

Amino acid–sensing mTOR signaling controls the homeostasis of skeletal myogenesis. The Rag GTPases negatively regulate differentiation by activating mTORC1 and subsequently suppressing the IRS1-PI3K-Akt pathway, whereas a Vps34-PLD1-PA-mTOR pathway activates the transcriptional regulation of Igf2 that is essential for myogenesis.
Abstract
Signaling through the mammalian target of rapamycin (mTOR) in response to amino acid availability controls many cellular and developmental processes. mTOR is a master regulator of myogenic differentiation, but the pathways mediating amino acid signals in this process are not known. Here we examine the Rag GTPases and the class III phosphoinositide 3-kinase (PI3K) Vps34, two mediators of amino acid signals upstream of mTOR complex 1 (mTORC1) in cell growth regulation, for their potential involvement in myogenesis. We find that, although both Rag and Vps34 mediate amino acid activation of mTORC1 in C2C12 myoblasts, they have opposing functions in myogenic differentiation. Knockdown of RagA/B enhances, whereas overexpression of active RagB/C mutants impairs, differentiation, and this inhibitory function of Rag is mediated by mTORC1 suppression of the IRS1-PI3K-Akt pathway. On the other hand, Vps34 is required for myogenic differentiation. Amino acids activate a Vps34-phospholipase D1 (PLD1) pathway that controls the production of insulin-like growth factor II, an autocrine inducer of differentiation, through the Igf2 muscle enhancer. The product of PLD, phosphatidic acid, activates the enhancer in a rapamycin-sensitive but mTOR kinase–independent manner. Our results uncover amino acid–sensing mechanisms controlling the homeostasis of myogenesis and underline the versatility and context dependence of mTOR signaling.
INTRODUCTION
Skeletal muscle differentiation is a highly ordered multistage process that includes mononucleated myoblasts exiting the cell cycle and fusing to form multinucleated myofibers/myotubes (Andres and Walsh, 1996). This myogenic process is controlled by a muscle-specific gene expression program under the regulation of numerous signaling pathways (Naya and Olson, 1999; Berkes and Tapscott, 2005). In recent years, the mammalian target of rapamycin (mTOR) has emerged as a key regulator of skeletal myogenesis by governing multiple stages of myogenic differentiation through distinct mechanisms (Ge and Chen, 2012).
mTOR is a Ser/Thr kinase that functions as a master regulator of cell growth, proliferation, and various types of cellular differentiation (Erbay et al., 2005; Laplante and Sabatini, 2012). Much of the current understanding of mTOR signaling mechanism is in the context of cell growth regulation. There exist two protein complexes, namely mTORC1 and mTORC2, that mediate rapamycin-sensitive and rapamycin-insensitive signaling of mTOR, respectively (Sarbassov et al., 2005). Raptor-associated mTORC1 assembles a signaling network that transduces amino acid availability and mitogenic signals. One of the best characterized downstream targets of mTORC1 is S6K1, the ribosomal S6 kinase that regulates protein synthesis at the translational initiation level (Ma and Blenis, 2009). Several mediators of amino acid signals have been reported to lie upstream of mTORC1, including the small GTPase Rag and the class III phosphoinositide 3-kinase (PI3K) Vps34. The Rag heterodimers, regulated by the guanine nucleotide exchange factor activity of the Ragulator protein complex and the GTPase-activating protein activity of the GATOR complex, bind raptor and translocate mTORC1 to the lysosomal surface in response to amino acid stimulation, which is necessary for the activation of mTORC1 (Sancak et al., 2008, 2010; Bar-Peled et al., 2012, 2013). Vps34 mediates amino acid signals to activate mTORC1 (Byfield et al., 2005; Nobukuni et al., 2005) by targeting phospholipase D1 (PLD1) for lysosomal translocation and activation (Xu et al., 2011; Yoon et al., 2011a). PLD1 and its product, phosphatidic acid (PA), are important for mitogenic as well as amino acid activation of mTORC1 (Sun and Chen, 2008; Xu et al., 2011; Yoon et al., 2011a).
Rapamycin-sensitive mTOR signaling regulates at least two distinct processes of myoblast differentiation. In a kinase-independent manner, mTOR controls the myogenic expression of insulin-like growth factor II (IGF-II) at a transcriptional level via a muscle-specific enhancer (Erbay et al., 2003) and at a posttranscriptional level via microRNA-125b (Ge et al., 2011a). IGFs are critically involved in skeletal muscle development and adult muscle regeneration and hypertrophy (Florini et al., 1991a; Barton-Davis et al., 1999). In cultured myoblasts, growth factor deprivation initiates the differentiation program owing to the induction of IGF-II (Tollefsen et al., 1989; Florini et al., 1991b) and subsequent activation of the IGF-I receptor and a major downstream pathway mediated by PI3K and Akt (Perry and Rudnicki, 2000). In addition, mTOR is also responsible for myocyte fusion through a MyoD/microRNA-1/follistatin pathway, which requires mTOR kinase activity (Sun et al., 2010).
It is presumed that, as a major nutrient signal transducer, mTOR mediates signals from amino acid sufficiency to regulate myogenesis. However, such a pathway has not been clearly delineated, and the roles of amino acid–sensing pathways identified in cell growth regulation have never been examined in myogenesis. In the present study, we investigate the involvement of Rag GTPases and Vps34 in mouse C2C12 myoblast differentiation.
RESULTS
Rag GTPases are negative regulators of myoblast differentiation
To identify amino acid–sensing pathways regulating myoblast differentiation, we first examined the potential involvement of Rag GTPases. Four Rag proteins form active heterodimers (GTP-bound RagA or B with GDP-bound RagC or D) to regulate mTORC1 (Sancak et al., 2008). Serum withdrawal induced differentiation of C2C12 myoblasts, as indicated by expression of the early myogenic marker myogenin and the later myogenic marker myosin heavy chain (MHC) in the course of 3 d. The protein levels of RagA, B, and C were markedly increased after 1 d of differentiation and remained elevated (Figure 1A). RagD protein was not detected by Western analysis in myoblasts or myotubes. Amino acid–stimulated mTORC1 activation in C2C12 cells, measured by S6K1 phosphorylation on T389, was blocked by lentivirally delivered shRNAs for RagA and RagB (Supplemental Figure S1), confirming that Rag is a critical mediator of amino acid–sensing mTORC1 signaling in these cells. Surprisingly, knockdown of RagA, RagB, or both enhanced myotube formation, evidenced by morphological observation and quantification of fusion index (Figure 1B), as well as by MHC expression (Figure 1C). Conversely, coexpression of two Rag mutants that are constitutively active toward mTORC1—RagB-Q99L (GTP-bound) and RagC-S75L (GDP-bound) (Sancak et al., 2008)—suppressed myogenin and MHC expression (Figure 1D) and impaired myotube formation (Figure 1E). Taken together, these results suggest that Rag GTPases are negative regulators of myogenic differentiation. Note that although Rag is necessary for S6K1 activation in these cells (Supplemental Figure S1), S6K1 is not involved in myoblast differentiation (Ge and Chen, 2012).
FIGURE 1:

Rag negatively regulates myogenic differentiation. (A) Confluent C2C12 cells were induced to differentiate by serum withdrawal. Cells were lysed every 24 h, and the lysates were subjected to Western analysis. (B) C2C12 cells were infected with lentiviruses expressing shRNAs for RagA, RagB, and negative control (scrambled hairpin) as indicated and then induced to differentiate for 3 d. The cells were stained for MHC (green) and DAPI (magenta) and quantified for fusion index. (C) Cells treated as in B were subjected to Western analysis. (D) Cells stably expressing RagB-GTP and RagC-GDP mutants were induced to differentiate for 3 d, followed by Western analysis. (E) Cells treated as in D were stained for MHC (green) and DAPI (magenta) and quantified for fusion index. All data shown are mean ± SD or representative blots from three independent experiments. A paired t test was performed to compare each sample to the control. *p < 0.05; **p < 0.01. Scale bars, 100 μm.
Rag negatively regulates myogenic differentiation through mTORC1 inhibition of IRS1-Akt signaling
Because Rag depletion suppressed mTORC1 activation in C2C12 cells (Supplemental Figure S1) and we previously reported that mTORC1 plays a negative role in myogenic differentiation through a feedback inhibition of IRS1-Akt signaling (Ge et al., 2011b), we considered the possibility of Rag acting via this feedback pathway. Indeed, IRS1 phosphorylation on Ser-307 was decreased by the knockdown of RagA/B in myoblasts, which was accompanied by increased Akt phosphorylation (Figure 2A), mirroring the results of Rheb and raptor knockdown (Ge et al., 2011b). Conversely, overexpression of constitutively active RagB/C decreased IRS1 levels and pAkt (Figure 2B). More important, knockdown of IRS1 eliminated the enhancement of differentiation resulted from Rag depletion, as assessed by both MHC expression (Figure 2C) and myotube formation (Figure 2D). Therefore, by activating mTORC1, Rag induces IRS1 serine phosphorylation and suppresses IRS1 signaling to Akt, subsequently inhibiting myogenic differentiation.
FIGURE 2:

Rag inhibits differentiation through the IRS1/Akt pathway. (A) C2C12 cells were infected with lentiviruses expressing shRNAs for RagA and RagB. Cell lysates were analyzed by Western blotting. (B) Cells stably expressing RagB-GTP and RagC-GDP mutants were induced to differentiate for 3 d, followed by Western analysis. (C) Cells were infected with lentiviruses expressing various shRNAs as indicated, followed by differentiation for 3 d and Western analysis of cell lysates. (D) Cells treated as in C were stained for MHC (green) and DAPI (magenta) and quantified for fusion index. Data shown are mean ± SD or representative blots from three independent experiments. A paired t test was performed to compare each sample to the control. *p < 0.05. Scale bar, 100 μm.
We wondered whether the protein level increase of Rag during differentiation (Figure 1A) served to enhance mTORC1 inhibition of the IRS1 pathway. To address that, we examined the activity of Rag toward mTORC1 using the association between RagA and mTORC1 as a readout. Of interest, despite increased levels of both mTOR and RagA, the relative amount of RagA associated with mTORC1 (raptor and mTOR) decreased by day 2 of differentiation (Figure 3A). Furthermore, we probed the subcellular localization of mTOR and active RagB/C. Whereas the majority of mTOR and RagB/C colocalized to the perinuclear region of the cell before induction of differentiation, a significant portion of mTOR distributed throughout the cytoplasm upon differentiation and no longer overlapped with RagB/C (Figure 3B). Therefore a sufficient amount of mTOR is likely free of active Rag and available for other modes of myogenic regulation. It is also possible that the Rag proteins may have other mTORC1-independent functions during myogenesis or in mature muscles.
FIGURE 3:

Activity and subcellular localization of Rag proteins during myoblast differentiation. (A) Confluent C2C12 cells were induced to differentiate by serum withdrawal. Cells were lysed at times indicated, and the lysates were subjected to raptor immunoprecipitation (IP). Western blot band intensities were analyzed using ImageJ (National Institutes of Health, Bethesda, MD), and the relative ratio of RagA and mTOR in the IP was calculated. The data shown are mean ± SD from three independent experiments. One-sample t test was performed to compare the data on days 1 and 2 to those of day 0. **p < 0.01. (B) Cells expressing HA-RagB-GTP and HA-RagC-GDP mutants were induced to differentiate. At times indicated, cells were fixed and immunostained for mTOR (green) and HA (magenta). Scale bar, 10 μm.
Vps34 is a positive regulator of myogenesis
Because Rag was excluded as a positive mediator of amino acid sensing in myogenesis, we turned to Vps34, which had also never been examined in this context. As shown in Figure 4A, the protein level of Vps34 was drastically increased during myoblast differentiation. The PI3K inhibitor 3-methyladenine (3-MA) inhibited C2C12 differentiation in a dose-dependent manner, as indicated by both myotube formation (Figure 4B) and expression of the myogenic markers MHC and myogenin (Figure 4C). As expected, rapamycin treatment completely blocked differentiation. More important, Vps34 knockdown by two independent short hairpin RNAs (shRNAs) also impaired differentiation (Figure 4, D and E). These results strongly suggest that Vps34 has a positive role in myogenic differentiation. Note that Vps34 is a well-established positive regulator of autophagy (Jaber et al., 2012; Simonsen and Tooze, 2009). However, the myogenic role of Vps34 is most likely independent of its autophagic function, because autophagy has been reported to suppress myoblast differentiation (Ciavarra and Zacksenhaus, 2010; Iovino et al., 2012).
FIGURE 4:

Vps34 is necessary for myogenic differentiation. (A) Confluent C2C12 cells were induced to differentiate and lysed every 24 h, followed by Western analysis. (B) Cells were induced to differentiate for 3 d in the presence or absence of 1 mM 3-MA, 5 mM 3-MA, or 100 nM rapamycin (Rap), followed by staining for MHC (green) and DAPI (magenta) and quantification for fusion index. (C) Cells treated as in B were subjected to Western analysis. (D) Cells were infected with lentiviruses expressing shRNAs for Vps34 or negative control (scrambled hairpin) and then induced to differentiate for 3 d. The cells were stained for MHC (green) and DAPI (magenta) and quantified for fusion index. (E) Cells treated as in D were subjected to Western analysis. Data shown are mean ± SD or representative blots from three to five independent experiments. A paired t test was performed to compare each sample to the control. *p < 0.05; **p < 0.01. Scale bars, 100 μm.
Vps34 mediates amino acid activation of PLD1 in myoblasts
Previously we reported that PLD1 is required for myoblast differentiation by acting upstream of mTOR (Yoon and Chen, 2008), but a role of PLD1 in mediating amino acid signals was unknown at that time. Given the recently discovered connection of Vps34 and PLD1 in amino acid sensing (Yoon et al., 2011a), and now a role of Vps34 in myogenesis, we asked whether Vps34 and PLD1 function together to mediate amino acid signals in myocytes. Indeed, both PLD1 and Vps34 are necessary for amino acid activation of mTORC1 in C2C12 cells, as their knockdown inhibited S6K1 phosphorylation (Figure 5A). PLD activity was increased during the course of differentiation, as we previously reported (Yoon and Chen, 2008), and this increase was abolished by amino acid deprivation (Figure 5B). The short duration (2 h) of amino acid withdrawal ensured that the effect would be the result of affecting a signaling cascade rather than a general stoppage of protein synthesis. In addition, acute amino acid stimulation activated PLD activity in myoblasts, and this activation was blocked by 3-MA treatment, as well as by Vps34 knockdown (Figure 5C). Depletion of Vps34 also decreased PLD activity in differentiated myotubes (Figure 5D). On the other hand, Vps34 knockdown did not affect IRS1 levels (Figure 5E). Taken together, our observations are consistent with a possible role of the Vps34-PLD pathway in mediating amino acid signals that govern myogenic differentiation.
FIGURE 5:

PLD is activated by amino acids in a Vps34-dependent manner. (A) C2C12 cells expressing lentivirus-delivered shRNAs were serum starved overnight and subjected to amino acid withdrawal for 2 h, followed by amino acid stimulation for 30 min before cell lysis and Western analysis. (B) Differentiating cells were deprived of amino acids (AA withdrawal) for 2 h at indicated times of differentiation (Diff.), followed by in vivo PLD assays. (C) Cells treated as in A with or without 3-MA (5 mM) or rapamycin (100 nM) during amino acid withdrawal and stimulation were subjected to in vivo PLD assay. (D) Cells expressing shVps34 were induced to differentiate for 3 d, followed by in vivo PLD assay. (E) Cells expressing shVps34 were analyzed by Western blotting. All data shown are mean ± SD or representative blots from three independent experiments. A one-sample t test was performed for data in B and D for comparison to control; a one-sample or paired t test was performed in C to compare the indicated pairs of data. *p < 0.05; **p < 0.01.
The MyoD-follistatin pathway is not regulated by Vps34, PLD1, or amino acids
Two distinct rapamycin-sensitive pathways positively regulate myoblast differentiation by controlling the expression of two secreted myogenic factors—follistatin and IGF-II, respectively (Ge and Chen, 2012). As we previously reported (Sun et al., 2010), follistatin was drastically induced at the mRNA level upon differentiation, which was partly blocked by rapamycin treatment (Figure 6A). Of interest, 3-MA had no effect on follistatin expression (Figure 6A). Furthermore, neither Vps34 knockdown nor PLD1 knockdown had any effect on follistatin levels (Figure 6B). mTOR regulates follistatin expression by stabilizing MyoD protein (Sun et al., 2010), and rapamycin treatment at the initiation of differentiation led to a loss of MyoD (Figure 6C). Again, neither 3-MA treatment nor knockdown of Vps34 or PLD1 affected MyoD levels (Figure 6, C and D). Finally, amino acid withdrawal also had no effect on MyoD stability (Figure 6E). Therefore mTOR regulation of follistatin expression is independent of amino acid signaling and the Vps34-PLD1 pathway.
FIGURE 6:

The MyoD/follistatin pathway is not regulated by Vps34, PLD1, or amino acids. (A) C2C12 cells were treated with rapamycin (100 nM) or 3-MA (5 mM) during the course of differentiation as indicated, followed by qRT-PCR analysis of relative levels of follistatin mRNA. (B) Cells expressing shRNA for Vps34 or PLD1 were differentiated for 3 d and then subjected to qRT-PCR analysis of follistatin mRNA levels. (C, D) Cells were differentiated for 4 h in the presence or absence of rapamycin (100 nM) or 3-MA (5 mM) or infected with various shRNAs as indicated before differentiation for 4 h. Cell lysates were subjected to Western analysis. (E) Cells were induced to differentiation in the presence or absence of amino acids for 4 h, followed by Western analysis. Data shown are mean ± SD or representative blots from three independent experiments. A one-sample or paired t test was performed. **p < 0.01.
Vps34 is necessary for amino acid–dependent transcriptional regulation of Igf2
Next we asked whether Vps34 regulates IGF-II expression during myogenic differentiation. A muscle-specific enhancer (ME) of Igf2 is activated at the initiation of differentiation, and it requires amino acid sufficiency and rapamycin-sensitive mTOR function (Erbay et al., 2003). PLD1 was also necessary for increased ME activity during differentiation, but its connection to amino acid signals was not examined (Yoon and Chen, 2008). As shown in Figure 7A, the activity of a luciferase reporter of the Igf2 ME was increased at 6 h after the induction of differentiation, and it was inhibited by amino acid withdrawal and restored by amino acid readdition. The reinstatement of ME activity by amino acids was inhibited by rapamycin. Of importance, 3-MA treatment also inhibited amino acid–restored ME activity (Figure 7A). Furthermore, knockdown of Vps34 and PLD1 significantly dampened amino acid–stimulated ME activity (Figure 7B), similar to the effect of mTOR knockdown. Vps34 was also necessary for increase of ME activity during differentiation, as were mTOR and PLD1 (Figure 7C). Consistent with their inhibition of the ME reporter, 3-MA treatment and knockdown of Vps34 led to decreased Igf2 mRNA levels during differentiation (Figure 7D). As expected, knockdown of RagA and RagB had no effect on the ME reporter activity (Supplemental Figure S2).
FIGURE 7:

Vps34 regulates differentiation by controlling Igf2 transcription. (A) C2C12 cells stably expressing the Igf2 enhancer reporter (H19-luc-ME) were deprived of amino acids (AA withdrawal) for 2 h in the presence of rapamycin (100 nM) or 3-MA (1 and 5 mM) and induced to differentiate for 6 h with or without amino acid readdition before cell lysis and luciferase assays. (B) Cells stably expressing H19-luc-ME were infected with lentiviruses expressing various shRNAs and then treated as in A. (C) Cells stably expressing H19-luc-ME were infected with various lentiviruses, induced to differentiate for 3 d, and then subjected to luciferase assays. (D) Cells expressing indicated shRNAs or treated with rapamycin (100 nM) or 3-MA (5 mM) were differentiated for 3 d. Total RNA was extracted, and Igf2 mRNA levels were measured by quantitative RT-PCR. (E) Cells infected with lentiviruses expressing Vps34 shRNA were induced to differentiate for 3 d with or without recombinant IGF-II (300 ng/ml), followed by staining for MHC (green) and DAPI (magenta). All data shown are mean ± SD from three independent experiments. A one-sample or paired t test was performed to compare the indicated pairs of data. *p < 0.05; **p < 0.01. Scale bar, 100 μm.
To further validate IGF-II as the functional target of Vps34 in myogenesis, we provided the cells expressing Vps34 shRNA with recombinant IGF-II. Remarkably, exogenous IGF-II completely reversed the negative effect of Vps34 knockdown on differentiation and fully rescued myotube formation (Figure 7E). Taken together with our previously reported observation of IGF-II rescuing differentiation from PLD1 knockdown (Yoon and Chen, 2008), these results strongly suggest that a Vps34-PLD1 pathway mediates amino acid signaling to control IGF-II expression, which is critical for myogenic differentiation.
Phosphatidic acid activates Igf2 ME
Previously we established that mTOR regulates myogenic IGF-II expression through a kinase-independent mechanism (Erbay and Chen, 2001; Erbay et al., 2003; Ge et al., 2009). However, we also found that the product of PLD, PA, regulates mTORC1 by directly activating the kinase activity in HEK293 cells in the context of cell growth (Yoon et al., 2011a). Thus a role of PLD1 in the kinase-independent mTOR-IGF-II pathway appears paradoxical. To further ascertain the function of PLD, and PA, in the regulation of IGF-II expression, we examined the effect of PA on Igf2 ME reporter activity. As shown in Figure 8, PA activated the ME reporter in the absence of amino acids to the same extent as amino acid addition, suggesting that PA may be a primary mediator of amino acid signaling to IGF-II expression. Because PA does not activate mTORC1 in the absence of amino acids (Yoon et al., 2011a), this result is also consistent with the notion that PA stimulates an mTOR function that is independent of its kinase activity.
FIGURE 8:
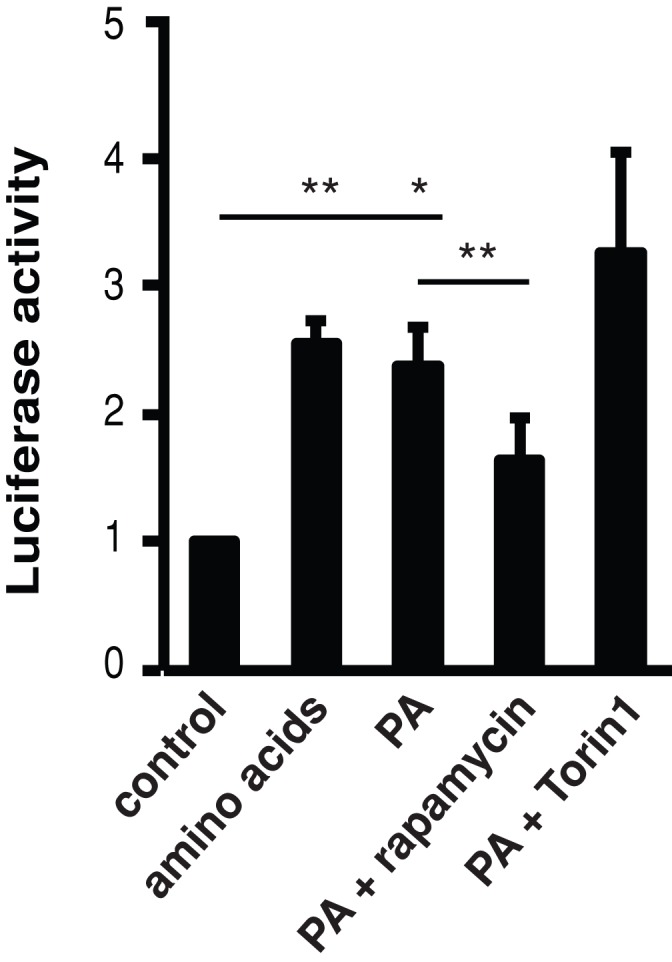
PA stimulates ME activity. C2C12 cells stably expressing the H19-luc-ME reporter were deprived of amino acids for 2 h in the absence or presence of rapamycin (200 nM) or Torin1 (200 nM), followed by induction of differentiation for 6 h in the presence of amino acids, PA (300 μM), or PA together with an inhibitor. Cells were lysed and subjected to luciferase assays. Data shown are mean ± SD of three independent experiments. A one-sample or paired t test was performed to compare the indicated pairs of data. *p < 0.05; **p < 0.01.
PA directly interacts with the rapamycin-FKBP12 binding (FRB) domain of mTOR, and rapamycin disrupts this interaction (Fang et al., 2001; Veverka et al., 2008). Although PA regulation of the Igf2 ME is most likely independent of mTOR kinase activity, rapamycin is still expected to interfere with this regulation if the mechanism of PA action is through binding to FRB. Indeed, we found that rapamycin inhibited PA-induced ME activity, whereas the mTOR kinase inhibitor Torin1 had no effect (Figure 8). Therefore it is highly likely that the amino acid–sensing Vps34-PLD pathway regulates IGF-II expression through PA interaction with mTOR during myogenic differentiation.
DISCUSSION
Amino acids have long been known to play a critical role in regulating skeletal muscle mass by stimulating protein synthesis and muscle hypertrophy, as well as by suppressing protein degradation and muscle atrophy (Nair et al., 1992; Louard et al., 1995; Kimball, 2007). As a major transducer of amino acid signals, mTORC1 is an established regulator of both anabolic and catabolic processes in skeletal muscles (Bodine et al., 2001; Rommel et al., 2001; Herningtyas et al., 2008; McCarthy and Esser, 2010). The role of amino acid signaling in muscle differentiation, however, has been far less explored, even though it is established that mTOR controls skeletal myogenesis at multiple stages (Ge and Chen, 2012). The present study is the first to decipher signaling pathways sensing amino acids upstream of mTOR in myogenesis. We found that a Vps34-PLD1-PA-mTOR pathway mediates amino acid signals to govern Igf2 transcriptional up-regulation that is essential for the initiation of myogenic differentiation. On the other hand, another major amino acid–sensing regulator of mTORC1, Rag, has no role in supporting differentiation. Instead, Rag suppresses differentiation through mTORC1-dependent down-regulation of IRS1 and subsequent inhibition of Akt signaling. Although the Vps34-PLD1 pathway is also necessary for amino acid activation mTORC1 in myocytes (Figure 5A), it does not appear to contribute to the negative regulation through IRS1 (Figure 5E). Hence amino acid–sensing mTOR signaling governs the homeostasis of myogenic differentiation through distinct upstream pathways and downstream effectors (Figure 9).
FIGURE 9:

A proposed model of mTOR myogenic signaling. Amino acids activate two pathways of opposing functions in myogenesis. The Rag GTPases activate mTORC1 and subsequently suppress the PI3K-Akt pathway through IRS1 phosphorylation. The Vps34-PLD1-PA pathway activates a kinase-independent function of mTOR that regulates myogenic expression of IGF-II. A third mTOR pathway, mediated by MyoD and follistatin, is rapamycin sensitive and kinase dependent but is independent of amino acids.
In regulating cell growth, the Rag pathway and the Vps34-PLD1 pathway converge on the lysosome in response to amino acid stimulation, and both are required for mTORC1 activation and cell size control (Wiczer and Thomas, 2012). Our results described here show that in myogenic differentiation, however, these two pathways play opposing roles even though both are necessary for amino acid activation of mTORC1 in myoblasts. In addition to Rag knockdown, we also observed enhanced differentiation in myoblasts with the depletion of P18 and MP1—components of Ragulator (unpublished data). This is in full agreement with the notion that mTORC1 is a negative regulator of myogenic differentiation, which was first suggested by the negative roles of raptor and Rheb in this context (Ge et al., 2011b). The complete lack of positive contribution of the canonical mTORC1 (raptor-mTOR complex) to the differentiation process is also consistent with its major target, S6K1, being dispensable (Ge and Chen, 2012). Most likely, mTORC1 itself, rather than S6K1, phosphorylates IRS1 on Ser-307 (Shah and Hunter, 2006) to effect the inhibition of PI3K-Akt signaling, subsequently suppressing myogenic differentiation. Indeed, it has been reported that activation of S6K1 does not affect the IRS1-PI3K pathway in myoblast differentiation (Hamilton et al., 2010). Although this mTORC1-IRS1 pathway is considered a negative feedback loop in other cellular contexts, it appears to be the major if not only function of mTORC1 in myogenesis. It is striking that the rapamycin-sensitive mTOR functions in myogenesis are completely independent of mTORC1.
We find that amino acids activate PLD1 through Vps34 in C2C12 myocytes. Others also reported that leucine, a branched-chain amino acid that activates mTORC1 (Fox et al., 1998; Patti et al., 1998; Xu et al., 1998), stimulates Vps34 in the same cells (MacKenzie et al., 2009). These results are consistent with observations in other types of cells. However, our conclusion that the Vps34-PLD1-PA pathway is a major mediator of amino acid signals upstream of mTOR in regulating Igf2 transcription could not have been deduced from what was already known. In cell growth regulation the amino acid–sensing Vps34-PLD1 pathway leads to direct activation of the mTORC1 kinase by PA (Yoon et al., 2011a; Wiczer and Thomas, 2012), but the myogenic IGF-II pathway is independent of mTOR kinase activity (Erbay and Chen, 2001; Erbay et al., 2003; Ge et al., 2009). Of interest, PA activates the Igf2 transcriptional enhancer in a rapamycin-sensitive and mTOR kinase activity–independent manner, which is consistent with a direct interaction between PA and the rapamycin-binding domain FRB in mTOR (Fang et al., 2001; Veverka et al., 2008), but it also suggests a unique mechanism of mTOR activation by PA that is yet to be deciphered. Another distinction between myogenic and mitogenic mTOR signaling is that amino acid sensing may be uncoupled from the lysosome in myogenesis, as neither the lysosomal v-ATPase inhibitor concanamycin A nor knockdown of v-ATPase subunits had any effect on amino acid–stimulated Igf2 transcriptional activation in myocytes (unpublished observations). Novel regulators of rapamycin-sensitive and mTORC1-independent myogenic signaling likely exist and await discovery by future investigations.
MATERIALS AND METHODS
Reagents
The MF20 anti-sarcomeric myosin heavy chain (MHC) and F5D anti-myogenin antibodies were obtained from the Developmental Studies Hybridoma Bank (developed under the auspices of the National Institute of Child Health and Human Development, National Institutes of Health, and maintained by the Department of Biological Sciences, University of Iowa, Iowa City, IA). Anti-MyoD was from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-raptor was from Bethyl Laboratory (Montgomery, TX). Alexa Fluor 594 anti-mouse immunoglobulin G (IgG) and 488 anti-rabbit IgG were from Life Technologies (Grand Island, NY). All other primary antibodies were from Cell Signaling Technology (Danvers, MA). All other secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). Recombinant IGF-II, 3-MA, and gelatin were obtained from Sigma-Aldrich (St. Louis, MO), rapamycin was from LC Labs (Woburn, MA), and all other cell culture reagents were from Life Technologies. Hemagglutinin (HA)–glutathione S-transferase (GST)–RagB-Q99L and HA-GST–RagC-S75L were obtained from Addgene (Sancak et al., 2008). 1,2-Dioctanoyl-sn-glycero-3–PA was obtained from Avanti Polar Lipids (Alabaster, AL). Torin1 was a generous gift from David Sabatini (Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, MA).
Cell Culture
C2C12 myoblasts were maintained in DMEM containing 1 g/l glucose with 10% fetal bovine serum at 37°C with 7.5% CO2. To induce differentiation, cells were plated on tissue culture plates coated with 0.2% gelatin and grown to 100% confluence, changed into differentiation medium (DMEM containing 2% horse serum), and replenished with fresh medium daily for 3 d. Transfection of myoblasts was performed using TransIT-LT1 (Mirus, Madison, WI) following the manufacturer's recommendations, and stable pools were obtained by selection in 1 mg/ml G418.
Determination of PLD activity
PLD activity was determined as previously described by measuring [3H]phosphatidylbutanol produced in [3H]oleic acid–labeled cells via PLD-catalyzed transphosphatidylation (Sun et al., 2008). C2C12 cells are subjected to various treatments as described in figure legends. After 1-butanol treatment, the cells were lysed, and lipids were extracted and analyzed by TLC.
Cell lysis, immunoprecipitation, and Western blot analysis
C2C12 cells were lysed in ice-cold lysis buffer containing 20 mM Tris-HCl (pH 7.5), 0.1 mM Na3VO4, 25 mM NaF, 25 mM β-glycerophosphate, 2 mM EDTA, 2 mM ethylene glycol tetraacetic acid, 0.3% Triton X-100, 1 mM dithiothreitol, and 0.5 mM phenylmethylsulfonyl fluoride. The supernatant after microcentrifugation at 13,000 rpm for 10 min was collected and then either subjected to immunoprecipitation or boiled in SDS sample buffer for 5 min. Immunoprecipitation was performed with anti-IRS1 or anti-raptor antibody followed by protein G agarose for 1 h at 4°C. For immunoprecipitation of raptor, the lysis buffer contained 40 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.4), 120 mM NaCl, 10 mM pyrophosphate, 50 mM NaF, 10 mM β-glycerophosphate, 2 mM EDTA, 1× Sigma protease inhibitor cocktail, and 0.3% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate. The beads were washed with lysis buffer and then boiled in SDS sample buffer for 5 min. Proteins were resolved on SDS–PAGE, transferred onto polyvinylidene fluoride membrane (Millipore, Billerica, MA), and incubated with various antibodies following the manufacturer's recommendations. Detection of horseradish peroxidase–conjugated secondary antibodies was performed with Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer Life Sciences, Waltham, MA).
Immunofluorescence microscopy and quantitative analysis of myocytes
C2C12 cells differentiated in 12-well plates were fixed and stained for MHC and 4′,6-diamidino-2-phenylindole (DAPI) as previously described (Yoon and Chen, 2008). The stained cells were examined with a Leica DMI 4000B fluorescence microscope, and the fluorescence images were captured using a RETIGA EXi camera and analyzed with Q-capture Pro51 software (QImaging, Redwood City, CA). The fusion index was calculated as the percentage of nuclei in myotubes with ≥2 nuclei. Each data point was generated from quantifying all cells in five randomly chosen microscopic fields. For mTOR and Rag B/C localization, cells were differentiated on coverslips 24 h after transfection with HA-GST–RagB-Q99L and HA-GST–RagC-S75L. The cells were fixed and stained for endogenous mTOR and HA as previously described (Yoon et al., 2011a). To analyze the fluorescence signal, a personal deconvolution microscope system (DeltaVision; Applied Precision, Issaquah, WA) was used with a 60×/numerical aperture 1.4 lens. Deconvolution used an enhanced ratio iterative-constrained algorithm (Agard et al., 1989). XY and Z optical displacement between different filter sets was determined experimentally using Tetraspeck fluorescent microsphere standards (Life Technologies).
Lentivirus-delivered RNA interference
All shRNAs were obtained from Sigma-Aldrich in the pLKO.1-puro vector (MISSION shRNA). Clone IDs were Vps34-1, TRCN0000025373; Vps34-2, TRCN0000322313; RagA, TRCN0000316855; and RagB, TRCN0000102657. The shRNAs for mTOR (TRCN0000054980), PLD1 (TRCN0000076820), and IRS1 (TRCN0000238269) were previously reported (Ge et al., 2011b; Yoon and Chen, 2008). Lentivirus packaging and testing were performed as previously described (Yoon and Chen, 2008). C2C12 cells were transduced with lentiviruses in growth medium containing 8 μg/ml Polybrene and selected in 3 μg/ml puromycin for 3 d, followed by plating into 12-well plates for differentiation.
qRT-PCR
C2C12 cells were lysed directly in TRIZOL (Life Technologies). Total RNA was isolated following the manufacturer's protocol. Quantitative reverse transcription-PCR (qRT-PCR) for Igf2 mRNA and follistatin mRNA was performed as previously described (Yoon and Chen, 2008; Sun et al., 2010).
Luciferase reporter assays
C2C12 cells stably expressing H19-luc-ME (Erbay et al., 2003) and infected by various lentiviruses were grown to 100% confluence and induced to differentiate. The cells were lysed at the indicated times in Passive Lysis Buffer (Promega, Madison, WI), and luciferase assays were performed using the Luciferase Assay Systems kit (Promega) following the manufacturer's protocol. PA vesicle was made as previously described (Yoon et al., 2011b).
Statistical analysis
All data are presented as mean ± SD of at least three sets of independent experiments. Whenever necessary, statistical significance of the data was analyzed by performing one-sample or paired t tests. The specific types of tests and the p values, when applicable, are indicated in the figure legends.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health to J.C. (AR048914 and GM089771).
Abbreviations used:
- IGF-II
insulin-like growth factor-II
- IRS1
insulin receptor substrate 1
- 3-MA
3-methyladenine
- ME
muscle-specific enhancer
- MHC
myosin heavy chain
- mTOR
mammalian target of rapamycin
- PA
phosphatidic acid
- PI3K
phosphoinositide 3-kinase
- PLD1
phospholipase D1
- S6K1
ribosomal S6 kinase 1
- shRNA
short hairpin RNA
- Vps34
vacuolar protein sorting 34
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-06-0353) on September 25, 2013.
REFERENCES
- Agard DA, Hiraoka Y, Shaw P, Sedat JW. Fluorescence microscopy in three dimensions. Methods Cell Biol. 1989;30:353–377. doi: 10.1016/s0091-679x(08)60986-3. [DOI] [PubMed] [Google Scholar]
- Andres V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis. J Cell Biol. 1996;132:657–666. doi: 10.1083/jcb.132.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton-Davis ER, Shoturma DI, Sweeney HL. Contribution of satellite cells to IGF-I induced hypertrophy of skeletal muscle. Acta Physiol Scand. 1999;167:301–305. doi: 10.1046/j.1365-201x.1999.00618.x. [DOI] [PubMed] [Google Scholar]
- Berkes CA, Tapscott SJ. MyoD and the transcriptional control of myogenesis. Semin Cell Dev Biol. 2005;16:585–595. doi: 10.1016/j.semcdb.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Bodine SC, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- Ciavarra G, Zacksenhaus E. Rescue of myogenic defects in Rb-deficient cells by inhibition of autophagy or by hypoxia-induced glycolytic shift. J Cell Biol. 2010;191:291–301. doi: 10.1083/jcb.201005067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbay E, Chen J. The mammalian target of rapamycin regulates C2C12 myogenesis via a kinase-independent mechanism. J Biol Chem. 2001;276:36079–36082. doi: 10.1074/jbc.C100406200. [DOI] [PubMed] [Google Scholar]
- Erbay E, Kim JE, Chen J. Amino acid-sensing mTOR signaling. In: Zempleni J, Dakshinamurti K, editors. Nutrient and Cell Signaling. Boca Raton, FL: CRC Press; 2005. pp. 353–380. [Google Scholar]
- Erbay E, Park IH, Nuzzi PD, Schoenherr CJ, Chen J. IGF-II transcription in skeletal myogenesis is controlled by mTOR and nutrients. J Cell Biol. 2003;163:931–936. doi: 10.1083/jcb.200307158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- Florini JR, Ewton DZ, Magri KA. Hormones, growth factors, and myogenic differentiation. Annu Rev Physiol. 1991a;53:201–216. doi: 10.1146/annurev.ph.53.030191.001221. [DOI] [PubMed] [Google Scholar]
- Florini JR, Magri KA, Ewton DZ, James PL, Grindstaff K, Rotwein PS. “Spontaneous” differentiation of skeletal myoblasts is dependent upon autocrine secretion of insulin-like growth factor-II. J Biol Chem. 1991b;266:15917–15923. [PubMed] [Google Scholar]
- Fox HL, Pham PT, Kimball SR, Jefferson LS, Lynch CJ. Amino acid effects on translational repressor 4E-BP1 are mediated primarily by L-leucine in isolated adipocytes. Am J Physiol. 1998;275:C1232–C1238. doi: 10.1152/ajpcell.1998.275.5.C1232. [DOI] [PubMed] [Google Scholar]
- Ge Y, Chen J. Mammalian target of rapamycin (mTOR) signaling network in skeletal myogenesis. J Biol Chem. 2012;287:43928–43935. doi: 10.1074/jbc.R112.406942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Sun Y, Chen J. IGF-II is regulated by microRNA-125b in skeletal myogenesis. J Cell Biol. 2011a;192:69–81. doi: 10.1083/jcb.201007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Wu AL, Warnes C, Liu J, Zhang C, Kawasome H, Terada N, Boppart MD, Schoenherr CJ, Chen J. mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am J Physiol Cell Physiol. 2009;297:C1434–C1444. doi: 10.1152/ajpcell.00248.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Yoon MS, Chen J. Raptor and Rheb negatively regulate skeletal myogenesis through suppression of insulin receptor substrate 1 (IRS1) J Biol Chem. 2011b;286:35675–35682. doi: 10.1074/jbc.M111.262881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DL, Philp A, MacKenzie MG, Baar K. Prolonged activation of S6K1 does not suppress IRS or PI-3 kinase signaling during muscle cell differentiation. BMC Cell Biol. 2010;11:37. doi: 10.1186/1471-2121-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herningtyas EH, Okimura Y, Handayaningsih AE, Yamamoto D, Maki T, Iida K, Takahashi Y, Kaji H, Chihara K. Branched-chain amino acids and arginine suppress MaFbx/atrogin-1 mRNA expression via mTOR pathway in C2C12 cell line. Biochim Biophys Acta. 2008;1780:1115–1120. doi: 10.1016/j.bbagen.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Iovino S, Oriente F, Botta G, Cabaro S, Iovane V, Paciello O, Viggiano D, Perruolo G, Formisano P, Beguinot F. PED/PEA-15 induces autophagy and mediates TGF-beta1 effect on muscle cell differentiation. Cell Death Differ. 2012;19:1127–1138. doi: 10.1038/cdd.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Lin RZ, Zhang J, Zong WX. Mammalian PIK3C3/VPS34: the key to autophagic processing in liver and heart. Autophagy. 2012;8:707–708. doi: 10.4161/auto.19627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR. The role of nutrition in stimulating muscle protein accretion at the molecular level. Biochem Soc Trans. 2007;35:1298–1301. doi: 10.1042/BST0351298. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louard RJ, Barrett EJ, Gelfand RA. Overnight branched-chain amino acid infusion causes sustained suppression of muscle proteolysis. Metabolism. 1995;44:424–429. doi: 10.1016/0026-0495(95)90047-0. [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- MacKenzie MG, Hamilton DL, Murray JT, Taylor PM, Baar K. mVps34 is activated following high-resistance contractions. J Physiol. 2009;587:253–260. doi: 10.1113/jphysiol.2008.159830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Esser KA. Anabolic and catabolic pathways regulating skeletal muscle mass. Curr Opin Clin Nutr Metab Care. 2010;13:230–235. doi: 10.1097/MCO.0b013e32833781b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair KS, Schwartz RG, Welle S. Leucine as a regulator of whole body and skeletal muscle protein metabolism in humans. Am J Physiol. 1992;263:E928–E934. doi: 10.1152/ajpendo.1992.263.5.E928. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Olson E. MEF2: a transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr Opin Cell Biol. 1999;11:683–688. doi: 10.1016/s0955-0674(99)00036-8. [DOI] [PubMed] [Google Scholar]
- Nobukuni T. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase; Proc Natl Acad Sci USA; 2005. pp. 14238–14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Brambilla E, Luzi L, Landaker EJ, Kahn CR. Bidirectional modulation of insulin action by amino acids. J Clin Invest. 1998;101:1519–1529. doi: 10.1172/JCI1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RLS, Rudnicki MA. Molecular mechanisms regulating myogenic determination and differentiation. Frontiers Biosci. 2000;5:d750–767. doi: 10.2741/perry. [DOI] [PubMed] [Google Scholar]
- Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Hunter T. Turnover of the active fraction of IRS1 involves raptor-mTOR- and S6K1-dependent serine phosphorylation in cell culture models of tuberous sclerosis. Mol Cell Biol. 2006;26:6425–6434. doi: 10.1128/MCB.01254-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7:3118–3123. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- Sun Y, Fang Y, Yoon MS, Zhang C, Roccio M, Zwartkruis FJ, Armstrong M, Brown HA, Chen J. Phospholipase D1 is an effector of Rheb in the mTOR pathway. Proc Natl Acad Sci USA. 2008;105:8286–8291. doi: 10.1073/pnas.0712268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ge Y, Drnevich J, Zhao Y, Band M, Chen J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J Cell Biol. 2010;189:1157–1169. doi: 10.1083/jcb.200912093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefsen SE, Sadow JL, Rotwein P. Coordinate expression of insulin-like growth factor II and its receptor during muscle differentiation. Proc Natl Acad Sci USA. 1989;86:1543–1547. doi: 10.1073/pnas.86.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veverka V, Crabbe T, Bird I, Lennie G, Muskett FW, Taylor RJ, Carr MD. Structural characterization of the interaction of mTOR with phosphatidic acid and a novel class of inhibitor: compelling evidence for a central role of the FRB domain in small molecule-mediated regulation of mTOR. Oncogene. 2008;27:585–595. doi: 10.1038/sj.onc.1210693. [DOI] [PubMed] [Google Scholar]
- Wiczer BM, Thomas G. Phospholipase D and mTORC1: nutrients are what bring them together. Sci Signal. 2012;5:pe13. doi: 10.1126/scisignal.2003019. [DOI] [PubMed] [Google Scholar]
- Xu G, Kwon G, Marshall CA, Lin TA, Lawrence JC, Jr, McDaniel ML. Branched-chain amino acids are essential in the regulation of PHAS-I and p70 S6 kinase by pancreatic beta-cells. A possible role in protein translation and mitogenic signaling. J Biol Chem. 1998;273:28178–28184. doi: 10.1074/jbc.273.43.28178. [DOI] [PubMed] [Google Scholar]
- Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1) J Biol Chem. 2011;286:25477–25486. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Chen J. PLD regulates myoblast differentiation through the mTOR-IGF2 pathway. J Cell Sci. 2008;121:282–289. doi: 10.1242/jcs.022566. [DOI] [PubMed] [Google Scholar]
- Yoon MS, Du G, Backer JM, Frohman MA, Chen J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol. 2011a;195:435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Sun Y, Arauz E, Jiang Y, Chen J. Phosphatidic acid activates mammalian target of rapamycin complex 1 (mTORC1) kinase by displacing FK506 binding protein 38 (FKBP38) and exerting an allosteric effect. J Biol Chem. 2011b;286:29568–29574. doi: 10.1074/jbc.M111.262816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.