same parameters: No enzyme was selected for digestion with no missed cleavages; precursor ion tolerance of 5 ppm; fragment ion tolerance 0.5 Da; variable modification was phosphorylation of serine/threonine. Decoy searching was selected to establish false discovery rates. Significance thresholds were set to give false discovery rates of 1%. The search results were manually filtered for rank 1 peptide identifications, and peptide assignments with incorrect phosphorylation site localizations were removed.
Results and Discussion
Analysis of Peptide Library
The peptide libraries contain peptides with the basic sequences GPSGXVpSXAQLX(K/R) and SXPFKXpSPLXFG(K/R), where X = ADEFGLSTVY. The combined library contains 4000 peptides in approximately equal abundance. Each peptide contains three variable amino acids resulting in between one and six sequence isomers. In addition, different combinations of amino acids can have equivalent masses, further extending the number of isomers. In total, there are 556 unique peptide masses contained within the library. The breakdown of these unique masses and the number of associated isomers is shown in Supplemental Table 1 in the Supporting Information. (In addition, there are 11 600 serine, threonine, and tyrosine residues in the peptide library. Of these, 4 000 are phosphorylated and the remaining 7 600 are “potential” phosphorylation sites. Although the phosphorylation sites are defined in the library and no actual phosphorylation localization variants exist, as all peptides contain at least two serines, every library member has theoretical localization variants. This aspect is explored further below).
The peptide library was first analyzed by reversed-phase (RP) LC–ETD-MS/MS without SCX prefractionation. That analysis resulted in 2556 ETD events. The protein database search identified 329 peptide spectral matches (PSMs) and a total of 322 unique peptides (Supplemental Table 2 in the Supporting Information), that is, 8% of the peptide library. The peptides identified corresponded to 247 unique masses. For 74% (183/247) of those unique masses, a single sequence was identified. For 21.5% (53/247), two peptide isomers were identified, and for 4.5% (11/247), three peptide isomers were identified. These results highlight the need for fractionation of complex peptide mixtures.
To compare the performance of gas-phase fractionation by use of FAIMS and prefractionation by use of strong cation exchange (SCXLC) for the analysis of peptide sequence isomers, equal amounts of the peptide library were analyzed by RPLC–FAIMS-ETD-MS/MS and SCXLC RPLC–ETD-MS/MS. Nine SCX fractions, containing a total of 9 μg of the peptide library, were analyzed by RPLC–ETD-MS/MS. An equivalent amount (9 μg) was analyzed by RPLC–FAIMS-ETD using the external CV stepping method. That is, nine analyses of 1 μg of the peptide library, at CVs of −45, −42.5, −40, −37.5, −35, −32.5, −30, −27.5, and −25 V, were performed. For both approaches, the total amount of instrument time was 9 h. Both data sets were searched against a custom database containing the synthetic peptides using the Mascot algorithm. Data were searched both as individual fractions (SCXLC or CV) and as complete data sets.
The number of nonredundant phosphopeptides identified per SCX fraction is shown in Figure 1a. A total of 1212 (redundant) peptides were identified with a 1% false discovery rate (Supplemental Table 3 in the Supporting Information). The histogram shows that the distribution of peptides across the fractions is not uniform: 45% of the peptides were identified from two fractions (2 and 6). The UV trace from the SCX fractionation (Supplemental Figure 3 in the Supporting Information) correlates well with the number of peptides identified per fraction. Combined analysis of the nine fractions resulted in the identification of 693 unique nonredundant peptides (978 peptide spectral matches) from a total of 28208 queries, a 2.5% identification rate. (See Supplemental Table 4 in the Supporting Information). That is, there was a 42.8% redundancy between SCX fractions, i.e., 42.8% of the peptide assigned were identified in multiple SCX fractions. Of the 978 PSMs, 63% corresponded to doubly charged precursor ions and 37% to triply charged precursor ions. No peptides were identified with higher charge states. Overall, 17.3% (693/4000) of the peptide library was identified in the SCX analysis.
Figure 1.

(a) Number of peptides identified per SCXLC fraction and (b) number of sequence isomers identified per unique precursor mass from the combined SCXLC analysis.
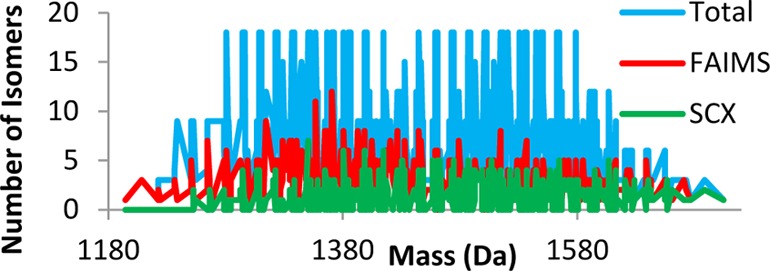
The number of isomers identified per unique precursor mass from the combined SCXLC RPLC–MS/MS analysis is shown in Figure 1b. The peptides identified correspond to 364 unique masses. For 177 of those unique masses, a single sequence was identified. For 102 unique masses, two sequence isomers were identified, and for 43, three isomers were identified. The maximum number of sequence isomers for a given precursor mass was 7. That was observed for m/z 1349.634 only (GPSGLVSGAQLER, GPSGAVSDAQLLR, GPSGVVSAAQLER, GPSGLVSDAQLAR, GPSGDVSLAQLAR, GPSGDVSAAQLLR, and GPSGEVSGAQLLR). The average number of sequence isomers identified per precursor mass was 1.9. (The average number of isomers per precursor mass for the library is 7.2). Figure 2 (green trace) shows the number of isomers which were detected for each precursor mass. The blue trace shows the actual number of sequence isomers present at each precursor mass.
Figure 2.

Number of sequence isomers versus unique precursor mass: blue trace, distribution of sequence isomers in peptide library; green trace, sequence isomers identified in the SCXLC analysis; red trace, sequence isomers identified in the FAIMS analysis.
Figure 3a shows the number of nonredundant peptides identified from each of the nine different FAIMS analyses. The total number of (redundant) peptides identified was 2097 (Supplemental Table 5 in the Supporting Information). The distribution of peptides across the nine CV fractions is more uniform than observed in the SCX fractionation, although 58% (1218) of the peptides were identified in the CV range −32.5 to −25 V, compared with 42% (879) in the range −45 to −35 V. None of the FAIMS CV fractions have fewer than 120 peptides, and on average 233 peptides were identified per CV fraction (compared with 135 peptides identified per SCXLC fraction). Figure 4 shows the distribution of the peptide m/z values versus compensation voltage. It is clear from this that doubly charged ions elute from the FAIMS over a different CV range compared to triply charged ions as shown by others with nonmodified peptides.9 Only one triply charged peptide was identified at a compensation voltage higher (less negative) than −35 V. When the combined data were searched, 1388 unique peptides were identified (Supplemental Table 6 in the Supporting Information) (2495 PSMs) from a total of 10 712 queries (23.2% identification rate). The redundancy rate between CV fractions was 34%, that is 34% of the peptide assignments were observed in multiple CV fractions. The charge state distribution for the FAIMS analysis was similar to the SCXLC analysis with 64% of the peptides identified as doubly charged and 36% identified as triply charged. No higher charge states were identified and this is not surprising given the sequences of the peptide libraries. Overall, 35% (1388/4000) of the peptide library was identified in the FAIMS analysis.
Figure 3.

(a) Number of peptides identified per FAIMS compensation voltage and (b) number of sequence isomers identified per unique precursor mass from the combined FAIMS analysis.
Figure 4.
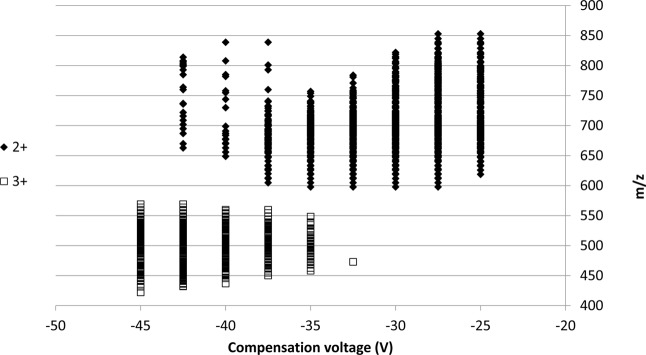
Mass-to-charge ratio of the peptides identified from the FAIMS analysis versus compensation voltage.
A manual analysis of the amino acid compositions of the peptide sequences identified in the FAIMS analysis was performed. Supplemental Figure 4a–d in the Supporting Information shows the variation in % of amino acid residues at each substitution site across the compensation voltages. The analysis shows that threonine is underrepresented at all substitution sites across all CVs. A decrease in % leucine between CV −32.5 and −40 V is observed for all three substitution sites. A similar trend is observed for aspartic acid on the site closest to the C-terminus. There are no other obvious trends at any of the sites.
The number of peptide isomers identified per precursor mass from the FAIMS data is shown in Figure 3b. Both the range and distribution are increased in comparison to the SCXLC analysis. In total, 493 unique masses were identified. Of those unique masses, 131 (26.6%) were identified as single sequences, 122 corresponded to two sequence isomers, and 104 to three sequence isomers. The maximum number of sequence isomers identified for a given precursor mass was 12 (1369.6643 Da). There are three different combinations of the variable amino acids for this precursor mass: SFL, TVF, and LAY. All six of the isomers with SFL residues were identified (SFL, SLF, FLS, FSL, LSF, and LFS), in addition to TVF, FVT, AYL, LAY, ALY, and LYA. The 12 peptides were identified over 8 of the 9 CV fractions. (None were identified at CV −37.5 V). Three of the SFL isomers were identified at CV −32.5 V. The remaining SFL isomers were identified at CVs of −25, −27.5, and −40 V. A total of 11 isomers were identified for precursor mass 1355.6486 Da. In that case, peptides were identified from the full range of CV values. The average number of isomers identified per precursor mass was 2.8 (cf., calculated average number of isomers per mass in the library of 7.2). The red trace in Figure 2 shows the distribution of the isomers across the precursor mass range compared to the SCXLC analysis (green) and the total number of isomers in the library (blue). There is no correlation between the precursor mass and the number of isomers identified.
Gas-phase fractionation by use of FAIMS clearly outperforms both 1-D and 2-D liquid chromatography for the separation of peptide isomers. The peptides contained within the library do not differ greatly, and similar chromatographic behavior might be expected. That is, the low identification rates observed for RPLC–MS/MS (8%) and SCXLC-RPLC–MS/MS (17.3%) may be due to coelution of isomeric peptides and the resulting composite MS/MS spectra. Figure 5a shows the ETD mass spectrum for the doubly charged precursor of m/z 710.8287 from the SCXLC analysis. Only one peptide sequence was identified with this mass: GPSGYVpSAAQLYK. There are two possible additional isomers with the same amino acids (AYY, YYA) and an additional six isomeric peptides with the substituted amino acids SVY. There are fragment ions identified that could derive from all nine of the possible isomers (fragment ion mass accuracy <0.5 Da) and unique fragments for three of the nine isomers (as shown in Supplemental Table 7 in the Supporting Information) contained within the mass spectrum. In the FAIMS analysis, seven of the nine peptides were identified over a CV range from −25 to −37.5 V (GPSGSVpSFAQLYK, GPSGSVpSYAQLFK, GPSGYVpSAAQLYK, GPSGYVpSFAQLSK, GPSGYVpSSAQLFK, GPSGAVpSYAQLYK, and GPSGFVpSYAQLSK). The peptide GPSGYVpSAAQLYK identified in the SCXLC analysis (Figure 5a) was identified in the FAIMS analysis at a CV of −25 V (see Figure 5b). All of the peaks in the spectrum correspond to fragments of this peptide. It should be noted that all of the fragments of this peptide have identical m/z to fragments from other isomers, however no fragments unique to other isomers are observed in this spectrum. This simplification of the ETD mass spectrum afforded by FAIMS separation may explain why the Mascot identification rate, that is, conversion of MS/MS spectrum to peptide assignment, is greater for the FAIMS analysis than the SCXLC analysis (23% cf., 2.5%, respectively).
Figure 5.

(Top) ETD mass spectrum of the doubly charged peptide ion with precursor m/z 710.8287 from the SCX analysis. The spectrum was assigned to peptide GPSGYVpSAAQLYK in the database search. Fragments from multiple isomers are observed. (Bottom) ETD mass spectrum of the peptide ion [GPSGYVpSAAQLYK]2+ from the FAIMS analysis (CV = −25 V).
We, and others, have previously shown the complementarity between SCXLC and FAIMS for the identification of peptides (11, 12). Similar complementarity is demonstrated here. Figure 6 shows the number of peptides identified in the two analyses. A total of 1690 peptides were identified, that is, 42.3% of the peptide library. A total of 56% (391) of the peptides identified in the SCX analysis were also identified in the FAIMS analysis. Of the total peptides identified, 82% were observed in the FAIMS analysis, with 18% (302) of the identifications deriving solely from the SCXLC analysis.
Figure 6.
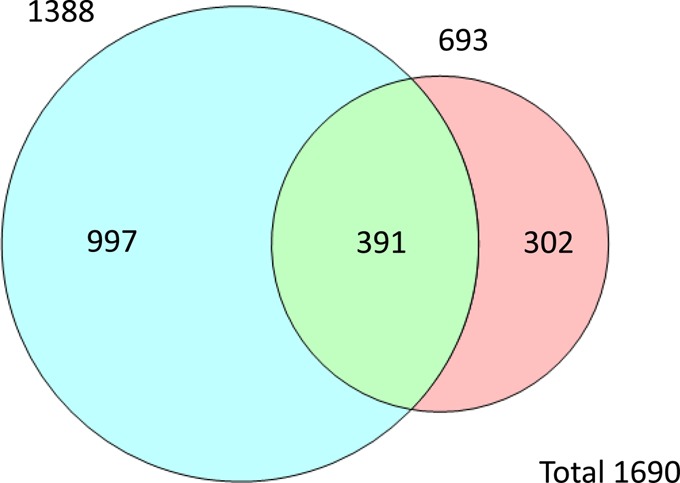
Venn diagram showing the number of unique peptides identified from the FAIMS (blue) and SCX analysis (pink).
To investigate whether any correlation exists between SCX elution profile and compensation voltage, the peptides identified at CV −27.5 V were considered in terms of their corresponding SCX fraction (if any). A total of 67 peptides identified at CV −27.5 V were also identified in the SCX analysis. Of these, 33 were identified in SCX fraction 6. No more than 10 were identified from any other single SCX fraction. While this result might suggest a correlation, it should be noted that fraction 6 was the most populated SCX fraction (230 out of 693 peptides identified), and this result may simply illustrate the limited separation capabilities of SCX.
In the searches described above, phosphorylation of serine/threonine was specified as a variable modification. This was necessary because although the site of phosphorylation in the library peptides was fixed, unmodified serine was also present. That is, all peptides contain both phosphorylated and unmodified serine. The search results were filtered for rank 1 peptides and a 1% FDR and further filtered manually to remove those assignments with incorrect phosphorylation site. However, the use of the phosphopeptide library provides an opportunity to assess the false localization rate (FLR) resulting from the protein database searches. As above the search results were filtered for rank 1 peptides and a 1% FDR, but peptide assignments with incorrect phosphorylation sites were retained. An additional 23 peptides were “assigned” from the FAIMS data set and an additional 22 peptides from the SCXLC data set. The FLR, calculated as the number of falsely localized PSMs/total number of PSMs, was 0.9% for the FAIMS data set and 2.2% for the SCXLC data set.
Conclusions
By use of a large and defined phosphopeptide library containing numerous sequence inversions and structural isomers, tryptic in nature, we have investigated a key challenge in mass spectrometry-based proteomics, that of peptide isomers. We have shown that reversed-phase LC alone provides inadequate separation of isomers. Only 8% of the library was identified. Prefractionation of the peptides by use of strong cation exchange chromatography results in a 2-fold increase in the number of peptide assignments (17% of the peptide library); however, the majority of the unique peptide masses were assigned to single sequences, indicating that many peptide isomers were not separated. Indeed, manual inspection reveals the composite nature of the MS/MS spectra. Further support for this conclusion comes from the conversion rate of MS/MS spectra to peptide assignment, just 2.5%. Gas-phase separation of the peptides by use of FAIMS resulted in a further 2-fold increase in peptide assignments (35% of the library). The proportion of single sequence isomers was reduced (from 49% to 27%) and the maximum number of isomers identified for a unique mass was 12. The ETD mass spectra were simplified and the “MS/MS to assignment” conversion rate was increased to 23%. In summary, we have demonstrated that use of FAIMS in the mass spectrometry workflow is the more suitable approach to address the problem of peptide isomers.
Acknowledgments
The Wellcome Trust (Grant 091892) (H.J.C.) and CRUK (A.J.C.) are gratefully acknowledged for funding. The authors thank Gavin Reid for the gift of the synthetic phosphopeptides LFtGHPESLER and LFTGHPEsLER. The Advion Triversa Nanomate, Dionex LC and Thermo Fisher Velos Orbitrap mass spectrometer used in this research were funded through the Birmingham Science City Translational Medicine: Experimental Medicine Network of Excellence Project, with support from Advantage West Midlands (AWM).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Rehman I.; Wright P. C. J. Proteome Res. 2009, 8, 5347–5355. [DOI] [PubMed] [Google Scholar]
- Ting L.; Rad R.; Gygi S. P.; Haas W. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C. A. S.; Hilderbrand A. E.; Valentine S. J.; Clemmer D. E. Anal. Chem. 2002, 74, 26–36. [DOI] [PubMed] [Google Scholar]
- Hudgins R. R.; Jarrold M. F. J. Am. Chem. Soc. 1999, 121, 3494–3501. [Google Scholar]
- Srebalus C. A.; Li J. W.; Marshall W. S.; Clemmer D. E. Anal. Chem. 1999, 71, 3918–3927. [DOI] [PubMed] [Google Scholar]
- Shvartsburg A. A.; Creese A. J.; Smith R. D.; Cooper H. J. Anal. Chem. 2011, 83, 6918–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buryakov I. A.; Krylov E. V.; Makas A. L.; Nazarov E. G.; Pervukhin V. V.; Rasulev U. K. Pisma Zh. Tekh. Fiz. 1991, 17, 60–65. [Google Scholar]
- Buryakov I. A.; Krylov E. V.; Nazarov E. G.; Rasulev U. K. Int. J. Mass Spectrom. Ion Processes 1993, 128, 143–148. [Google Scholar]
- Saba J.; Bonneil E.; Pomies C.; Eng K.; Thibault P. J. Proteome Res. 2009, 8, 3355–3366. [DOI] [PubMed] [Google Scholar]
- Swearingen K. E.; Hoopmann M. R.; Johnson R. S.; Saleem R. A.; Aitchison J. D.; Moritz R. L. Mol. Cell. Proteomics 2012, 11, M111.014985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese A. J.; Shimwell N. J.; Larkins K. P. B.; Heath J. K.; Cooper H. J. J. Am. Soc. Mass Spectrom. 2013, 24, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridon G.; Bonneil E.; Muratore-Schroeder T.; Caron-Lizotte O.; Thibault P. J. Proteome Res. 2012, 11, 927–940. [DOI] [PubMed] [Google Scholar]
- Sweet S. M. M.; Bailey C. M.; Cunningham D. L.; Heath J. K.; Cooper H. J. Mol. Cell. Proteomics 2009, 8, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courcelles M.; Bridon G.; Lemieux S.; Thibault P. J. Proteome Res. 2012, 11, 3756–3765. [DOI] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H. J.; Hakansson K.; Marshall A. G. Mass Spectrom. Rev. 2005, 24, 201–222. [DOI] [PubMed] [Google Scholar]
- Mann M.; Ong S. E.; Gronborg M.; Steen H.; Jensen O. N.; Pandey A. Trends Biotechnol. 2002, 20, 261–268. [DOI] [PubMed] [Google Scholar]
- Sweet S. M. M.; Creese A. J.; Cooper H. J. Anal. Chem. 2006, 78, 7563–7569. [DOI] [PubMed] [Google Scholar]
- Swaney D. L.; McAlister G. C.; Coon J. J. Nat. Methods 2008, 5, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.