

Abstract
The brain is rich in metals and has a high metabolic rate, making it acutely vulnerable to the toxic effects of endogenously produced free radicals. The abundant metals, iron and copper, transfer single electrons as they cycle between their reduced (Fe2+, Cu1+) and oxidized (Fe3+, Cu2+) states making them powerful catalysts of reactive oxygen species (ROS) production. Even redox inert zinc, if present in excess, can trigger ROS production indirectly by altering mitochondrial function. While metal chelators seem to improve the clinical outcome of several neurodegenerative diseases, their mechanisms of action remain obscure and the effects of long‐term use are largely unknown. Most chelators are not specific to a single metal and could alter the distribution of multiple metals in the brain, leading to unexpected consequences over the long‐term. We show here how X‐ray fluorescence will be a valuable tool to examine the effect of chelators on the distribution and amount of metals in the brain.
Keywords: Chelator, Copper, Iron, Metal, Neurodegeneration, Synchrotron, X‐ray fluorescence, Zinc
Introduction
The brain acquires metals and exploits their unique biochemistry as part of its normal functioning [1, 2]. Metals such as iron, copper and zinc act as essential cofactors in metalloproteins required for the normal functioning of the nervous tissue (Table 1) with unique importance in myelin synthesis and neurotransmission while heavy metals such as mercury and lead are known neurotoxins. There is increasing evidence that dysregulation of iron, copper, and zinc homeostasis contributes to a vast range of neurodegenerative diseases. Here we describe how synchrotron x‐ray fluorescence (XRF) imaging can be used to quantitatively assess how the distribution and chemistry of multiple brain metals in experimental animal models are affected by chelators currently used for the treatment of neurodegenerative disease.
Table 1.
Important metalloenzymes and roles of metals.
| Role | Metal | Enzyme |
|---|---|---|
| Neurotransmission | ||
| Dopamine synthesis | Iron | Tyrosine hydroxylase |
| Norepinephrine synthesis | Copper | Dopamine β hydroxylase |
| Neuromodulation at excitatory synapses (down‐regulates glutamate response) | Zinc | N/A (vesicular Zn2+) |
| Antioxidant defense | ||
| Dismutation of O2 −· to H2O2 | Copper, zinc | Cu/Zn SOD |
| Dismutation of O2 −· to H2O2 | Manganese | Mn SOD |
| Reduction of H2O2 | Selenium | Glutathione peroxidase |
| Reduction of H2O2 | Iron | Catalase |
| Protection of mtDNA against ROS | Iron | Aconitase |
| Iron metabolism | ||
| Registration of cellular iron status and translational regulation of expression of iron metabolism proteins | Iron | IRP1/aconitase |
| Ferrous iron oxidation | Copper | Ceruloplasmin |
| Ferrous iron oxidation | Copper | Hephaestin |
| Heme synthesis | ||
| Conversion of δ ALA to porphobilinogen | Zinc | δ ALA dehydratase |
| Insertion of ferrous iron into protoporphyrin IX with formation of heme | Iron | Ferrochelatase |
| Pentose phosphate shunt | ||
| Conversion of glucose‐6‐phosphate to 6‐phosphoglucono‐δ‐lactone with the formation of NADPH | Iron | Glucose‐6‐phoshate dehydrogenase |
| Glycolysis | ||
| Conversion of glucose to pyruvate, NADH and ATP | Magnesium, potassium | Kinases (the substrate is a Mg2+‐ATP complex; Mg2+ is essential for kinase activity) |
| Synthesis of acetyl‐CoA | ||
| Activation of pyruvate dehydrogenase complex | Calcium | Pyruvate dehydrogenase phosphatase (activated by Ca2+) |
| Citric acid cycle | ||
| Isomerization of citrate to isocitrate | Iron | Aconitase |
| Dehydrogenation of succinate to fumarate | Iron | Succinate dehydrogenase |
| Decarboxylation of isocitrate to α‐ketoglutarate | Calcium | Isocitrate dehydrogenase (activated by Ca2+) |
| Decarboxylation of α‐ketoglutarate to succinyl‐CoA | Calcium | α‐ketoglutarate dehydrogenase (activated by Ca2+) |
| Electron transport | ||
| Transport of e− to CoQ | Iron | Complex I |
| Transport of e− to CoQ | Iron | Complex II |
| Transport of e− from CoQ to cytochrome c | Iron | Complex III |
| Transport of e− to complex IV | Heme | Cytochrome c |
| Four e− reduction of O2 to H2O | Iron, copper, zinc, magnesium | Complex IV |
| Other | ||
| Cholesterol synthesis | Iron | HMG‐CoA reductase |
| Synthesis of NO | Iron | NO synthase |
| Synthesis of deoxyribonucleotides | Iron | Ribonucleotide reductase |
| Conversion of toxic sulfite to sulfate | Iron, molybdenum | Sulfite oxidase |
| Oxygen transport | Iron | Hemoglobin |
| Purine symthesis | Iron | Amido phosphoribosyl transferase |
| DNA binding | Zinc | NFkβ, Sp1, PARP |
| Formation and compaction of myelin | Zinc | MBP, MAG |
| Initiation and propagation of action potentials | Sodium, potassium, calcium | N/A |
| Long‐term potentiation | Magnesium, calcium, zinc | N/A |
| Second messenger | Calcium | N/A |
| Apoptosis | Calcium, zinc | N/A |
N/A not applicable; O2 −· superoxide anion; H2O2 hydrogen peroxide; SOD superoxide dismutase; IRP iron regulatory protein; ALA aminolevulinic acid; NADPH nicotinamide adenine dinucleotide phosphate (reduced); NADH nicotinamide adenine dinucleotide (reduced); ATP adenosine triphosphate; CoA coenzyme A; CoQ coenzyme; NO nitric oxide; NFkβ nuclear factor kβ; Sp1 specificity protein 1; PARP poly (adenosine diphosphate‐ribose) polymerase; MBP myelin basic protein; MAG myelin‐associated glycoprotein.
Mapping and Quantification of Metals in the Brain
While analytic techniques such as atomic absorption spectrometry and inductively coupled mass spectrometry have been used to quantify metals in selected brain regions [3, 4, 5, 6, 7, 8, 9, 10, 11] and some magnetic resonance imaging (MRI) sequences, such as susceptibility weighted imaging are used to image iron in vivo[12], histochemistry has long been the gold standard to localize metals in brain slices. Laser ablation inductively coupled plasma mass spectrometry (LA‐ICP‐MS) is another very powerful mapping and quantification tool that has been applied to brain (3, reviewed in 4). XRF is in some respects a ‘poor man's’ LA‐ICP‐MS because little or no sample preparation is required, no equipment needs to be purchased and the use of the synchrotron is free of charge. XRF can map multiple metals and is also nondestructive so samples can be used for other purposes following mapping.
While Perls’ and Turnbull's methods detect only nonheme iron [13, 14, 15, 16] they are useful in brain and comparable to XRF because most of the iron in the brain is nonheme iron. Detection of copper by histochemistry lacks sensitivity and specificity which has hindered studies, e.g. of Wilson's disease [17, 18, 19]. Complex techniques have been developed to distinguish protein‐bound from free zinc [20, 21]. In contrast, XRF detects total zinc which can now be quantified to allow comparison between individual brain samples. Unlike XRF which can map multiple metals simultaneously, each histochemical method employs a different chemistry, and so they cannot be combined or used sequentially on the same tissue section. XRF removes one of the barriers to elucidation of the functional interrelationships between metals by making metal co‐localization easy and quantitative.
X‐ray fluorescence spectroscopic imaging (XRF) is element specific and can simultaneously map multiple metals in all chemical forms. Our laboratory has applied rapid scanning XRF (RS‐XRF) to map the global distribution of iron, copper, and zinc in whole slices of both normal and diseased brain [22, 23, 24]. Traditional point‐to‐point X‐ray microprobe has previously been successfully employed to map multiple metals in small areas of brain tissue sections at cellular or subcellular resolutions [25, 26, 27, 28, 29], but the time required to map whole brain slices with the microprobe is prohibitive. For example, a human brain hemisphere can be mapped in 6 h at 100 μm resolution [22] but at 2 μm resolution it would take days. Therefore, RS‐XRF is ideal to get quick anatomic views of global metal distribution in slices of whole organs.
Used as a quantification tool, RS‐XRF is a cost‐effective alternative to conventional methods in which excised cubes of tissue are dissolved for bulk analytical analysis. Recently, new tools have been developed that enable the user to calibrate metal maps using XRF quantification standards to yield concentrations in μg/cm2. Standard quantification foils with known amounts of deposited metal are mapped and then average fluorescence, concentration of the foil and other values (e.g. gains) are used to create a parameter file that yields a concentration scale bar for each image. While concentration per cm2 is very useful for thin sections that are essentially two‐dimensional, we have developed quantification algorithms that take into account the fluorescence escape depth for each element and the x‐ray density of the brain to yield concentration in μg/g w/w (Hopp et al., unpublished). Values for iron using this method were comparable to published iron concentrations using inductively coupled plasma mass spectrometry (Table 2).
Table 2.
XRF Quantification of brain iron. (Hopp et al., in press)
| Anatomic Region | XRF (counts) | Iron μg/g ww (ave foil) |
|---|---|---|
| Periventricular white matter | 21.8 | 46 |
| Subcortical white matter, precentral gyrus | 39.6 | 84 |
| Superior temporal gyrus | 41.2 | 88 |
| Insular cortex | 43.0 | 92 |
| Middle frontal gyrus | 43.0 | 92 |
| Cortex, precentral gyrus | 47.3 | 101 |
| Internal capsule | 52.0 | 111 |
| External capsule and claustrum | 54.8 | 117 |
| Middle temporal gyrus | 57.5 | 122 |
| Ventricular wall, superior to caudate nucleus | 67.5 | 144 |
| Superior caudate nucleus | 91.5 | 195 |
| Superior globus pallidus | 121.7 | 259 |
| Superior putamen | 122.5 | 261 |
| Inferior caudate nucleus | 123.1 | 262 |
| Anterior commissure | 144.0 | 306 |
| Inferior globus pallidus | 157.6 | 335 |
| Inferior putamen | 163.5 | 348 |
Used as an imaging method, RS‐XRF shows all metals simultaneously thus surpassing all “single‐metal” histochemistry. Unlike conventional histology sections, XRF samples retain metals because they do not have to be dehydrated or embedded, and after nondestructive XRF mapping, the same sample can be processed for histology or immunohistochemistry. RS‐XRF is the only way to nondestructively and simultaneously visualize multiple metals in the same section to gauge the effect of therapeutic drugs on brain metals. Synchrotrons around the world accept proposals to access beamtime on a peer‐reviewed basis and once awarded, beamtime is free for researchers. Access can also be purchased by companies at a reasonable cost. Software to visualize and quantify images, Sam's microanalysis toolkit (SMAK), can be downloaded free of charge. Therefore XRF is a useful tool to assess the effect of drugs on preventing or reversing metal‐related brain pathology. We have also shown that it can be combined with MRI analysis of iron [30].
RS‐XRF has shown iron, copper, and zinc localized to discrete regions within the normal substantia nigra [22] and dentate nucleus of the cerebellum [23], high copper content in the normal olivary region of the medulla [24] and shown changes in metal content and distribution associated with neurodegeneration [24]. By combining RS‐XRF with X‐ray Absorption Near Edge Structure (XANES) analysis of regions of interest, we have determined the chemical form of copper in the medullary olive (data not shown).
RS‐XRF makes systematic studies of the effects of therapeutic agents on brain metals practical.
X‐ray Fluorescence Imaging and Spectroscopy Background
Rapid‐scanning X‐ray fluorescence imaging (RS‐XRF) and X‐ray absorption spectroscopy (XAS) are well‐established, element‐specific, quantitative techniques used to determine the location and chemical form of elements in a broad range of tissue samples. X‐rays, focused to a small spot or passed through a pinhole, interrogate the sample as it is raster‐scanned in the beam. For imaging, the incident X‐ray beam energy is fixed so as to excite all elements with K‐shell absorption edges below the incident energy. In our experiments 13 keV is suitable to excite K‐shell electrons of iron, copper, and zinc, but would also excite the L‐shell electrons of gold or mercury (if present) and if the detector is very close to the sample, even sulfur, calcium, and phosphorus can be mapped. After a K‐shell electron is excited and ejected into the continuum, an electron from an outer shell falls in to fill the core hole, emitting a fluorescent photon of a specific energy. These X‐ray fluorescent photons are detected and by carefully windowing on the highest energy X‐ray fluorescence emission line for each element, the location of multiple elements can be simultaneously mapped. Because XRF relies upon the physics of the atom rather than on a chemical reaction, every atom of a metal that is interrogated by the x‐ray beam emits X‐ray fluorescence regardless of its chemical form, oxidation state or physical state (bound or free). The depth from which X‐ray fluorescence, arising from a specific element, can escape from a sample is known. For example, zinc is measured from a greater depth than iron because it has a more energetic X‐ray fluorescence.
Because the X‐ray beam interrogates the sample for only milliseconds per point, radiation damage is low and the sample can be used for other analytic techniques, such as histology or MRI. After metals are mapped, points of interest can be chemically analyzed using near edge spectroscopy (XANES). Formalin can alter the chemical form of many metals, but near edge spectroscopy can still yield clues about the chemical form (e.g. heme iron vs ferritin iron).
Distribution of Metals in the Normal Adult Brain
In the adult brain, regions associated with motor functions contain two to three times more iron than non motor related regions [2, 8, 31]. Gray matter structures generally contain more iron than white matter [10, 22]. Substantia nigra, globus pallidus, putamen, caudate nucleus, red nucleus, dentate nucleus, and locus coeruleus contain the highest iron concentrations in the adult brain explaining why in movement disorders these regions are vulnerable to effects of iron imbalance [5, 8, 9, 10, 11, 12, 22, 23, 24, 31, 32].
While iron is highest in the extrapyramidal regions, copper concentrations are highest in the substantia nigra, locus coeruleus, dentate nucleus, and cerebellum [8, 10, 22, 23, 33, 34]. The basal ganglia also have a high copper content, but the difference between their copper levels and those of other brain regions is not as striking as for iron [8, 10, 22, 23, 24, 33, 34].
The white matter of the adult brain contains more zinc than the gray matter [22, 23, 24] because myelin is rich in zinc which stabilizes myelin structure. The zinc content of myelin is about 50 μM in vivo[35, 36, 37]. While most iron‐ and copper‐rich brain structures are low in zinc [7, 10, 22, 23, 24] the hippocampus and amygdala are rich in zinc because these brain regions have numerous zincergic neurons [10, 38, 39].
Thus, the brain is a major metal repository where each brain region has a unique complement of metals that is vital for their normal functioning.
Therapeutic Strategies for Combating Metal‐Associated Neurodegeneration
Iron and copper transfer single electrons as they cycle between their reduced (Fe2+, Cu1+) and oxidized (Fe3+, Cu2+) states and this redox cycling can catalyze the production of reactive oxygen species (ROS). Reduced cations promote the one electron reduction of O2 with formation of superoxide (O2 −·) and of hydrogen peroxide (H2O2) with formation of hydroxyl (OH·), the most reactive of all endogenously generated free radicals. ROS have the ability to damage carbohydrates, lipids, proteins, and DNA [40, 41].
Moreover, even redox inert metals such as zinc, if in excess, can trigger ROS production. Zn2+ inhibits cellular respiration and facilitates the leakage of electrons from the mitochondrial electron transport chain by inhibiting complex I and III [42]. Zinc is also able to increase production of O2 −· (via activation of protein kinase C (PKC) and increase activity of NADPH oxidase) and of nitric oxide (NO) (via induction of NO synthase) [42]. NO can react with O2 −· to produce peroxynitrite (ONOO−), a free radical that can cause oxidation and nitration of proteins, lipids, and DNA [39, 42].
Cells have developed a system of antioxidant defenses aimed at eliminating ROS. Superoxide dismutases (SOD) are metalloproteins that accelerate the dismutation of O2 −· to H2O2 and O2. Mammalian Cu/Zn SOD is present in the cytosol and mitochondrial intermembrane space of all cells and Mn SOD serves the same function in the mitochondrial matrix. H2O2 is removed by catalase or by the selenoprotein glutathione peroxidase with the concomitant oxidation of glutathione [41]. Cellular antioxidant defense is further bolstered by melatonin, flavonoids, α tocopherol, β carotene, ascorbic acid, and ubiquinol [40, 41].
Since excessive amounts of metal‐catalyzed ROS can rapidly overcome these defense systems, cells have metal chaperones and storage proteins that keep metals in a nontoxic, but bioavailable form (Table 3). The cytosolic ferritin [43, 44], mitochondrial ferritin (MtFt) [45], and mitochondrial frataxin [46, 47] have ferroxidase centers that promote the oxidation of excess ferrous iron and its storage as ferrihydrite (a biomineral formed of ferric oxides/hydroxides octahedra) [48, 49, 50]. Ferritin, frataxin, and the iron–sulfur cluster‐containing aconitase protect mitochondrial and nuclear DNA, heme, and iron–sulfur cluster proteins against ROS damage [43, 46, 51, 52].
Table 3.
Important metal storage proteins.
| Cell | Protein | Role |
|---|---|---|
| Neuronsa | Mostly H‐chain ferritin | Ferroxidase activity (cytosol) |
| DNA protection from ROS (nucleus) | ||
| Frataxinb2 | Iron chaperone, ferroxidase activity and iron storage (mitochondria) | |
| Metallothionein (MT) III | Sequestration of copper and zinc | |
| Neurogliaa,c | Mostly L‐chain ferritin (microglia) | Nucleation of iron and iron storage |
| H and L‐chain ferritin (oligodendrocytes) | Ferroxidase activity (cytosol) and iron storage | |
| DNA protection from ROS (nucleus) | ||
| Frataxinb | Iron chaperone, ferroxidase activity and iron storage (mitochondria) | |
| MT I and II (astrocytes, microglia in response to injury) | Sequestration of copper and zinc |
aIn the human mitochondrial ferritin (MtFt) is expressed at high levels in erythroblasts from patients with sideroblastic anemia and testis. In mouse, MtFt expression is also detected in pancreatic islets of Langerhans, brain, spinal cord and adipocytes [146, 147].
bIn the CNS, frataxin mRNA expression is the highest in the spinal cord, with less expression in cerebellum and very little in cerebral cortex. Further study is needed to determine the level of frataxin expression in different cells types of the nervous tissue.
cVery little ferritin expression is seen in astrocytes.
Intracellular copper is very tightly regulated through an elegant system of chaperones that carry and insert copper ions into specific copper metalloproteins [53]. There are no known chaperones for zinc, and free zinc actually contributes to normal brain function [38, 39]. The only protein thought to function as a zinc chaperone is metallothionein (MT), a cysteine‐rich cytoplasmic protein that can sequester both copper and zinc. Three MTs are expressed in the brain, protecting it against zinc and copper toxicity [39, 54].
Both redox‐inert metal ions such as zinc and redox‐active metal ions like iron and copper can associate with proteins and exacerbate or cause, the aggregation of Aβ‐amyloid, α‐synuclein, prion protein or ataxin‐3 [2, 39, 54, 55, 56, 57, 58] triggering neurodegeneration.
Iron [59, 60], copper [54], and zinc [61, 62] are essential for myelin synthesis, stability, and maintenance by oligodendrocytes. Many enzymes involved in the production of ATP and myelin precursors (cholesterol and fatty acids) are metal dependent (Table 1). Metal dyshomeostasis leads to impaired function and degradation of metalloenzymes causing a decrease in myelin production, improper brain maturation and associated impairments [41, 54, 58, 63, 64].
Metal chelators have been proposed as potential therapeutic agents to scavenge excess brain metals by the formation a nontoxic metal‐chelator complexes and their subsequent excretion in urine. Important requirements for effective metal chelators are their ability to cross the blood brain barrier and their selectivity not only for a specific metal, but for specific chemical forms and oxidation states of that metal [65].
Indiscriminate metal chelation can induce a generalized metal deficiency with chelator‐induced inhibition of metalloenzymes, metal redistribution, interference with neurotransmitter metabolism and cytotoxicity [2, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74]. Cuprizone (bis‐cyclohexanone oxaldihydrazone) is an example of a toxic metal chelator, used to induce a reversible demyelination in rodents that mimics multiple sclerosis in humans. It is not known why oligodendrocytes are more vulnerable than other cell types to cuprizone toxicity [75].
Clioquinol, a small hydrophobic metal chelator with affinity for iron, copper, and zinc [2, 39, 58, 65], was used as an antiparasitic until 1970 when it was withdrawn because of its association with subacute myelo‐optic neuropathy (SMON) [70], but the mechanism of its toxicity remains unknown. We have recently described a case of undefined progressive spinocerebellar ataxia characterized by degeneration of olivocerebellar fibers associated with decreased copper levels in the olivary region [24]. Although there was no history of exposure to chelators, the neuropathology of this case [24] is identical to the neuropathology reported in clioquinol intoxication [74]. There is also a striking clinical resemblance between copper deficiency and clioquinol intoxication [74] and clioquinol‐induced SMON [75, 76, 77, 78, 79]. All these findings now suggest that an abnormality in copper homeostasis is responsible for clioquinol‐induced SMON. The definitive confirmation of the role of copper in SMON is essential since clioquinol is proposed for use in patients with AD and PD [2, 39, 58, 65].
The rationale for the use of clioquinol in the treatment of Alzheimer's disease (AD) arose from studies showing that copper and zinc bound to amyloid β[80, 81, 82] and that clioquinol inhibited its accumulation on amyloid [83]. Fe3+ and Zn2+ can modulate the ability of α‐secretase to cleave the amyloid protein precursor (APP) [2, 39]. Both APP and Aβ can bind Fe3+ and Zn2+ in addition to Cu2+ and induce precipitation and aggregation of Aβ, while Aβ‐bound Fe2+ and Cu1+ are primarily involved in the chronic oxidative damage that characterizes AD. Metals promote both the precipitation and deposition of amyloid‐β (Aβ) and oxidative stress which is associated with the neuritic plaques [2, 38, 39, 54, 55, 57, 58]. It is not known if the metal deposition is the cause or the consequence of the oxidative stress.
Clinical trials indicate that clioquinol slows cognitive decline in AD patients [84], but its mechanism of action is controversial [83]. The beneficial effects of clioquinol may relate more to its ability to redistribute metals than remove them. White et al. have shown that clioquinol, in both its metal‐free or complexed forms, increases the synthesis of metalloproteinases that degrade amyloid β secreted by APP transfected Chinese hamster ovary cells in culture [85]. This suggests that while some of the clioquinol‐bound copper is excreted in the urine, clioquinol can also transport copper into neurons, thereby relieving neuronal copper deficiency.
XRF microprobe has the micron to submicron resolution necessary to examine metal distribution at the cellular level and so has been applied to examine metals in AD [25, 28, 86, 87, 88, 89, 90, 91]. With respect to therapeutics, RS‐XRF and XRF microprobe simultaneously map multiple metals and should be able to distinguish between metal chelators that remove metals from the brain and those that redistribute metals within the brain. When combined with X‐ray absorption spectroscopy, subtle changes in metal chemistry can be detected [49].
Regions of the brain affected in AD, the inferior parietal cortex, motor cortex, olfactory cortex and cortex of the frontal pole [92, 93, 94, 95, 96, 97, 98], accumulate iron at a rate that may exceed the capacity of ferritin to sequester it [31]. XRF is a valuable tool to localize abnormal iron or zinc. As shown in Figure 1, the metal accumulation in the AD hippocampus does not rival the normally high iron and zinc content of the substantia nigra and white matter, respectively. This suggests that inappropriate metal sequestration rather than the absolute metal amount is partially responsible for the AD pathology.
Figure 1.
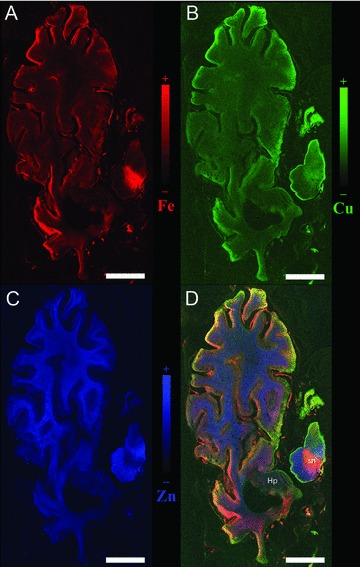
Metals in the brain of a subject with AD. A. Iron map; B. Copper map; C. Zinc map; D. Overlay of iron, copper and zinc; color scales represent the normalized total Kα fluorescence counts, proportional to the total metal present, from black (lowest) to color (highest); sn substantia nigra; Hp hippocampus; axial section; scale bar 10 mm.
While iron chelators are not currently advised to treat patients with AD [99], new chelators are under development and RS‐XRF would be an excellent tool to examine their selectivity in vivo.
The most common and consistent finding in Parkinson's disease (PD) is the accumulation of iron in substantia nigra and basal ganglia (Figure 2) [8, 22, 100, 101, 102, 103, 104, 105] and this has led many researchers to seek for a potential benefit of metal chelators in PD. Ferric iron deposits have been found in dopaminergic neurons, microglia, oligodendrocytes, astrocytes, and Lewy bodies in the substantia nigra of PD patients [106]. Copper is decreased [7, 8, 22] and zinc is increased [7, 8] in the PD substantia nigra, although at least one PD case characterized by decreased zinc has been reported [22]. The caudate, putamen, and globus pallidus have also been reported to accumulate metals in PD [7, 8, 22] (Figure 2).
Figure 2.
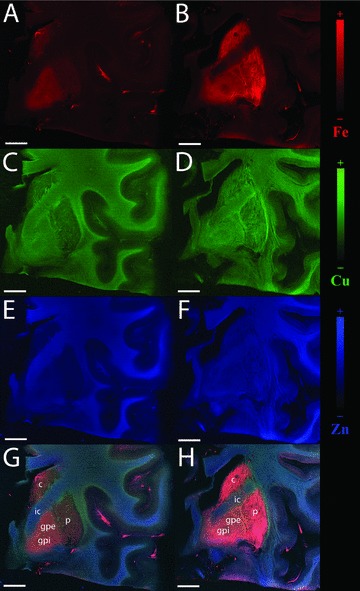
The caudate, putamen and globus pallidus of a subject with PD (B, D, F, H) and matched control (A, C, E, G); PD shows accumulation of iron, copper and zinc when compared to control. A, B. Iron maps; C, D. Copper maps; E, F. Zinc maps; G, H. Overlay of iron, copper and zinc; color scales represent the normalized total Kα fluorescence counts, proportional to the total metal present, from black (lowest) to color (highest); c caudate; p putamen; gpe globus pallidus pars externa; gpi globus pallidus pars interna; ic internal capsule; coronal section; scale bar 10 mm.
The mechanisms of the metal changes in PD are not well understood, but the iron accumulation in the substantia nigra may trigger oxidative stress [107], followed by a protective increase in the iron binding capacity of ferritin [108] and iron chelation by neuromelanin [109, 110, 111]. The neuromelanin‐iron complexes activate microglia to release cytokines and neurotoxic compounds inducing a vicious cycle of chronic inflammation and neuronal loss in the PD substantia nigra [112, 113]. Metallothionein expression also increases in the substantia nigra [114] either as a result of the release of pro‐inflammatory cytokines [115] or in order to accommodate the dyshomeostasis of copper and zinc and scavenge free radicals [114, 116]. However, metallothioneins may not always represent a protective factor since it has been proven that they can release zinc in toxic amounts in neurons [117].
Clioquinol and desferrioxamine have been shown to reduce the iron content of the substantia nigra of PD animal models, decrease the oxidative stress markers and glutathione depletion, and prevent the iron‐induced neurotoxicity [118, 119, 120, 121, 122]. All these studies suggest a potential benefit of metal chelators in PD, but their use has not been validated in clinical trials as yet. However, it is also possible that the iron build‐up seen in PD brains is simply the consequence of neuronal loss and replacement of neurons by cells with higher iron content [2]. To date, no data are available to prove whether iron accumulation in PD brains is an early event causing the degeneration of the dopaminergic neurons or a late event caused by the neurodegenerative process.
Using animal models, RS‐XRF has great potential to improve our understanding of global metal metabolism and the interrelationships between metals. Metal maps of brains from various animal models for neurodegeneration at various stages of the disease will likely demonstrate whether metal accumulation is the cause or the consequence of the neurodegeneration. Understanding the metal dyshomeostasis and its relation to the neuronal loss and disease progression would allow for the development of nontoxic and more selective metal chelators targeted to removing those metal species that are most reactive.
Friedreich's ataxia (FRDA) is caused by deficiency of a nuclear encoded mitochondrial targeted protein named frataxin [123, 124] which can function as both an iron chaperone for biosynthesis of heme and iron sulfur clusters and as an iron storage protein [45, 46, 47, 48, 125, 126]. FRDA patients have increased iron levels in the heart [127, 128, 129] and dentate nucleus [130, 131] and it has been suggested that the severe degeneration of the cardiomyocytes in the heart and neurons in the dentate nucleus is the result of oxidative damage caused by mitochondrial iron accumulation. Deferiprone has been shown to selectively reduce the iron accumulation in dentate nuclei of FRDA patients [130] most likely because of its ability to chelate both the cytosolic and mitochondrial labile iron and redistribute it between different cellular compartments and even to different cell populations [132].
Our laboratory has used XANES to speciate the chemical form of iron in mitochondria of fibroblasts from FRDA patients. We have shown that most of the mitochondrial iron is stored as the nontoxic, but also poorly bioavailable ferrihydrite of mitochondrial ferritin (MtFt) and little ferrous redox‐active iron is present [49]. These data support the new concept that once MtFt is upregulated in FRDA (by whatever means) it competes for and sequesters the available mitochondrial iron. Since MtFt mineralizes this iron, but does not readily release it, our data support the hypothesis that the lack of iron available for heme and iron–sulfur cluster synthesis is more important than oxidative stress in FRDA pathophysiology. Like FRDA, most of the excess iron in other neurodegenerative diseases is also stored as relatively nontoxic ferrihydrite [47, 48] in ferritin [2, 48, 101] and chelation strategies designed for these diseases should take into account that at least in cells that are able to upregulate their ferritin and MtFt production, chelating the small nonferrihydrite iron pool could prevent the cells from carrying out even minimal functions such as heme synthesis.
Wilson's disease (WD) is one of the few neurodegenerative diseases where the success of the chelation therapy is proven over many years of clinical use. In WD, mutations in the gene encoding the copper transporter Atp7b result in toxic copper build‐up in hepatocytes [133]. Copper accumulation leads to copper‐mediated oxidative damage of the liver cells, hepatic necrosis, release of large amounts of free copper in the bloodstream and copper overload in all tissues, including basal ganglia [134–138].
D‐penicillamine was the first drug to be successfully employed in WD. D‐penicillamine not only chelates the tissue copper and promotes its excretion in the urine, but also detoxifies it by promoting the synthesis of metallothionein, which forms a nontoxic combination with copper [65, 139, 140]. Trientine is a less toxic alternative to D‐penicillamine also able to chelate the tissue copper and increase its urinary excretion [65, 139, 140]. Other chelating agents that can be used are ammonium tetrathiomolybdate that complexes with copper in the intestine and blood [65, 139, 141]. The advantage of tetrathiomolybdate is its specificity for copper and not other metals. Zinc sulfate has been used to prevent the absorption of copper from the intestine by inducing the expression of copper‐binding metallothioneins in enterocytes [65, 139, 142, 143]. While the benefit of these copper chelators in removing the toxic deposits of copper is beyond any doubt, the long‐term effects of copper depletion on copper‐rich brains areas, such as the olivary nucleus, have not been explored.
Excellent reviews on therapeutic chelators have been recently published [2, 65, 73, 139, 144], but many questions remain to be answered. How specific is a “specific” chelator? How does the chelation of a metal affect the distribution of other metals? Are metals chelated only in brain regions where they accumulate? RS‐XRF is an ideal, inexpensive and relatively straightforward way to compare the amount and distribution of brain metals in groups of animals (or even pathological human brain specimens) with and without chelation therapy. When combined with chemical speciation using XANES [145], RS‐XRF has the potential to become the tool of choice in mapping and speciating brain metals in situ.
Conclusion
The brain is a major metal repository [8, 9, 10, 11, 32], where large amounts of metals coexist and colocalize. The high metal content of the CNS makes it particularly susceptible to metal‐catalyzed oxidative damage, protein aggregation, neurotoxicity, and neurodegeneration [2, 53, 54].
RS‐XRF is a new and powerful tool that makes systematic studies of brain metals practical. It can be applied to any human or animal tissue and we foresee important applications in testing whether a metal chelator designed to remove a particular metal disrupts the levels of other metals. Using animal models, RS‐XRF also has great potential to improve our understanding of metal metabolism and the interrelationships between metals in the brain. With rapidly advancing beamline technology at third‐generation synchrotrons, it should be possible to map very large sections in a matter of minutes, making systematic RS‐XRF studies on serial slices of whole human brain practical.
Conflict of Interest
The authors have no conflict of interest.
References
- 1. Bartzokis G, Tishler TA, Lu PH, et al Brain ferritin iron may influence age‐ and gender‐related risks of neurodegeneration. Neurobiol Aging 2007;28:414–423. [DOI] [PubMed] [Google Scholar]
- 2. Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 2004;5:863–873. [DOI] [PubMed] [Google Scholar]
- 3. Becker JS, Zoriy MV, Pickhardt C, Palomero‐Gallagher N, Zilles K. Imaging of copper, zinc, and other elements in thin section of human brain samples (hippocampus) by laser ablation inductively coupled plasma mass spectrometry. Anal Chem 2005;77:3208–3216. [DOI] [PubMed] [Google Scholar]
- 4. Dexter DT, Carayon A, Javoy‐Agid F, et al Alterations in the levels of iron, ferritin and other trace metals in Parkinson's disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991;114(Pt 4):1953–1975. [DOI] [PubMed] [Google Scholar]
- 5. Dexter DT, Jenner P, Schapira AH, Marsden CD. Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. The Royal Kings and Queens Parkinson's Disease Research Group. Ann Neurol 1992;32(Suppl):S94–S100. [DOI] [PubMed] [Google Scholar]
- 6. Dexter DT, Sian J, Jenner P, Marsden CD. Implications of alterations in trace element levels in brain in Parkinson's disease and other neurological disorders affecting the basal ganglia. Adv Neurol 1993;60:273–281. [PubMed] [Google Scholar]
- 7. Dexter DT, Wells FR, Agid F, et al Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987;2:1219–1220. [DOI] [PubMed] [Google Scholar]
- 8. Dexter DT, Wells FR, Lees AJ, et al Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease. J Neurochem 1989;52:1830–1836. [DOI] [PubMed] [Google Scholar]
- 9. Hock A, Demmel U, Schicha H, Kasperek K, Feinendegen LE. Trace element concentration in human brain. Activation analysis of cobalt, iron, rubidium, selenium, zinc, chromium, silver, cesium, antimony and scandium. Brain 1975;98:49–64. [DOI] [PubMed] [Google Scholar]
- 10. Duflou H, Maenhaut W, De Reuck J. Regional distribution of potassium, calcium, and six trace elements in normal human brain. Neurochem Res 1989;14:1099–1112. [DOI] [PubMed] [Google Scholar]
- 11. Rajan MT, Jagannatha Rao KS, Mamatha BM, et al Quantification of trace elements in normal human brain by inductively coupled plasma atomic emission spectrometry. J Neurol S 1997;146:153–166. [DOI] [PubMed] [Google Scholar]
- 12. Haacke EM, Cheng NY, House MJ, et al Imaging iron stores in the brain using magnetic resonance imaging. Magn Reson Imaging 2005;23:1–25. [DOI] [PubMed] [Google Scholar]
- 13. Perls M. Nachweis von Eisenoxyd in gewissen Pigmenten. Virchows Arch Path Anat 1867;39:42–48. [Google Scholar]
- 14. Zaleski SS. Das Eisen der Organe bei Morbus maculosus Werlhoffi. Arch Experiment Patholol Pharmacol 1887;23:77–90. [Google Scholar]
- 15. Gomori G. Microtechnical demonstartion of iron: A criticism of its methods. Am J Pathol 1936;12:655–663. [PMC free article] [PubMed] [Google Scholar]
- 16. Meguro R, Asano Y, Odagiri S, Li C, Iwatsuki H, Shoumura K. Nonheme‐iron histochemistry for light and electron microscopy: A historical, theoretical and technical review. Arch Histol Cytol 2007;70:1–19. [DOI] [PubMed] [Google Scholar]
- 17. Pilloni L, Lecca S, Van Eyken P, et al Value of histochemical stains for copper in the diagnosis of Wilson's disease. Histopathology 1998;33:28–33. [DOI] [PubMed] [Google Scholar]
- 18. Henwood A. Current applications of orcein in histochemistry. A brief review with some new observations concerning influence of dye batch variation and aging of dye solutions on staining. Biotech Histochem 2003;78:303–308. [DOI] [PubMed] [Google Scholar]
- 19. Ferenci P, Steindl‐Munda P, Vogel W, et al Diagnostic value of quantitative hepatic copper determination in patients with Wilson's Disease. Clin Gastroenterol Hepatol 2005;3:811–818. [DOI] [PubMed] [Google Scholar]
- 20. Danscher G, Howell G, Perez‐Clausell J, Hertel N. The dithizone, Timm's sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry 1985;83:419–422. [DOI] [PubMed] [Google Scholar]
- 21. Lopez‐Garcia C, Varea E, Palop JJ, et al Cytochemical techniques for zinc and heavy metals localization in nerve cells. Microsc Res Tech 2002;56:318–331. [DOI] [PubMed] [Google Scholar]
- 22. Popescu BF, George MJ, Bergmann U, et al Mapping metals in Parkinson's and normal brain using rapid‐scanning x‐ray fluorescence. Phys Med Biol 2009;54:651–663. [DOI] [PubMed] [Google Scholar]
- 23. Popescu BF, Robinson CA, Rajput A, Rajput AH, Harder SL, Nichol H. Iron, copper, and zinc distribution of the cerebellum. Cerebellum 2009;8:74–79. [DOI] [PubMed] [Google Scholar]
- 24. Popescu BF, Robinson CA, Chapman LD, Nichol H. Synchrotron X‐ray fluorescence reveals abnormal metal distributions in brain and spinal cord in spinocerebellar ataxia: A case report. Cerebellum 2009. [DOI] [PubMed] [Google Scholar]
- 25. Collingwood J, Dobson J. Mapping and characterization of iron compounds in Alzheimer's tissue. J Alzheimers Dis 2006;10:215–222. [DOI] [PubMed] [Google Scholar]
- 26. Ishihara R, Ide‐Ektessabi A, Ikeda K, et al Investigation of cellular metallic elements in single neurons of human brain tissues. Neuroreport 2002;13:1817–1820. [DOI] [PubMed] [Google Scholar]
- 27. Linkous DH, Flinn JM, Koh JY, et al Evidence that the ZNT‐3 protein controls the total amount of elemental zinc in synaptic vesicles. J Histochem Cytochem 2008;56:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. Synchrotron‐based infrared and X‐ray imaging shows focalized accumulation of Cu and Zn co‐localized with beta‐amyloid deposits in Alzheimer's disease. J Struct Biol 2006;155:30–37. [DOI] [PubMed] [Google Scholar]
- 29. Yoshida S, Ektessabi A, Fujisawa S. XANES spectroscopy of a single neuron from a patient with Parkinson's disease. J Synchrotron Radiat 2001;8(Pt 2):998–1000. [DOI] [PubMed] [Google Scholar]
- 30. McCrea RP, Harder SL, Martin M, Buist R, Nichol H. A comparison of rapid‐scanning X‐ray fluorescence mapping and magnetic resonance imaging to localize brain iron distribution. Eur J Radiol 2008;68(3 Suppl):S109–S113. [DOI] [PubMed] [Google Scholar]
- 31. Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron‐regulatory proteins in the brain in aging and Alzheimer's disease. J Neurosci Res 1992;31:327–335. [DOI] [PubMed] [Google Scholar]
- 32. Morris CM, Candy JM, Oakley AE, Bloxham CA, Edwardson JA. Histochemical distribution of non‐haem iron in the human brain. Acta Anat (Basel) 1992;144:235–257. [DOI] [PubMed] [Google Scholar]
- 33. Goldberg WJ, Allen N. Determination of Cu, Mn, Fe, and Ca in six regions of normal human brain, by atomic absorption spectroscopy. Clin Chem 1981;27:562–564. [PubMed] [Google Scholar]
- 34. Warren PJ, Earl CJ, Thompson RH. The distribution of copper in human brain. Brain 1960;83:709–717. [DOI] [PubMed] [Google Scholar]
- 35. Altman PL, Dittmer DS. Biology data book. Maryland : FASEB, 1973. [PubMed] [Google Scholar]
- 36. Harauz G, Ishiyama N, Hill CM, Bates IR, Libich DS, Fares C. Myelin basic protein‐diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron 2004;35:503–542. [DOI] [PubMed] [Google Scholar]
- 37. Iyengar GV, Koomer WE, Bown HJM. The elemental composition of human tissues and body fluids. New York : Verlag‐Chemie, 1978. [Google Scholar]
- 38. Cuajungco MP, Lees GJ. Zinc metabolism in the brain: Relevance to human neurodegenerative disorders. Neurobiol Dis 1997;4:137–169. [DOI] [PubMed] [Google Scholar]
- 39. Mocchegiani E, Bertoni‐Freddari C, Marcellini F, Malavolta M. Brain, aging and neurodegeneration: Role of zinc ion availability. Prog Neurobiol 2005;75:367–390. [DOI] [PubMed] [Google Scholar]
- 40. Winterbourn CC. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol Lett 1995;82–83:969–974. [DOI] [PubMed] [Google Scholar]
- 41. Halliwell B. Role of free radicals in the neurodegenerative diseases: Therapeutic implications for antioxidant treatment. Drugs Aging 2001;18:685–716. [DOI] [PubMed] [Google Scholar]
- 42. Frazzini V, Rockabrand E, Mocchegiani E, Sensi SL. Oxidative stress and brain aging: Is zinc the link? Biogerontology 2006;7:307–314. [DOI] [PubMed] [Google Scholar]
- 43. Arosio P, Ingrassia R, Cavadini P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta 2009;1790:589–599. [DOI] [PubMed] [Google Scholar]
- 44. Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1996;1275:161–203. [DOI] [PubMed] [Google Scholar]
- 45. Bou‐Abdallah F, Santambrogio P, Levi S, Arosio P, Chasteen ND. Unique iron binding and oxidation properties of human mitochondrial ferritin: A comparative analysis with Human H‐chain ferritin. J Mol Biol 2005;347:543–554. [DOI] [PubMed] [Google Scholar]
- 46. O’Neill HA, Gakh O, Park S, et al Assembly of human frataxin is a mechanism for detoxifying redox‐active iron. Biochemistry 2005;44:537–545. [DOI] [PubMed] [Google Scholar]
- 47. O’Neill HA, Gakh O, Isaya G. Supramolecular assemblies of human frataxin are formed via subunit–subunit interactions mediated by a non‐conserved amino‐terminal region. J Mol Biol 2005;345:433–439. [DOI] [PubMed] [Google Scholar]
- 48. Nichol H, Gakh O, O’Neill HA, Pickering IJ, Isaya G, George GN. Structure of frataxin iron cores: An X‐ray absorption spectroscopic study. Biochemistry 2003;42:5971–5976. [DOI] [PubMed] [Google Scholar]
- 49. Popescu BF, Pickering IJ, George GN, Nichol H. The chemical form of mitochondrial iron in Friedreich's ataxia. J Inorg Biochem 2007;101:957–966. [DOI] [PubMed] [Google Scholar]
- 50. Cavadini P, O’Neill HA, Benada O, Isaya G. Assembly and iron‐binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum Mol Genet 2002;11:217–227. [DOI] [PubMed] [Google Scholar]
- 51. Shadel GS. Mitochondrial DNA, aconitase ‘wraps’ it up. Trends Biochem Sci 2005;30:294–296. [DOI] [PubMed] [Google Scholar]
- 52. Campanella A, Rovelli E, Santambrogio P, Cozzi A, Taroni F, Levi S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum Mol Genet 2009;18:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O’Halloran TV, Culotta VC. Metallochaperones, an intracellular shuttle service for metal ions. J Biol Chem 2000;275:25057–25060. [DOI] [PubMed] [Google Scholar]
- 54. Madsen E, Gitlin JD. Copper and iron disorders of the brain. Annu Rev Neurosci 2007;30:317–337. [DOI] [PubMed] [Google Scholar]
- 55. Molina‐Holgado F, Hider RC, Gaeta A, Williams R, Francis P. Metals ions and neurodegeneration. Biometals 2007;20:639–654. [DOI] [PubMed] [Google Scholar]
- 56. Ricchelli F, Fusi P, Tortora P, et al Destabilization of non‐pathological variants of ataxin‐3 by metal ions results in aggregation/fibrillogenesis. Int J Biochem Cell Biol 2007;39:966–977. [DOI] [PubMed] [Google Scholar]
- 57. Bush AI. Metals and neuroscience. Curr Opin Chem Biol 2000;4:184–191. [DOI] [PubMed] [Google Scholar]
- 58. Zatta P, Frank A. Copper deficiency and neurological disorders in man and animals. Brain Res Rev 2007;54:19–33. [DOI] [PubMed] [Google Scholar]
- 59. Ortiz E, Pasquini JM, Thompson K, et al Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J Neurosci Res 2004;77:681–689. [DOI] [PubMed] [Google Scholar]
- 60. Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR. Oligodendrocytes and myelination: The role of iron. Glia 2009;57:467–478. [DOI] [PubMed] [Google Scholar]
- 61. Tsang D, Tsang YS, Ho WK, Wong RN. Myelin basic protein is a zinc‐binding protein in brain: Possible role in myelin compaction. Neurochem Res 1997;22:811–819. [DOI] [PubMed] [Google Scholar]
- 62. Kursula P, Merilainen G, Lehto VP, Heape AM. The small myelin‐associated glycoprotein is a zinc‐binding protein. J Neurochem 1999;73:2110–2118. [PubMed] [Google Scholar]
- 63. Snyder AM, Connor JR. Iron, the substantia nigra and related neurological disorders. Biochim Biophys Acta 2009;1790:606–614. [DOI] [PubMed] [Google Scholar]
- 64. Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr 2003;23:41–58. [DOI] [PubMed] [Google Scholar]
- 65. Bolognin S, Drago D, Messori L, Zatta P. Chelation therapy for neurodegenerative diseases. Med Res Rev 2009;29:547–570. [DOI] [PubMed] [Google Scholar]
- 66. Hider RC. Potential protection from toxicity by oral iron chelators. Toxicol Lett 1995;82–83:961–967. [DOI] [PubMed] [Google Scholar]
- 67. Singh S, Khodr H, Taylor MI, Hider RC. Therapeutic iron chelators and their potential side‐effects. Biochem Soc Symp 1995;61:127–137. [DOI] [PubMed] [Google Scholar]
- 68. Cooper CE, Lynagh GR, Hoyes KP, Hider RC, Cammack R, Porter JB. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J Biol Chem 1996;271:20291–20299. [DOI] [PubMed] [Google Scholar]
- 69. Liu ZD, Lockwood M, Rose S, Theobald AE, Hider RC. Structure‐activity investigation of the inhibition of 3‐hydroxypyridin‐4‐ones on mammalian tyrosine hydroxylase. Biochem Pharmacol 2001;61:285–290. [DOI] [PubMed] [Google Scholar]
- 70. Tsubaki T, Honma Y, Hoshi M. Neurological syndrome associated with clioquinol. Lancet 1971;1:696–697. [DOI] [PubMed] [Google Scholar]
- 71. Oakley GP, Jr . The neurotoxicity of the halogenated hydroxyquinolines. A commentary. Jama 1973;225:395–397. [PubMed] [Google Scholar]
- 72. Schaumburg H, Herskovitz S. Copper deficiency myeloneuropathy: A clue to clioquinol‐induced subacute myelo‐optic neuropathy Neurology 2008;71:622–623. [DOI] [PubMed] [Google Scholar]
- 73. Marmolino D, Acquaviva F. Friedreich's ataxia: From the (GAA)(n) repeat mediated silencing to new promising molecules for therapy. Cerebellum 2009;8:245–259. [DOI] [PubMed] [Google Scholar]
- 74. Koga M, Tsutsumi A, Shirabe T. The pathogenesis of olivary changes in clioquinol intoxication. Neuropathology 1997;17:290–294. [Google Scholar]
- 75. Torkildsen O, Brunborg LA, Myhr KM, Bo L. The cuprizone model for demyelination. Acta Neurologica Scandinavica 2008;188:72–76. [DOI] [PubMed] [Google Scholar]
- 76. Kumar N, Gross JB, Jr ., Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology 2004;63:33–39. [DOI] [PubMed] [Google Scholar]
- 77. Kumar N, Ahlskog JE, Klein CJ, Port JD. Imaging features of copper deficiency myelopathy: A study of 25 cases. Neuroradiology 2006;48:78–83. [DOI] [PubMed] [Google Scholar]
- 78. Nations SP, Boyer PJ, Love LA, et al Denture cream: an unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology 2008;71:639–643. [DOI] [PubMed] [Google Scholar]
- 79. Schleper B, Stuerenburg HJ. Copper deficiency‐associated myelopathy in a 46‐year‐old woman. J Neurol 2001;248:705–706. [DOI] [PubMed] [Google Scholar]
- 80. Atwood CS, Scarpa RC, Huang X, et al Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar‐affinity copper binding site on amyloid beta1–42. J Neurochem 2000;75:1219–1233. [DOI] [PubMed] [Google Scholar]
- 81. Danielsson J, Pierattelli R, Banci L, Graslund A. High‐resolution NMR studies of the zinc‐binding site of the Alzheimer's amyloid beta‐peptide. FEBS J 2007;274:46–59. [DOI] [PubMed] [Google Scholar]
- 82. Syme CD, Nadal RC, Rigby SE, Viles JH. Copper binding to the amyloid‐beta (Abeta) peptide associated with Alzheimer's disease: Folding, coordination geometry, pH dependence, stoichiometry, and affinity of Abeta‐(1–28): Insights from a range of complementary spectroscopic techniques. J Biol Chem 2004;279:18169–18177. [DOI] [PubMed] [Google Scholar]
- 83. Cherny RA, Atwood CS, Xilinas ME, et al Treatment with a copper‐zinc chelator markedly and rapidly inhibits beta‐amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 2001;30:665–676. [DOI] [PubMed] [Google Scholar]
- 84. Ritchie CW, Bush AI, Mackinnon A, et al Metal‐protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch Neurol 2003;60:1685–1691. [DOI] [PubMed] [Google Scholar]
- 85. White AR, Du T, Laughton KM, et al Degradation of the Alzheimer disease amyloid beta‐peptide by metal‐dependent up‐regulation of metalloprotease activity. J Biol Chem 2006;281:17670–17680. [DOI] [PubMed] [Google Scholar]
- 86. Collingwood JF, Chong RK, Kasama T, et al Three‐dimensional tomographic imaging and characterization of iron compounds within Alzheimer's plaque core material. J Alzheimers Dis 2008;14:235–245. [DOI] [PubMed] [Google Scholar]
- 87. Collingwood JF, Mikhaylova A, Davidson M, et al In situ characterization and mapping of iron compounds in Alzheimer's disease tissue. J Alzheimers Dis 2005;7:267–272. [DOI] [PubMed] [Google Scholar]
- 88. Gossuin Y, Hautot D, Muller RN, et al Looking for biogenic magnetite in brain ferritin using NMR relaxometry. NMR Biomed 2005;18:469–472. [DOI] [PubMed] [Google Scholar]
- 89. House E, Collingwood J, Khan A, Korchazkina O, Berthon G, Exley C. Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Abeta42 in a manner which may have consequences for metal chelation therapy in Alzheimer's disease. J Alzheimers Dis 2004;6:291–301. [DOI] [PubMed] [Google Scholar]
- 90. Leskovjan AC, Lanzirotti A, Miller LM. Amyloid plaques in PSAPP mice bind less metal than plaques in human Alzheimer's disease. NeuroImage 2009;47:1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rajendran R, Minqin R, Ynsa MD, et al A novel approach to the identification and quantitative elemental analysis of amyloid deposits – Insights into the pathology of Alzheimer's disease. Biochem Biophys Res Commun 2009;382:91–95. [DOI] [PubMed] [Google Scholar]
- 92. Cornett CR, Markesbery WR, Ehmann WD. Imbalances of trace elements related to oxidative damage in Alzheimer's disease brain. Neurotoxicology 1998;19:339–345. [PubMed] [Google Scholar]
- 93. Corrigan FM, Reynolds GP, Ward NI. Hippocampal tin, aluminum and zinc in Alzheimer's disease. Biometals 1993;6:149–154. [DOI] [PubMed] [Google Scholar]
- 94. Deibel MA, Ehmann WD, Markesbery WR. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer's disease: Possible relation to oxidative stress. J Neurol Sci 1996;143:137–142. [DOI] [PubMed] [Google Scholar]
- 95. Plantin LO, Lying‐Tunnel U, Kristensson K. Trace elements in the human central nervous system studied with neutron activation analysis. Biol Trace Elem Res 1987;13:69–75. [DOI] [PubMed] [Google Scholar]
- 96. Samudralwar DL, Diprete CC, Ni BF, Ehmann WD, Markesbery WR. Elemental imbalances in the olfactory pathway in Alzheimer's disease. J Neurol Sci 1995;130:139–145. [DOI] [PubMed] [Google Scholar]
- 97. Thompson CM, Markesbery WR, Ehmann WD, Mao YX, Vance DE. Regional brain trace‐element studies in Alzheimer's disease. Neurotoxicology 1988;9:1–7. [PubMed] [Google Scholar]
- 98. Ward NI, Manson JA. Neutron activation analysis techniques for identifying elemental status in Alzheimer's disease. J Radioanalytical Nucl Chem 1987;113:515–526. [Google Scholar]
- 99. Crapper McLachlan DR, Dalton AJ, Kruck TP, et al Intramuscular desferrioxamine in patients with Alzheimer's disease. Lancet 1991;337:1304–1308. [DOI] [PubMed] [Google Scholar]
- 100. Wallis LI, Paley MN, Graham JM, et al MRI assessment of basal ganglia iron deposition in Parkinson's disease. J Magn Reson Imaging 2008;28:1061–1067. [DOI] [PubMed] [Google Scholar]
- 101. Gorell JM, Ordidge RJ, Brown GG, Deniau JC, Buderer NM, Helpern JA. Increased iron‐related MRI contrast in the substantia nigra in Parkinson's disease. Neurology 1995;45:1138–1143. [DOI] [PubMed] [Google Scholar]
- 102. Griffiths PD, Crossman AR. Distribution of iron in the basal ganglia and neocortex in postmortem tissue in Parkinson's disease and Alzheimer's disease. Dementia 1993;4:61–65. [DOI] [PubMed] [Google Scholar]
- 103. Mann VM, Cooper JM, Daniel SE, et al Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann Neurol 1994;36:876–881. [DOI] [PubMed] [Google Scholar]
- 104. Sofic E, Riederer P, Heinsen H, et al Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm 1988;74:199–205. [DOI] [PubMed] [Google Scholar]
- 105. Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MB. Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem 1991;56:978–982. [DOI] [PubMed] [Google Scholar]
- 106. Zecca L, Stroppolo A, Gatti A, et al The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A 2004;101:9843–9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Connor JR, Snyder BS, Arosio P, Loeffler DA, LeWitt P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer's diseased brains. J Neurochem 1995;65:717–724. [DOI] [PubMed] [Google Scholar]
- 108. Griffiths PD, Dobson BR, Jones GR, Clarke DT. Iron in the basal ganglia in Parkinson's disease. An in vitro study using extended X‐ray absorption fine structure and cryo‐electron microscopy. Brain 1999;122:667–673. [DOI] [PubMed] [Google Scholar]
- 109. Zecca L, Shima T, Stroppolo A, et al Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience 1996;73:407–415. [DOI] [PubMed] [Google Scholar]
- 110. Jellinger K, Kienzl E, Rumpelmair G, et al Iron‐melanin complex in substantia nigra of parkinsonian brains: An x‐ray microanalysis. J Neurochem 1992;59:1168–1171. [DOI] [PubMed] [Google Scholar]
- 111. Good PF, Olanow CW, Perl DP. Neuromelanin‐containing neurons of the substantia nigra accumulate iron and aluminum in Parkinson's disease: A LAMMA study. Brain Res 1992;593:343–346. [DOI] [PubMed] [Google Scholar]
- 112. Zecca L, Zucca FA, Wilms H, Sulzer D. Neuromelanin of the substantia nigra: A neuronal black hole with protective and toxic characteristics. Trends Neurosci 2003;26:578–580. [DOI] [PubMed] [Google Scholar]
- 113. Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, Lucius R. Activation of microglia by human neuromelanin is NF‐kappaB dependent and involves p38 mitogen‐activated protein kinase: Implications for Parkinson's disease. Faseb J 2003;17:500–502. [DOI] [PubMed] [Google Scholar]
- 114. Ebadi M, Pfeiffer RF, Murrin LC, Shiraga H. Metallothionein and oxidation reactions in Parkinson's disease. Proc West Pharmacol Soc 1991;34:285–290. [PubMed] [Google Scholar]
- 115. Barcia C, Fernandez Barreiro A, Poza M, Herrero MT. Parkinson's disease and inflammatory changes. Neurotox Res 2003;5:411–418. [DOI] [PubMed] [Google Scholar]
- 116. Ebadi M, Hiramatsu M, Burke WJ, Folks DG, el‐Sayed MA. Metallothionein isoforms provide neuroprotection against 6‐hydroxydopamine‐generated hydroxyl radicals and superoxide anions. Proc West Pharmacol Soc 1998;41:155–158. [PubMed] [Google Scholar]
- 117. Lee JY, Kim JH, Palmiter RD, Koh JY. Zinc released from metallothionein‐iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol 2003;184:337–347. [DOI] [PubMed] [Google Scholar]
- 118. Ben‐Shachar D, Eshel G, Finberg JP, Youdim MB. The iron chelator desferrioxamine (Desferal) retards 6‐hydroxydopamine‐induced degeneration of nigrostriatal dopamine neurons. J Neurochem 1991;56:1441–1444. [DOI] [PubMed] [Google Scholar]
- 119. Ben‐Shachar D, Zuk R, Gazawi H, Ljubuncic P. Dopamine toxicity involves mitochondrial complex I inhibition: Implications to dopamine‐related neuropsychiatric disorders. Biochem Pharmacol 2004;67:1965–1974. [DOI] [PubMed] [Google Scholar]
- 120. Kaur D, Yantiri F, Rajagopalan S, et al Genetic or pharmacological iron chelation prevents MPTP‐induced neurotoxicity in vivo: A novel therapy for Parkinson's disease. Neuron 2003;37:899–909. [DOI] [PubMed] [Google Scholar]
- 121. Lan J, Jiang DH. Desferrioxamine and vitamin E protect against iron and MPTP‐induced neurodegeneration in mice. J Neural Transm 1997;104:469–481. [DOI] [PubMed] [Google Scholar]
- 122. Youdim MB, Fridkin M, Zheng H. Novel bifunctional drugs targeting monoamine oxidase inhibition and iron chelation as an approach to neuroprotection in Parkinson's disease and other neurodegenerative diseases. J Neural Transm 2004;111:1455–1471. [DOI] [PubMed] [Google Scholar]
- 123. Campuzano V, Montermini L, Lutz Y, et al Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet 1997;6:1771–1780. [DOI] [PubMed] [Google Scholar]
- 124. Campuzano V, Montermini L, Molto MD, et al Friedreich's ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996;271:1423–1427. [DOI] [PubMed] [Google Scholar]
- 125. Gakh O, Adamec J, Gacy AM, Twesten RD, Owen WG, Isaya G. Physical evidence that yeast frataxin is an iron storage protein. Biochemistry 2002;41:6798–6804. [DOI] [PubMed] [Google Scholar]
- 126. Rouault TA, Tong WH. Iron‐sulfur cluster biogenesis and human disease. Trends Genet 2008;24:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sanchez‐Casis G, Cote M, Barbeau A. Pathology of the heart in Friedreich's ataxia: Review of the literature and report of one case. Can J Neurol Sci 1976;3:349–354. [DOI] [PubMed] [Google Scholar]
- 128. Lamarche JB, Cote M, Lemieux B. The cardiomyopathy of Friedreich's ataxia morphological observations in 3 cases. Can J Neurol Sci 1980;7:389–396. [DOI] [PubMed] [Google Scholar]
- 129. Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH. Clinical, biochemical and molecular genetic correlations in Friedreich's ataxia. Hum Mol Genet 2000;9:275–282. [DOI] [PubMed] [Google Scholar]
- 130. Boddaert N, Le Quan Sang KH, Rotig A, et al Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007;110:401–408. [DOI] [PubMed] [Google Scholar]
- 131. Waldvogel D, van Gelderen P, Hallett M. Increased iron in the dentate nucleus of patients with Friedrich's ataxia. Ann Neurol 1999;46:123–125. [DOI] [PubMed] [Google Scholar]
- 132. Sohn YS, Breuer W, Munnich A, Cabantchik ZI. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008;111:1690–1699. [DOI] [PubMed] [Google Scholar]
- 133. Bibudhendra S. Copper transport and its defect in Wilson disease: Characterization of the copper‐binding domain of Wilson disease ATPase. J Inorg Biochem 2000;79:187–191. [DOI] [PubMed] [Google Scholar]
- 134. Gitlin JD. Wilson disease. Gastroenterology 2003;125:1868–1877. [DOI] [PubMed] [Google Scholar]
- 135. Tao TY, Gitlin JD. Hepatic copper metabolism: Insights from genetic disease. Hepatology 2003;37:1241–1247. [DOI] [PubMed] [Google Scholar]
- 136. Mufti AR, Burstein E, Csomos RA, et al XIAP Is a copper binding protein deregulated in Wilson's disease and other copper toxicosis disorders. Mol Cell 2006;21:775–785. [DOI] [PubMed] [Google Scholar]
- 137. Schilsky ML. Wilson disease: Genetic basis of copper toxicity and natural history. Semin Liver Dis 1996;16:83–95. [DOI] [PubMed] [Google Scholar]
- 138. Strickland GT, Frommer D, Leu ML, Pollard R, Sherlock S, Cumings JN. Wilson's disease in the United Kingdom and Taiwan. I. General characteristics of 142 cases and prognosis. II. A genetic analysis of 88 cases. Q J Med 1973;42:619–638. [PubMed] [Google Scholar]
- 139. Das SK, Ray K. Wilson's disease: An update. Nat Clin Pract 2006;2:482–493. [DOI] [PubMed] [Google Scholar]
- 140. Medici V, Rossaro L, Sturniolo GC. Wilson disease—A practical approach to diagnosis, treatment and follow‐up. Dig Liver Dis 2007;39:601–609. [DOI] [PubMed] [Google Scholar]
- 141. Gooneratne SR, Howell JM, Gawthorne JM. An investigation of the effects of intravenous administration of thiomolybdate on copper metabolism in chronic Cu‐poisoned sheep. Br Nutr 1981;46:469–480. [DOI] [PubMed] [Google Scholar]
- 142. Brewer GJ, Dick RD, Johnson VD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson's disease with zinc: XV long‐term follow‐up studies. J Lab Clin Med 1998;132:264–278. [DOI] [PubMed] [Google Scholar]
- 143. Brewer GJ, Hill GM, Prasad AS, Cossack ZT, Rabbani P. Oral zinc therapy for Wilson's disease. Ann Intern Med 1983;99:314–319. [DOI] [PubMed] [Google Scholar]
- 144. Hider RC, Ma Y, Molina‐Holgado F, Gaeta A, Roy S. Iron chelation as a potential therapy for neurodegenerative disease. Biochem Soc Trans 2008;36(Pt 6):1304–1308. [DOI] [PubMed] [Google Scholar]
- 145. Zhang L, Lichtmannegger J, Summer KH, Webb S, Pickering IJ, George GN. Tracing copper‐thiomolybdate complexes in a prospective treatment for Wilson's disease. Biochemistry 2009;48:891–897. [DOI] [PubMed] [Google Scholar]
- 146. Drysdale J, Arosio P, Invernizzi R, et al Mitochondrial ferritin: A new player in iron metabolism. Blood Cells Mol Dis 2002;29:376–383. [DOI] [PubMed] [Google Scholar]
- 147. Levi S, Arosio P. Mitochondrial ferritin. Int J Biochem Cell Biol 2004;36:1887–1889. [DOI] [PubMed] [Google Scholar]