

Abstract
Background
Given the high technical reproducibility and orders of magnitude greater resolution than microarrays, next-generation sequencing of mRNA (RNA-Seq) is quickly becoming the de facto standard for measuring levels of gene expression in biological experiments. Two important questions must be taken into consideration when designing a particular experiment, namely, 1) how deep does one need to sequence? and, 2) how many biological replicates are necessary to observe a significant change in expression?
Results
Based on the gene expression distributions from 127 RNA-Seq experiments, we find evidence that 91% ± 4% of all annotated genes are sequenced at a frequency of 0.1 times per million bases mapped, regardless of sample source. Based on this observation, and combining this information with other parameters such as biological variation and technical variation that we empirically estimate from our large datasets, we developed a model to estimate the statistical power needed to identify differentially expressed genes from RNA-Seq experiments.
Conclusions
Our results provide a needed reference for ensuring RNA-Seq gene expression studies are conducted with the optimally sample size, power, and sequencing depth. We also make available both R code and an Excel worksheet for investigators to calculate for their own experiments.
1. Introduction
Progress in mRNA sequencing is rapidly advancing our understanding of gene expression far beyond that of conventional microarray analysis. As RNA-Seq rapidly develops and costs continue to decrease, more samples will continue to be sequenced and experiments performed, rather than run on a standard microarray.
A key difference from microarray expression measurement is that sequencing is a digital counting process, and the total amount of sequence can vary significantly both between runs and between genes within a given run, with some genes being invisible (0 counts) in a given run, whereas microarrays always have a fixed number of fluorescent probes and therefore have a constant amount of data per run (a given probe can saturate or fall to background level, however). Therefore, the amount of information in a sequencing run can change between experiments, and is a critical variation that needs to be accounted for in sample size estimates.
Recent studies have attempted to estimate the appropriate depth of RNA-Sequencing for measurements to be technically precise. Toung et al. (2011) pooled reads from 20 B-cell samples to create a dataset of 879 million reads. They concluded that only 6% of genes are within 10% of their true expression level when 100 million reads are sequenced, but the percentage of genes jumped to 72% when five-fold more reads are sequenced. In contrast, Wang et al. (2011) suggested that only 30 million reads are necessary to quantify gene expression in chicken lungs, and that 10 million reads could reliably estimate the level of expression of 80% of genes. This broad range of estimates, and the consequences for planning experiments, provides an attractive research opportunity to clarify the influence of variability.
To capture the influence of both biological and technical variability we base our calculations on a negative binomial (NB) distribution, since it accounts for both aspects. The NB model is also well suited to model count data such as RNA-Seq (Verbeke, 2001), and is used by several differential expression measurement methods including edgeR and DESeq (Anders and Huber, 2010; Oberg and others, 2012; Robinson and others, 2010). We derive an explicit sample size formula which includes both sequence based counting (i.e., Poisson) error and biological variability, while avoiding the rapidly diminishing returns (and expense) of over-increasing sequencing depth. To better understand important components of the formula and of a study's resultant statistical power, we used a collection of 12 human and 2 model organism experiments.
2. Implementation
There are two ways to use the formula described below, depending on the user's skill set. For most investigators, we provide a simple Excel sheet that allows users to ask one of two questions: How many samples do I need per group? and How small of fold change can I detect given a fixed number of samples? (Supplementary Data; supplementary material is available online at www.liebertpub.com/cmb). This type of analysis should yield a rough idea sufficient for grant preparations. For complex queries and advanced usage, we have also provided an R package available via Bioconductor (http://bioconductor.org/packages/release/bioc/html/RNASeqPower.html).
3. Results
Our basic formulas for the required number of samples per group is:
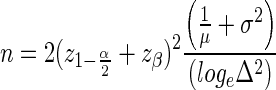 |
(1) |
The parameters ∝ and β are size and power of the test; z the corresponding cut points, and Δ the testing target. These three parameters will be fixed across genes or a given study, and are often dictated by external requirements. Typical values might be Δ = 1.5 (a.k.a fold change), corresponding to detection of a 50% change in gene expression between the two groups; 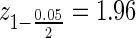 , corresponding to a two sided test at ∝ = 0.05; and zβ = 1.28 corresponding to 90% power. The other two variables will be gene and experiment dependent: the depth of coverage μ of the gene, and the coefficient of variation σ in this gene between biological replicates. (Note: throughout this article, we refer to depth and coverage interchangeably—both meaning how many reads are assigned to a particular gene) The technical variation of the comparison is inversely proportional to the number of sequenced reads for the gene and therefore decreases with sequencing depth. The biological variation is a property of the particular gene/model system/condition under study. One would expect it to be smaller for uniform systems such as cell culture and/or products that are under tight regulatory control, and larger for less uniform replicates such as human subject samples.
, corresponding to a two sided test at ∝ = 0.05; and zβ = 1.28 corresponding to 90% power. The other two variables will be gene and experiment dependent: the depth of coverage μ of the gene, and the coefficient of variation σ in this gene between biological replicates. (Note: throughout this article, we refer to depth and coverage interchangeably—both meaning how many reads are assigned to a particular gene) The technical variation of the comparison is inversely proportional to the number of sequenced reads for the gene and therefore decreases with sequencing depth. The biological variation is a property of the particular gene/model system/condition under study. One would expect it to be smaller for uniform systems such as cell culture and/or products that are under tight regulatory control, and larger for less uniform replicates such as human subject samples.
3.1. Rate of sequencing
To understand possible variation in depth of coverage between genes in the context of an mRNA experiment, we first empirically examined the relationship between the overall sequencing depth of an experiment and individual gene coverage (i.e. number of reads mapped to the gene). Table 1 provides a summary of 12 human and 2 model organism data sets. Each line represents a set of biological replicates that were subjected to the same experimental conditions, and lists the average sequencing depth for the set, the percent mapping to a pre-specified list of 21,214 genes, followed by the distribution of coverage for individual genes. The rate of sequencing for a gene is simply the number of times a DNA element derived from that gene is actually sequenced in a given experiment divided by the number of reads (in millions). As shown in Figure 1, the number of undetected genes is relatively constant across our data at 1%–2%; 216 genes were not found in any of the samples. The majority of the detected genes were sequenced at a rate of 0.1 to 100 counts per million mapped reads. Genes with counts below 0.1 per million reads sequenced represented 6%–5% of the annotated genes. Genes with a frequency less than 0.01 per million reads could only be detected in the most deeply sequenced samples, and represented less than 4% of all mapped reads. Overall, 85%–96% of all annotated genes were sequenced at a rate of 0.1 reads per million or greater, regardless of the biospecimen or depth. In other words, a sequencing depth of 10 million reads will ensure that approximately 90% of all genes will be covered by at least 10 reads.
Table 1.
Counts per Gene per Million Reads Mapped
| |
|
|
|
|
|
Counts per million mapped |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Sample | Type | n | Type | Avg Reads | % mapped | <0.01 | 0.01–.1 | 0.1–1 | 1–10 | 10–100 | 100–1000 | >1000 |
| a | Brain_A | diseased tissue | 7 | PE | 345 | 42 | 1.7 | 9.1 | 12.6 | 22.7 | 42.3 | 11.2 | 0.4 |
| b | Brain_B | diseased tissue | 8 | PE | 353 | 41 | 1.2 | 9.6 | 12.6 | 23.0 | 41.7 | 11.5 | 0.5 |
| c | Brain_C | diseased tissue | 4 | PE | 300 | 18 | 0.0 | 6.8 | 19.0 | 28.7 | 34.3 | 10.7 | 0.5 |
| d | Lung_A | cancer cell line | 8 | PE | 39 | 67 | 0.0 | 4.2 | 12.9 | 22.2 | 47.8 | 12.4 | 0.5 |
| e | Lung_B | cancer cell line | 8 | PE | 36 | 62 | 0.0 | 3.8 | 13.0 | 22.1 | 48.3 | 12.3 | 0.5 |
| f | Pancreas_A | non-cancer cell line | 8 | PE | 121 | 35 | 0.0 | 6.0 | 15.7 | 33.6 | 40.1 | 4.3 | 0.3 |
| g | Pancreas_B | cancer cell line | 8 | PE | 126 | 37 | 0.0 | 5.5 | 12.4 | 22.7 | 48.2 | 10.8 | 0.4 |
| h | PBMC_A | peripheral blood | 16 | SE | 190 | 62 | 3.5 | 11.1 | 14.4 | 19.4 | 38.0 | 13.0 | 0.5 |
| i | PBMC_B | peripheral blood | 20 | PE | 368 | 69 | 3.2 | 10.5 | 15.3 | 19.0 | 38.5 | 13.1 | 0.4 |
| j | PBMC_C | peripheral blood | 20 | PE/SE | 374 | 72 | 3.5 | 11.6 | 16.0 | 20.3 | 36.4 | 11.8 | 0.5 |
| k | TCL | cancer cell line | 12 | PE | 182 | 65 | 1.2 | 9.6 | 14.2 | 19.8 | 42.0 | 12.7 | 0.4 |
| l | TNBC | cancer cell line | 7 | PE | 129 | 33 | 0.0 | 6.2 | 15.0 | 28.5 | 37.4 | 12.4 | 0.5 |
| z | Zebrafish_B | whole fish | 4 | PE | 260 | 78 | 0.8 | 3.0 | 8.2 | 32.8 | 46.0 | 8.1 | 1.1 |
| z | Zebrafish_A | whole fish | 4 | PE | 287 | 63 | 0.6 | 2.8 | 7.7 | 32.3 | 46.9 | 8.6 | 1.1 |
PE, paired end; SE, single-end;
n = number of subjects;
id = label used in figures.
FIG. 1.

The number of unexpressed genes is relatively consistent. For each group, the number of genes with zero reads (i.e., not expressed) is plotted as a function of total sequencing depth.
We also looked at a model organism to ensure that the expression profile assumptions we made are not specific to humans. Expression data from the zebrafish show a shorter left-hand tail, possibly due to as yet undiscovered genes in this animal model. This hypothesis is supported by the larger number of unmapped reads observed (approx. 20%) compared to human (approx. 5%).
3.2. Estimation of the biological variation
For each gene in each data set of Table 1 with average counts of at least 5, we estimated the coefficient of variation (CV) in expression across samples in the data set using a negative binomial model (edgeR). Figure 2 displays the result for each as a cumulative distribution function. Marked on the plot are the σ90 values for each sample (i.e., the value such that 90% of the genes have a smaller variation). Values for the zebrafish are 0.19 and 0.26, and for the human samples range from 0.32 to 0.74 with a median of 0.43. These are consistent with the suggested default values in the edgeR user guide of 0.1 and 0.4 for inbred animal and human studies, in the case where no replicates are available. Such values can serve as conservative limits in Equation 1, since most genes will display lower variation.
FIG. 2.

Empirical Cumulative Distribution plots of CV for 12 human RNA-Seq datasets. Using the read counts for ∼24,000 human genes, we plotted the cumulative distribution function of the CV estimates. The Y-axis is the percentage of genes at or below a given CV value (X-axis). The 90th quintile value is shown at the bottom for each group of samples.
3.3. Biological variation versus technical variation
Technical variation is inversely correlated to the coverage of a gene (1/μ), which in turn is related to the depth of sequencing and the level of expression of that gene in the sample. In contrast, biological variation is a gene-specific constant depending upon the type of biospecimen studied. The global variance is defined as the sum of the technical and biological variance (1/μ+σ).
One way to decrease the technical variation is to increase sequencing depth. However, the impact of sequencing depth on the global variance of the experiment is limited by the amplitude of the biological variation. Consider a gene experiment with σ=1 and sequencing depths of 1, 5, 10, and 100; the relevant multiplier in the sample size calculation is (1+1), (1/5+1), (1/10+1), and (1/100+1), respectively. Once technical variation becomes 1/10 the biological variation, any gain in precision through increased depth will be negligible, and even the step from 5 to 10 is minor. Figure 3 demonstrates the required sample size needed to detect a two-fold difference in expression with 80% power at alpha=0.01 for three different CV values and a range of sequencing depths. These results reveal that gains are minimal for depths greater than 10.
FIG. 3.

Sample size estimates for identifying a two-fold change vary by CV, not coverage. The y-axis is the sample size needed to detect a two-fold difference in expression with 80% power, and 5% type 1 error, given at alpha = 0.01 for three different biological CV's and sequencing depths.
If we assume that further gain in technical variation is not worthwhile once it is at least 5 times lower than the biological variation, then for a gene with biological variation of σ=0.7 and detection rate of 0.1 read per million the technical variation threshold is 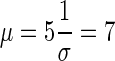 reads. To get 90% of genes sequenced at least 7 times, 7*0.1=70 million mapped reads is sufficient to provide optimal sequencing depth. Any further gains in study power require an increase in the number of biological samples. In the case of the zebrafish where σ90 is closer to 0.2, performance improves up to a depth of
reads. To get 90% of genes sequenced at least 7 times, 7*0.1=70 million mapped reads is sufficient to provide optimal sequencing depth. Any further gains in study power require an increase in the number of biological samples. In the case of the zebrafish where σ90 is closer to 0.2, performance improves up to a depth of 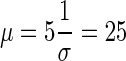 . Looking at Table 1, a depth of 25 million would suffice for 89% of the genes and 200 million would be sufficient for nearly all.
. Looking at Table 1, a depth of 25 million would suffice for 89% of the genes and 200 million would be sufficient for nearly all.
3.4. Gene populations
Our sample size formula describes the behavior of the study for any single gene. We can extend this to predict the result profile for the collection of all genes in an experiment. To illustrate this point, we looked at the data for PBMC A and B, the two largest experiments. Figure 4A shows the overall distribution of fold change. The calculation is restricted to the 15,980 genes that had at least one count in each of the 40 samples, as fold change estimates will be imprecise when zeros are present. Overlaid on the plot is the power curve for n=20 per group, coverage of 100, σ=0.32 (60th percentile of observed) and α=0.001. Genes with a fold change of 3 or greater have near certainty of detection, with lesser sensitivity for smaller changes. The total Figure 4B shows the distribution of all fold changes that were significant at 0.01, along with our prediction as a dashed line. The peak is for genes at about 1.5 fold changes. A more nuanced prediction would also have accounted for the differences in CV from gene to gene, though our model appears sufficient. Collections of “significant” genes, however, will be weighted towards the lower end of the fold change distribution since many are present in this region.
FIG. 4.

Distribution of observed fold changes between two groups of n subjects, along with the power curve for n = 20, CV = 0.32, and ∝ = 0.05 for detecting a two-fold change (top). The observed distribution of fold change among genes that were significant at p < 0.05. The dashed line is the predicted distribution (bottom).
4. Discussion
In this study, we have taken advantage of the properties of the NB distribution to determine the optimal statistical experimental design for RNA-Seq studies. Unlike previous studies, we took advantage of a large number of samples from diverse sources and conditions to get precise estimates of biological variability. Using the information gleaned from altering n and knowing the true CV of biological samples, we were able to successfully predict the effect of adding more sequencing depth or biological replicates on calling differential expressed genes.
We have also derived a sample size formula for RNA-Seq that highlights the important relationship between technical and biological variability. More importantly, we have been able to use a collection of studies from a wide variety of conditions to quantify the expected amount of biological variability in a study. This will allow investigators to intelligently choose sample size and depth values for sequencing studies.
Our results are in general agreement with Wang et al. (2011) that 30–40 million reads is sufficient to be technically precise in measuring gene expression for most genes, which is not surprising given the methods used. We and Wang et al. (2011) used raw count data, rather than the “fragments per kilobase of exon model per million mapped reads” (FPKM) normalized data as implemented in the Cufflinks software (Trapnell and others, 2010, 2012). The mathematical derivations necessary for sample size computation depend on using a valid model for the count data, a task that is tractable for the raw count data but would be much more difficult after the per-isoform scaling of FPKM. Our work is directly applicable to differential expression models that work on the raw count scale such as edgeR and DESeq. Nevertheless, the results should also apply, at least approximately, to the rescaled data. Using the FPKM method, the expression level for a gene is the sum of the FPKM values of its isoforms. Because the accuracy of gene–level expression is reliant on transcript level estimation, any error in accurately quantitating transcripts will also propagate to genes. Clearly, there are additional factors influencing technical precision of FPKM calculations that are not apparent when working with raw gene counts. Using non-normalized values of gene counts is also more intuitive. Normalization by gene length removes information about the original count value and decreases power to detect many genes (Oshlack and Wakefield, 2009). Many alternative normalization strategies have been proposed (Oshlack and others, 2010), but there is no consensus as of yet for the most optimal normalization strategy. Therefore, we support the notion that raw read counts are the preferred starting point for differential expression analysis.
Finally, we provide tools to help investigators optimally design RNA-Seq experiments: an R package “RNASeqPower” and an Excel spread sheet. Users can modify parameters such as Type I error rate, power, desired fold change, CV, and sequencing depth to see how the interaction of those parameters will affect final results. Taken together, we believe that the results presented herein will offer important insights into the design of appropriately powered RNA–Seq expression experiments, while minimizing the need for oversequencing. This should reduce costs while maintaining study power.
5. Derivation
The formal derivation of sample size is based on representing the problem as a generalized linear model (GAM). We then work through the algebraic steps found in Chapters 2.2 and 2.5 of McCullagh and Nelder (1989), the classic text on this subject. The notation of this section intentionally mimics the notation used in said reference. This includes the mean parameter μ, linear predictor η, variance function V, and the link function g. For most studies, the final analysis will focus on relative changes in the counts (e.g., “group 2 has 55% greater expression of the gene than group 1”). This implies using the logarithm as our link function g. Per other authors, we assume a negative binomial form for the variance V. If yij is the read count for gene i from sample j and dj is the total reads for that sample this leads to
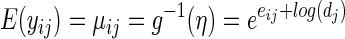 |
(2) |
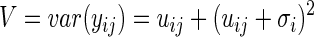 |
(3) |
The parameter σ is the biological CV for the gene within each group.
Using Equation 2.12, we can write the GLM algorithm as an iteratively reweighted least squared computation with weights of 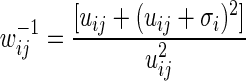 . With the usual starting estimates
. With the usual starting estimates  a one-step update is equivalent to a score test and can be used to estimate sample size and power. Assuming a sample size of n subjects in each group and sufficient sequencing depth to give an expected count of μ, the variance matrix X`WX is diagonal with elements niwi, the adjusted dependent variable is
a one-step update is equivalent to a score test and can be used to estimate sample size and power. Assuming a sample size of n subjects in each group and sufficient sequencing depth to give an expected count of μ, the variance matrix X`WX is diagonal with elements niwi, the adjusted dependent variable is 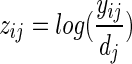 and the one-step estimate of ei is a weighted mean
and the one-step estimate of ei is a weighted mean 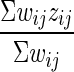 . The score test is a ratio whose numerator is ei – e2i(a.k.a. Δ), an estimate of the log-difference in expression of a gene between the two groups, and whose denominator is
. The score test is a ratio whose numerator is ei – e2i(a.k.a. Δ), an estimate of the log-difference in expression of a gene between the two groups, and whose denominator is
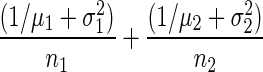 |
(4) |
Then the statistical properties of the test satisfy the formula
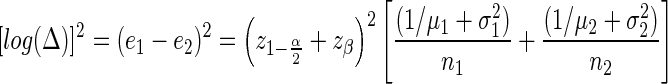 |
(5) |
where Δ is ratio of expression levels (a.k.a., fold change), μ1 and μ2 are the average expected count in the two samples, and σ1, σ2 are the coefficients of variation (i.e., biological variation) for samples from each population. The term 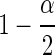 corrects the type 1 error for a two-sided test.
corrects the type 1 error for a two-sided test.
A formal sample size calculation for comparison of two groups will then involve five factors
The depth of sequencing and consequent expected count μ for a given transcript;
The coefficient of variation of counts within each of the two groups, σ1 and σ2;
The size of difference (fold-change) that we wish to detect Δ;
The target false positive rate α and false negative rate (or power);
The number of samples in each group n1 and n2.
If both samples are to be sequenced at the same depth and we assume that the CV (σ) is the same in both populations, then Equation 1 can be used to give the number of samples per group. As an example, assuming the usual false positive rate of 0.05 (two-sided), and power of 90% then z1-α/2+zβ=1.96+1.28 with an average of 20 sequence reads aligning to the gene, and within group variance σ of 0.6, in order to target a two-fold change (log(2)=0.69), then 18 samples per group are needed.
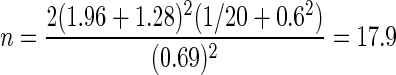 |
(6) |
6. Datasets
We collected 12 datasets of human tissue and cell lines and an additional 2 datasets of zebrafish samples. Each datasets corresponds to another condition (treated, untreated) or disease state and therefore considered as biologically different.
Data sets are summarized in Table I.
7. Read Alignment and Mapping
The reads from Illumina were aligned to the human genome build 37.1 using TopHat(1.3.3)(Trapnell and others, 2009) and Bowtie (0.12.7) (Langmead and others, 2009). HTSeq (0.5.3p3) [http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html] was used to perform gene counting.
Supplementary Material
Acknowledgments
We would like to thank the following investigators for providing their data: A Feldman, R Rademaker, A Thompson, and R Kumar. Thanks to J Bhavsar and the Bioinformatics Core at Mayo Clinic for pre-processing the datasets. This work was funded through the Center for Individualized Medicine at Mayo Clinic (112222) and federal funds from the National Institute of Health, under contract number U01 AI089859. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authors' contributions: SNH and JPK conceived the study, coordinated the design, and drafted the manuscript, TMT derived the formal statistics and helped in drafting the manuscript, YZ participated in the data analysis, GAP helped to draft the manuscript. All authors read and approved the final manuscript.
Author Disclosure Statement
The authors declare that there are no conflicting financial interests.
References
- Anders S. Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. Trapnell C. Pop M. Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P. Nelder JA. Generalized Linear Models. Chapman and Hall; London: 1989. [Google Scholar]
- Oberg AL. Bot BM. Grill DE. Poland GA. Therneau TM. Technical and biological variance structure in mRNA-Seq data: Life in the real world. BMC Genom. 2012;13:304. doi: 10.1186/1471-2164-13-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A. Robinson MD. Young MD. From RNA-seq reads to differential expression results. Genome Biol. 2010;11:220. doi: 10.1186/gb-2010-11-12-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshlack A. Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct. 2009;4:14. doi: 10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD. McCarthy DJ. Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toung JM. Morley M. Li M. Cheung VG. RNA-sequence analysis of human B-cells. Genome Res. 2011;21:991–998. doi: 10.1101/gr.116335.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. Pachter L. Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. Roberts A. Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. Williams BA. Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke G. In: Regression Analysis of Count Data. Cameron A. C., editor; Trivedi P. K., editor. Cambridge University Press; Cambridge: 2001. 1998. [Google Scholar]
- Wang Y. Ghaffari N. Johnson CD, et al. Evaluation of the coverage and depth of transcriptome by RNA-Seq in chickens. BMC Bioinform. 2011;12:S5. doi: 10.1186/1471-2105-12-S10-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.