

Background: The mechanisms that regulate cytokine secretion from platelets are undefined.
Results: Inhibiting the expression or function of the Wiskott-Aldrich syndrome protein (WASp) increases TGF-β1 release from platelets.
Conclusion: WASp curtails the secretion of TGF-β1 from human and mouse platelets.
Significance: Identifying the biochemical mechanisms of cytokine secretion will improve our understanding of platelets vis-à-vis the host immune response.
Keywords: Cytokine, Extracellular Matrix, Platelets, Signal Transduction, Signaling
Abstract
Platelets are immunologically competent cells containing cytokines such as TGF-β1 that regulate cell-mediated immunity. However, the mechanisms underlying cytokine secretion from platelets are undefined. The Wiskott-Aldrich syndrome protein (WASp) regulates actin polymerization in nucleated hematopoietic cells but has other role(s) in platelets. WASp-null (WASp−/−) platelets stimulated with a PAR-4 receptor agonist had increased TGF-β1 release compared with WT platelets; inhibiting WASp function with wiskostatin augmented TRAP-induced TGF-β1 release in human platelets. TGF-β1 release is dissociated from α-granule secretion (P-selectin up-regulation) and occurs more gradually, with ∼10–15% released after 30–60 min. Blockade of Src family kinase-mediated WASp Tyr-291/Tyr-293 phosphorylation increased TGF-β1 release, with no additive effect in WASp−/− platelets, signifying that phosphorylation is critical for WASp-limited TGF-β1 secretion. Inhibiting F-actin assembly with cytochalasin D enhanced secretion in WT platelets and further increased TGF-β1 release in WASp−/− platelets, indicating that WASp and actin assembly independently regulate TGF-β1 release. A permeabilized platelet model was used to test the role of upstream small GTPases in TGF-β1 release. N17Cdc42, but not Rac1 mutants, increased TGF-β1 secretion and abrogated WASp phosphorylation. We conclude that WASp function restricts TGF-β1 secretion in a Cdc42- and Src family kinase-dependent manner and independently of actin assembly.
Introduction
Platelets have a well established role in hemostasis, and the mechanisms underlying clot formation and thrombosis have been extensively studied (1). By contrast, much less is known about how platelets influence the host immune response. Platelets contain more than 30 cytokines, chemokines, and growth factors that are secreted upon binding of cell surface receptors with their cognate ligands (2). Platelet α granules are a rich source of TGF-β1 (3), a cytokine pivotal to wound healing, cancer, inflammation, and cell-mediated immunity (4). It is therefore of significant interest to elucidate the signaling mechanisms governing the release of TGF-β1 from platelets.
In platelets, recent evidence indicates that growth factors and cytokines may be organized and packaged into distinct subgroups of α-granules that are selectively targeted for release in response to specific signals (5). For example, Battinelli et al. (5) determined that pro- and anti-angiogenic factors are differentially released from platelets in an agonist-specific manner. In contrast, another group reported that α-granule content is released in a random fashion upon activation and that it is agonist potency that primarily determines the rate and extent of cargo release (6). In either case, the signals that regulate α-granule secretion are likely integrated with the actin cytoskeleton, which is extensively remodeled upon platelet activation (7). In platelets, actin assembly and disassembly are tightly orchestrated by a number of accessory proteins including vinculin (8), cortactin (9), filamin A (10), talin (11), gelsolin (12), cofilin (13), ARP2/3 complex (14), and WAVE (15).
The Wiskott-Aldrich syndrome protein (WASp) is another actin accessory protein that is expressed only in cells of hematopoietic origin (16). In humans, Wiskott-Aldrich syndrome (WAS)2 is characterized by eczema, thrombocytopenia, and severe dysfunction of the immune system (17). WAS-associated immunodeficiency has been traced to mutations of the WASp gene in hematopoietic cells and confirmed in studies using WASp knock-out animals. Defective migration and phagocytosis have been observed in WASp-null neutrophils (18) and macrophages (19), and the directed migration of B-lymphocytes is also WASp-dependent (20). In T-lymphocytes, the formation of critical cell/cell contact points known as immune synapses is contingent on WASp-mediated actin polymerization (21) as is cell polarization and cytokine secretion (22). However, WASp is not required for platelet spreading or its accompanying actin assembly reaction (23), although the hemostatic function in WASp-null mice is reduced because the platelets circulate poorly (24).
In light of the emerging role of platelets as cytokine-secreting immune cells (2), we evaluated the role of WASp in regulating TGF-β1 secretion from platelets. The recognized role of WASp in immune cell function (17), coupled with an overall lack of understanding of WASp function in platelets, highlighted WASp as an ideal protein to study. We report here that WASp-null mice have 4-fold higher circulating levels of TGF-β1 than wild-type controls. WASp normally restricts PAR-mediated secretion of TGF-β1 in a Cdc42- and Src family kinase (SFK)-dependent manner. Finally, WASp mediates platelet TGF-β1 secretion independently of the actin cytoskeleton.
EXPERIMENTAL PROCEDURES
Reagents
The PAR-4 receptor agonist peptide (PAR4-AP) was purchased from Bachem (King of Prussia, PA). The PAR-1 receptor-activating peptide (TRAP), FITC-phalloidin, poly-l-lysine, octyl glucopyranoside (OG), and the anti-β-tubulin antibody were from Sigma. Antibodies against GAPDH, WASp, phospho-WASp, TGF-β1, and GST were obtained from Abcam (Cambridge, MA) and from Millipore (Billerica, MA). The anti-mouse and anti-human P-selectin antibodies were from BD Biosciences (San Jose, CA). The anti-gelsolin antibody was previously described (25). Rhodamine-phalloidin and Alexa-647-phalloidin were from Molecular Probes (Eugene, OR). Cytochalasin D, wiskostatin, PP1, PP2, and the Rac1 inhibitor NSC23766 were obtained from Calbiochem (Billerica, MA). GST-tagged dominant negative (N17) Rac1 and Cdc42 recombinant proteins were purchased from Cytoskeleton (Denver, CO).
Animals
Wild-type, WASp-null (26), and gelsolin-null (27) mice were maintained on a 129Sv background. Knock-out of platelet WASp and gelsolin expression was verified by immunoblotting as previously described (24, 27). Approval for animal research was granted by the Harvard Medical Area Standing Committee, according to National Institutes of Health standards and as outlined in the Institute for Laboratory Animal Research Guide for Care and Use of Laboratory Animals.
Human and Mouse Platelet Preparation
Blood was obtained from healthy human volunteers with institutional review board approval from Brigham and Women's Hospital and informed consent in accordance with the Declaration of Helsinki. Human blood was collected by venipuncture into 1/10th volume of Aster-Jandl anticoagulant buffer (28). Blood was collected from wild-type, WASp-null, and gelsolin-null mice by retro-orbital plexus bleeding. Human and mouse platelets were isolated by sequential centrifugation of whole blood and the resultant platelet-rich plasma as previously described (29). The platelet pellet was washed twice with a washing buffer (140 mm NaCl, 5 mm KCl, 12 mm trisodium citrate, 10 mm glucose, 12.5 mm sucrose, pH 6.0) supplemented with prostaglandin-1 (Sigma). Washed platelets were resuspended in resuspension buffer (10 mm HEPES, 140 mm NaCl, 3 mm KCl, 0.5 mm MgCl2, 5 mm NaHCO3, 10 mm glucose, pH 7.4) containing apyrase (Sigma) and allowed to rest for 30 min at 37 °C prior to use.
Measurement of TGF-β1 Secretion
For quantification of TGF-β1 release, resting platelets were activated with 10 μm TRAP (human) or 500 μm PAR4-AP peptide (mouse). The reactions were stopped at the specified time points by centrifugation at 4 °C, and the ensuing pellets (cell fractions) and supernatants (releasate fractions) were separated. The concentration of TGF-β1 in the supernatants was determined using a Quantikine ELISA kit (R & D Systems, Minneapolis, MN) according to the manufacturer's instructions. The corresponding pellets were solubilized in lysis buffer supplemented with a protease/phosphatase inhibitor mixture (Thermo Fisher, Waltham, MA) and retained for SDS-PAGE and immunoblot analysis.
FITC-Phalloidin Binding Assay and Analysis of Surface P-selectin Expression
To determine platelet F-actin content, resting or activated platelets were fixed and permeabilized using CytoFix/CytoPerm solution (BD Biosciences) and incubated with a 1 μm solution of FITC-phalloidin (Sigma). The amount of bound FITC-phalloidin was then quantified by FACS analysis using a Becton-Dickinson FACSCalibur flow cytometer and CellQuestPro software. A minimum of 10,000 events were analyzed per sample. For determination of surface P-selectin expression, resting or activated platelets were labeled with a FITC-conjugated anti-P-selectin antibody for 30 min prior to FACS analysis.
Immunocytochemistry
To examine the spatial distribution of platelet proteins, glass coverslips were coated with fibrinogen and blocked for 1 h in 1% (w/v) BSA. Platelets were activated with TRAP (human) or PAR4-AP (mouse), plated on to the fibrinogen-coated coverslips, and allowed to spread for the indicated times. Platelets were then fixed in 4% paraformaldehyde, permeabilized with 0.1% (v/v) Triton-X-100, and blocked with 1% (w/v) BSA. Platelets were then incubated with an appropriate primary antibody for 1 h at room temperature, followed by a FITC-conjugated secondary antibody for 1 h, and then stained with rhodamine- or Alexa-647-phalloidin. For double immunofluorescence experiments, platelets were blocked a second time prior to incubating with a second primary antibody for 1 h at room temperature, followed by a TRITC-conjugated secondary antibody for 1 h at room temperature. In some experiments, resting platelets were centrifuged onto poly-l-lysine-coated coverslips, fixed immediately with 4% paraformaldehyde, and processed as described above. Confocal images were captured and processed using a Zeiss spinning disk confocal microscope and SlideBook software (Intelligent Imaging Innovations, Denver, CO). To facilitate micrograph interpretation, confocal images supplied as grayscale by the SlideBook program were colorized in green, red, or blue using the color levels function in Adobe Photoshop.
Purification of Recombinant Proteins
Vectors encoding GST-CA (amino acids 450–505 of N-WASp) and GST-VCA (amino acids 392–505 of N-WASp) constructs were previously described (14). The constitutively active GST-Rac1V12 and GST-Cdc42V12 constructs were also previously described (30). GST proteins were expressed in Escherichia coli and purified on glutathione-agarose beads (Amersham Biosciences). Protein purity was verified by SDS-PAGE and Coomassie Blue staining of the PAGE gels.
OG Permeabilization of Human Platelets
Permeabilization of human platelets using OG was performed essentially as described (14, 30, 31). Briefly, platelets were isolated from human platelet-rich plasma by gel filtration through a Sepharose 2B column (Amersham Biosciences). Platelet concentration was adjusted to 2 × 108 platelets/ml in platelet buffer (145 mm NaCl, 10 mm HEPES, 0.6 mm NaH2PO4, 2.5 mm KCl, 4 mm MgCl2, 10 mm glucose, 0.3% BSA, pH 7.4). Cells were permeabilized with 0.25% OG in PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA, 0.2 mm MgCl2, pH 7.0) and incubated with the GST-tagged recombinant proteins described above. For secretion studies, platelets reconstituted with the recombinant GST-tagged proteins were activated with TRAP (10 μm), and the TGF-β1 content of the corresponding releasates was analyzed by ELISA as described above.
Statistical Analysis
For time course experiments, a two-way analysis of variance and Bonferroni post hoc multiple comparison tests were employed to determine the effects of 1) genotype/treatment and 2) time on the secretion of platelet TGF-β1. Statistical significance was set at p < 0.05.
RESULTS
WASp Restricts PAR4-AP-mediated Secretion of TGF-β1 by Mouse Platelets
The levels of TGF-β1 in the platelet-poor plasma (following platelet isolation) were 4-fold higher in WASp−/− mice (43.12 ± 3.48 ng/ml, n = 4) relative to WT controls (10.61 ± 1.86 ng/ml, n = 4). Because platelet α-granules are the primary source of TGF-β1 in blood (3), we determined the kinetics of TGF-β1 release (Fig. 1A) and compared it with the surface up-regulation of P-selectin in activated mouse platelets (Fig. 1B). To restrict the signaling pathway to the mouse PAR-4 receptor, we employed its peptide agonist (PAR4-AP) (32). The release of TGF-β1 by PAR-4 ligation was gradual, because only ∼10–15% of the total platelet TGF-β1 content was secreted within 30–60 min of PAR4 stimulation (Fig. 1A). By contrast, 60% of total P-selectin translocated to the cell surface within 1 min of PAR4 ligation, as expected (Fig. 1B). These distinct release patterns are consistent with the separate distribution of P-selectin and TGF-β1 in human platelets (Fig. 2H). To evaluate the role of WASp in platelet secretion of TGF-β1, we studied wild-type and WASp−/− mice. Because only a fraction of the total TGF-β1 is released by PAR4 ligation, we were able to monitor the spatial distribution of TGF-β1-containing granules following platelet activation and spreading on fibrinogen. In both WT and WASp−/− platelets, TGF-β1-containing granules were evenly dispersed throughout the cytoplasm at rest (Fig. 1, C and D). In activated WASp−/− platelets, TGF-β1-containing granules centralized prematurely relative to wild-type platelets (Fig. 1, C and D), which is indicative of accelerated granule fusion into the open canalicular system (33). We quantified this observation in two ways: 1) by measuring the centralized/aggregated granule area relative to total cell area (Fig. 1E) and 2) by determining the percentage of cells exhibiting granule centralization (Fig. 1F). Both approaches showed that WASp−/− platelets had a significantly (p < 0.05) higher degree of granule centralization relative to controls. We therefore hypothesized that WASp may influence the PAR4-mediated secretion of TGF-β1 by platelets. Accordingly, the amount of TGF-β1 released from activated WASp−/− platelets was 2–5-fold higher than that from wild-type platelets (Fig. 1A), indicating that WASp curtails platelet secretion of TGF-β1. Statistical analysis showed that both genotype and time had significant (p < 0.0001) effects on the secretion of platelet TGF-β1; individual differences were evaluated by Bonferroni multiple comparison tests (Fig. 1A). The surface expression of P-selectin, a classical marker for α-granule secretion (34), was identical (p > 0.05) in wild-type and WASp−/− mouse platelets following PAR4-AP stimulation (Fig. 1B). Collectively, these data indicate that WASp constrains platelet secretion of TGF-β1 and that cytokine secretion can be altered independently of surface P-selectin expression.
FIGURE 1.

WASp restricts PAR-4 receptor-mediated secretion of TGF-β1 in mouse platelets. A, time course for PAR4-AP-mediated release of TGF-β1 from wild-type (white bars) and WASp−/− (black bars) mouse platelets. The data are expressed as means ± S.E. and represent three independent experiments. *, p < 0.01; **, p < 0.001, based on Bonferroni multiple comparison tests. B, time course of surface P-selectin expression by flow cytometry in wild-type (white bars) and WASp−/− (black bars) mouse platelets stimulated by PAR4-AP. The data are expressed as means ± S.E. and represent three independent experiments. C, confocal micrographs showing the distribution of TGF-β1 in wild-type mouse platelets at rest (far left panel) and after stimulation with 500 μm PAR4-AP and spreading on fibrinogen for 2 min (center left panel), 5 min (center right panel), and 15 min (far right panel). Bar, 10 μm; n = 3. D, distribution of TGF-β1 in WASp−/− platelets at rest (far left panel) and after stimulation with 500 μm PAR4-AP and spreading on fibrinogen for 2 min (center left panel), 5 min (center right panel), and 15 min (far right panel). Bars, 10 μm; n = 3. TGF-β1-containing granules are concentrated at the center more rapidly in WASp−/− platelets (arrows) than in WT platelets. E, time course for the centralization of TGF-β1-containing granules, relative to total cell area (%) in wild-type (white bars) and WASp−/− (black bars) platelets. The data are expressed as means ± S.E. and represent three independent experiments. *, p < 0.01, based on Bonferroni multiple comparison tests. F, quantification of the proportion of wild-type (white bars) and WASp−/− (black bars) platelets showing centralized TGF-β1-containing granules. The data are expressed as means ± S.E. and represent three independent experiments. *, p < 0.01, based on Bonferroni multiple comparison tests.
FIGURE 2.

WASp restricts PAR-1 receptor-mediated TGF-β1 secretion in human platelets. A, time course of TRAP-mediated release of TGF-β1 by human platelets treated with vehicle (white bars) or the WASp inhibitor wiskostatin (black bars). The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors. *, p < 0.05, based on Bonferroni multiple comparison tests. B, time course of surface P-selectin expression in TRAP-stimulated human platelets treated with either vehicle (white bars) or wiskostatin (black bars). The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors. C, representative confocal micrographs comparing the distribution of WASp (left-hand panel, green) and F-actin (center panels, red) in resting human platelets. WASp is concentrated at the plasma membrane (left-hand panel). Bar, 5 μm; n = 3. D, representative confocal micrographs comparing the distribution of WASp (left-hand panel, green) and F-actin (center panel, red) in human platelets, stimulated with TRAP (10 μm) and spread for 15 min on fibrinogen. WASp is located at the platelet center (right-hand panel, arrow and dotted circle, depicted at higher magnification in bottom right-hand panel). Bar, 10 μm; n = 3. Bottom right panel, high magnification of selected area (dotted circle) from the top right-hand panel, illustrating the centralization of WASp (green) relative to peripheral actin (red). Bar, 4 μm. E, quantification of the proportion of platelets showing centralized P-selectin (white bars), TGF-β1 (gray bars), and WASp (black bars). The data are expressed as means ± S.E. and represent three independent experiments. *, significantly (p < 0.001) different from TGF-β1 and WASp, based on Bonferroni multiple comparison tests. F, representative confocal micrographs comparing the distribution of WASp (left-hand panel, green) and F-actin (center panel, red) in human platelets treated with wiskostatin, stimulated with 10 μm TRAP, and spread for 15 min on fibrinogen. WASp remains near the platelet edge (right-hand panel, arrow). Bar, 10 μm; n = 3. G, spatial distribution of TGF-β1 (left-hand panel, green) and WASp (center panel, red) in human platelets stimulated by 10 μm TRAP and spread on fibrinogen for 15 min. Yellow indicates co-localization (right-hand panel). Bar, 10 μm; n = 3. H, representative confocal micrographs illustrate the separate distributions of P-selectin (left panel, green) and TGF-β1 (center panel, red) in resting human platelets. Bar, 5 μm; n = 3.
WASp Restricts TRAP-mediated Secretion of TGF-β1 by Human Platelets
To test the hypothesis that WASp function regulates PAR1-mediated secretion of TGF-β1 in human platelets, we assessed TRAP-induced TGF-β1 release in the presence or absence of wiskostatin, a chemical inhibitor with a documented inhibitory effect on WASp function (19). Consistent with our observations on WASp−/− mouse platelets, there was a significant (p < 0.05) increase in TGF-β1 release from wiskostatin-treated human platelets compared with vehicle-treated controls (Fig. 2A). In contrast, TRAP-induced release of platelet factor 4, another α-granule cytokine, was unaffected by wiskostatin (data not shown). Also consistent with the WASp−/− mouse platelets, the surface expression of P-selectin in human platelets was unaffected (p > 0.05) by wiskostatin treatment (Fig. 2B). The specificity of wiskostatin for WASp is suggested by the finding that wiskostatin treatment had no significant effect (p > 0.05) on TGF-β1 release from WASp−/− platelets (data not shown). WASp was primarily localized at the plasma membrane in resting platelets (Fig. 2C) but progressively aggregated at the platelet center after 15 min of platelet spreading on immobilized fibrinogen (Fig. 2, D and E). Wiskostatin treatment, however, impeded the centralization of WASp in TRAP-stimulated platelets (Fig. 2F). Notably, TGF-β1-positive granules uniformly co-localized with WASp in the activated platelet (Fig. 2G). Taken together, these data suggest that wiskostatin prevents the targeting of TGF-β1-containing granules by WASp. These data also indicate that WASp constrains TRAP-mediated TGF-β1 secretion in human platelets independently of surface P-selectin expression.
WASp and the Actin Cytoskeleton Regulate Platelet TGF-β1 Secretion Independently
WASp regulates actin polymerization in neutrophils, macrophages, and lymphocytes (17) but is not required for platelet spreading (23), a process that remodels and doubles cytoskeletal F-actin (7, 31). We therefore set out to dissect the individual and collective contributions of WASp and actin assembly to TGF-β1 secretion. To determine whether WASp affects PAR4-AP-mediated actin assembly, we used a FITC-phalloidin binding assay (35) to measure F-actin content in wild-type and WASp−/− platelets at rest and after stimulation with PAR4-AP. Consistent with previous reports, the actin assembly response of wild-type and WASp−/− mouse platelets was identical (p > 0.05) after PAR4-AP stimulation (Fig. 3A). There was also no significant difference (p > 0.05) in TRAP-induced actin assembly in vehicle- or wiskostatin-treated human platelets (Fig. 3B). An inhibition of actin assembly with cytochalasin D significantly (p < 0.05) increased the amount of TGF-β1 in the releasate of activated human and mouse platelets (Fig. 3, C and D), consistent with previous reports (36). Importantly, the combined stimulatory effects of cytochalasin D treatment and WASp−/− were additive (p < 0.05) on TGF-β1 secretion (Fig. 3D), indicating that WASp and the actin assembly can function independently in regulating TGF-β1 release. Moreover, both TGF-β1 and WASp showed little co-localization with F-actin in TRAP-activated human platelets spread on fibrinogen (Fig. 3, E and F).
FIGURE 3.

TGF-β1 secretion by mouse and human platelets is regulated independently by WASp and the actin cytoskeleton. A, PAR4-AP-induced actin assembly by wild-type (white bars) or WASp−/− (black bars) mouse platelets as determined by FITC-phalloidin binding. The data are expressed as means ± S.E. and represent three independent experiments. NS, difference not statistically significant (p > 0.05). B, TRAP-induced actin assembly by human platelets treated with vehicle (white bars) or wiskostatin (black bars) as determined by FITC-phalloidin binding. The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors. NS, difference not statistically significant (p > 0.05). C, TRAP-mediated release of TGF-β1 by human platelets treated with either vehicle (white bar) or 10 μm cytochalasin D (black bar). The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors. *, p < 0.05. D, PAR4-AP-mediated release of TGF-β1 by wild-type (white bars) or WASp−/− (black bars) mouse platelets pretreated with either vehicle or 10 μm cytochalasin D. The data are expressed as means ± S.E. and represent three independent experiments. *, significantly (p < 0.05) different from vehicle-treated wild-type platelets; **, significantly (p < 0.05) different from vehicle-treated WASp−/− platelets or cytochalasin-treated wild-type platelets. E, comparison of the spatial distribution of TGF-β1 (left-hand panels, green) and F-actin (center panels, red) in human platelets stimulated by TRAP (10 μm) and spread on fibrinogen for 15 min. Bar, 10 μm; n = 3. F, comparison of the distribution of WASp (left-hand panels, green) and F-actin (center panels, red) in human platelets stimulated by TRAP (10 μm) and spread on fibrinogen for 15 min. Bar, 10 μm; n = 3. G, effect of cytochalasin D or wiskostatin on PAR4-AP-mediated release of TGF-β1 by wild-type (white bars) or gelsolin-null (black bars) mouse platelets. The data are expressed as means ± S.E. and represent three independent experiments. *, significantly (p < 0.05) different from vehicle-treated wild-type platelets; **, significantly (p < 0.05) different from wild-type platelets (treated with either cytochalasin D or wiskostatin) or Gsn−/− platelets (treated either with vehicle or cytochalasin D). Cyto D, cytochalasin D.
As a complementary approach to dissecting the individual and collective roles of WASp and actin in the regulation of TGF-β1 secretion, we used mice deficient in gelsolin, a protein that promotes cytoskeletal reorganization and actin polymerization by severing actin filaments and generating additional barbed ends (12). PAR4-AP induced significantly (p < 0.05) more TGF-β1 release from gelsolin-null than from WT platelets (Fig. 3G). However, in the gelsolin-null platelets, cytochalasin D treatment did not amplify secretion (p > 0.05) (Fig. 3G). By contrast, wiskostatin treatment induced an additional 2-fold increase in TGF-β1 secretion in gelsolin-null platelets (Fig. 3G). Based on these data, it can be inferred that although both WASp and the actin cytoskeleton combine to restrict TGF-β1 release from platelets, their mechanisms of action are independent.
Platelet Secretion of TGF-β1 Is Regulated by Tyrosine Phosphorylation of WASp
WASp Tyr-291/Tyr-293 phosphorylation by SFK, a prerequisite for WASp function (37), is known to occur rapidly following human platelet activation (38–40). Accordingly, WASp in both human and mouse platelets was tyrosine-phosphorylated following activation with TRAP or PAR4-AP, respectively (Fig. 4, A and B). The phosphorylated form of WASp preferentially localized with TGF-β1 in both human and mouse platelets (Fig. 4, D and F). To test the significance of WASp phosphorylation in the context of TGF-β1 secretion, we abrogated activation-induced WASp phosphorylation in human and mouse platelets using PP1 and PP2 (41), two cell-permeant inhibitors of Src family kinases (Fig. 4, E and G). Relative to vehicle-treated controls, human platelets pretreated with PP1 and PP2 released more TGF-β1 after TRAP stimulation (Fig. 4H). Similarly, mouse platelets pretreated with PP1 and PP2 exhibited greater PAR4-AP-induced release of TGF-β1 relative to vehicle-treated controls (Fig. 4I). Notably, neither PP1 nor PP2 significantly affected (p > 0.05) TGF-β1 release from WASp-null platelets (Fig. 4I), indicating that the negative influence of WASp on TGF-β1 secretion is regulated through SFK-mediated tyrosine phosphorylation.
FIGURE 4.

WASp-mediated platelet secretion of TGF-β1 is regulated by tyrosine phosphorylation. A and B, tyrosine phosphorylation of WASp (Tyr-291 or Tyr-293) in activated human platelets activated by 10 μm TRAP (A) or mouse platelets activated by 500 μm PAR4-AP (B). Total WASp is shown as a loading control (n = 3). C, representative confocal micrographs comparing the distribution of Tyr-291 phospho-WASp (left-hand panel, green) to total WASp (center panel, red) in human platelets stimulated by TRAP (10 μm) and spread on fibrinogen for 15 min. Yellow indicates co-localization (right-hand panel). Bar, 10 μm; n = 3. D, co-localization of TGF-β1 (left-hand panel, green) and Tyr-291 phospho-WASp (center panel, red) in human platelets stimulated by TRAP (10 μm) and spread on fibrinogen for 15 min. Yellow indicates co-localization (right-hand panel). Bar, 10 μm; n = 3. E, the Src family kinase inhibitors PP1 and PP2 abrogate WASp phosphorylation in TRAP (10 μm)-stimulated human platelets. Total WASp is shown as a loading control (n = 3). F, co-localization of TGF-β1 (left-hand panel, green) and Tyr-293 phospho-WASp (center panel, red) in mouse platelets stimulated by PAR4-AP (500 μm) and spread on fibrinogen for 15 min. Yellow indicates co-localization (right-hand panel). Bar, 10 μm; n = 3. G, the Src family kinase inhibitors PP1 and PP2 abrogate WASp phosphorylation in PAR4-AP (500 μm)-stimulated mouse platelets. Total WASp is shown as a loading control (n = 3). H, TRAP-mediated release of TGF-β1 in human platelets treated with either vehicle, PP1, or PP2. The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors (*, p < 0.05). I, PAR4-AP-mediated release of TGF-β1 by wild-type (white bars) or WASp−/− (black bars) mouse platelets treated with either vehicle, PP1, or PP2. The data are expressed as means ± S.E. and represent three independent experiments. *, significantly (p < 0.05) different from vehicle-treated wild-type platelets. IB, immunoblot.
WASp-dependent TGF-β1 Secretion Is Regulated by Cdc42 but Not by Rac1
The Rho GTPases Rac1 and Cdc42 are activated in platelets (42) and have been shown to interact with WASp (17). To determine whether Rac1 contributes to WASp-mediated TGF-β1 secretion, we used the cell-permeant Rac1 inhibitor (NSC23766). Treatment of human or mouse platelets with NSC23766 had no effect on agonist-induced WASp phosphorylation (Fig. 5, A and B) but decreased TGF-β1 secretion by ∼25–30% in these platelets (Fig. 5, C and D). NSC23766 affected wild-type and WASp−/− mouse platelets equally, although the effect of NSC23766 was not statistically significant (p > 0.05) in mouse platelets (Fig. 5D). Taken together, these data indicate that Rac1 activity does influence platelet TGF-β1 secretion, but independently of WASp.
FIGURE 5.

Cdc42 influences WASp phosphorylation and platelet secretion of TGF-β1. A and B, tyrosine phosphorylation of WASp (Tyr-291 or Tyr-293) in human platelets activated by 10 μm TRAP (A) or mouse platelets activated by 500 μm PAR4-AP (B) in the presence or absence of the Rac1 inhibitor NSC23766. Total WASp is shown as a loading control. C, TRAP-mediated release of TGF-β1 by human platelets in the absence (white bar) or presence (black bar) of NSC23766. The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors (*, p < 0.05). D, PAR4-AP-mediated release of TGF-β1 by wild-type (white bars) or WASp−/− (black bars) mouse platelets treated with or without NSC23766. The data are expressed as means ± S.E. and represent three independent experiments. *, significantly (p < 0.05) different from vehicle-treated wild-type platelets. E, TRAP stimulation of OG-permeabilized human platelets increases P-selectin exposure (i.e., causes secretion). F, incorporation of recombinant GST protein into OG-permeabilized human platelets. GST in the OG-treated platelets was stained with a FITC-conjugated anti-GST antibody. Successful reconstitution is indicated by increased fluorescence. G, confocal micrographs reveal successful reconstitution of OG-permeabilized human platelets with GST protein (left panel, GST, green; center panel, F-actin, blue; right panel, overlay). Bar, 10 μm. H, TRAP (10 μm)-mediated tyrosine phosphorylation of Tyr-291 on WASp in OG-permeabilized human platelets reconstituted with: GST, the CA and VCA domains of N-WASp, and the constitutively active (V12) and dominant negative (N17) forms of Rac1 and Cdc42. Total WASp is shown as a loading control (n = 3). I, TRAP-mediated release of TGF-β1 from OG-permeabilized human platelets reconstituted with GST, the CA domain of N-WASp (CA), the VCA domains of N-WASp (VCA), constitutively active Rac1 (V12 Rac1), dominant negative Rac1 (N17 Rac1), constitutively active Cdc42 (V12 Cdc42), and dominant negative Cdc42 (N17 Cdc42). The data are expressed as means ± S.E. and represent three independent experiments using platelets from different blood donors. *, significantly (p < 0.05) different from OG-permeabilized platelets reconstituted with GST. IB, immunoblot.
To further investigate GTPase function, active and dominant negative Rac1 and Cdc42 mutant proteins were reconstituted into secretion-capable, permeabilized platelets. Permeabilization is a well established approach for reconstituting platelets with recombinant proteins (14, 30, 31), particularly for secretion assays (43). We chose OG as the permeabilizing agent because OG-permeabilized platelets retain their sensitivity to TRAP stimulation (30, 31), as confirmed by increased surface P-selectin expression (Fig. 5E). We then introduced GST-tagged recombinant proteins into the permeabilized platelets and analyzed the resultant effects on TRAP-mediated TGF-β1 secretion. To verify successful introduction of the recombinant proteins, platelets were stained with a FITC-conjugated anti-GST antibody; successful reconstitution was indicated by increased fluorescence (Fig. 5F); this was also verified by confocal microscopy (Fig. 5G). Although the constitutively active Rac1 mutant had no appreciable effect on TGF-β1 release, the dominant negative (N17) Rac1 mutant reduced TGF-β1 release by ∼50% (Fig. 5I), consistent with data obtained using the Rac1 inhibitor NSC23766 (Fig. 5C). Neither Rac1 mutant affected TRAP-induced WASp phosphorylation. By contrast, the constitutively active V12Cdc42 protein caused a decrease in TGF-β1 secretion (Fig. 5I). Strikingly, the dominant negative N17Cdc42 mutant completely abrogated WASp phosphorylation (Fig. 5H) and caused a 2-fold increase in TGF-β1 secretion (Fig. 5I). Collectively, these data show that Cdc42, not Rac1, promotes WASp phosphorylation and regulates WASp-mediated secretion of TGF-β1 from platelets.
The CA and VCA domains of N-WASp were used to manipulate TRAP-induced Arp2/3 activation and actin assembly (31). Platelets reconstituted with the Arp2/3 stimulatory VCA domain secreted ∼60% of the TGF-β1 compared with controls (Fig. 5I), consistent with the notion that actin polymerization impedes TGF-β1 secretion (Fig. 3C). Neither the CA or the VCA domain of N-WASp affected TRAP-induced WASp phosphorylation (Fig. 5H).
DISCUSSION
Platelets house pro- and anti-inflammatory cytokines and chemokines that contribute to the host immune response (2), but the mechanisms regulating their release are undefined. In the present study, we demonstrate that WASp is a key modulator of TGF-β1 secretion by platelets and that WASp regulates this process in a Cdc42- and SFK-dependent manner. Because WASp is a nonessential regulator of the actin cytoskeleton in platelets, we propose that its main role is to control the delivery rate of cytokines like TGF-β1 following platelet activation.
This conclusion is based on the finding that TGF-β1 secretion is increased significantly in platelets lacking WASp or after treating platelets with chemical inhibitors of WASp. Platelets are an abundant source of TGF-β1, which releases in a latent form bound to the latency-associated peptide following activation in blood clots (44). Because TGF-β1 levels in blood are increased in the WASp KO mice, it is likely that platelets are an abundant source of TGF-β1 and that WASp KO platelets prematurely release their TGF-β1 as they circulate in blood. Release of TGF-β1 occurs at a leisurely pace relative to the bulk of P-selectin up-regulation to the platelet surface, a classic marker for α-granule secretion (34) that was not greatly affected by the loss of WASp expression or function (24). There are two possible explanations for these distinct release patterns. Based on the model proposed by Jonnalagadda et al. (6), the PAR/WASp signaling pathway may uniformly regulate the secretion of all α-granules; the differing release kinetics of P-selectin and TGF-β1 would be due to differences in their relative solubility and not the result of differential granule packaging or agonist specificity. Alternatively, TGF-β1 may be concentrated and packaged into a compartment distinct from the bulk of the α-granules that is specifically targeted by WASp. Localization of WASp and TGF-β1 together in the activated platelet is consistent with this notion that WASp targets specific granules, impeding their centralization and subsequent fusion with the OCS. This hypothesis is supported by the enhanced centralization of TGF-β1-containing granules in WASp-null platelets. In either case, whereas the precise mechanism underlying the release of α-granular content remains unclear, WASp appears to act as a gatekeeper for the delivery of TGF-β1. Consequently, studying the release patterns of specific cytokines may represent a more elegant and informative approach to studying platelet secretion than by examining levels of surface P-selectin alone.
We also found that TGF-β1 secretion is increased by the actin polymerization toxin, cytochalasin D, in agreement with published observations on the secretions from α- and dense granules (33, 36). TGF-β1 secretion was also increased in gelsolin-null platelets. In activated platelets, gelsolin promotes actin rearrangements and polymerization by severing actin filaments, thus increasing the availability of short filaments for Arp2/3 amplification (12, 14). Consequently, gelsolin-null platelets are profoundly deficient in their ability to mount an agonist-stimulated actin assembly reaction (12, 35). Similarly, TGF-β1 secretion was markedly inhibited in platelets reconstituted with the VCA domain of N-WASp, which would promote barbed end generation and actin polymerization (31), data suggesting that F-actin networks pose a barrier to cytokine secretion. Because the effects of cytochalasin D and WASp deletion were additive, as was the effect of wiskostatin on gelsolin-null platelets, it would appear that actin and WASp regulate TGF-β1 secretion by independent but additive pathways.
We found that both mouse and human WASp phosphorylation increases after ligation of the PAR receptors, consistent with published observations (38–40). In its inactive form, WASp is in a closed, autoinhibited conformation (17, 45). Phosphorylation of tyrosine 291 (human) or tyrosine 293 (mouse) is a key regulatory step for opening of the WASp molecule and its subsequent activation (17, 37, 45). In our study, the Src family kinase inhibitors PP1 and PP2 prevented WASp phosphorylation and increased TGF-β1 secretion to the same extent as that found in the WASp-null platelets. Because phospho-WASp co-localized with TGF-β1-containing granules, we conclude that Src family kinases mediate their effects on platelet secretion via WASp phosphorylation. Conceivably, other signaling pathways that promote WASp phosphorylation, e.g., the glycoprotein VI (GPVI) pathway (38), may contribute to TGF-β1 secretion in a similar manner. This premise is supported, for example, by our finding that GPVI-mediated TGF-β1 release is elevated in WASp-inactivated human platelets via a pattern analogous to that of PAR-mediated TGF-β1 release (data not shown).
The Rho GTPases, Rac1 and Cdc42, are putative WASp binding partners (17, 46) that are rapidly GTP-charged after thrombin stimulation of platelets (30, 42). Both GTPases can modulate platelet secretion (47–49), and several reports have described impaired secretion in Rac1-null platelets (49) or in NSC23766-treated platelets (48). In our study, TGF-β1 secretion diminished when platelets were treated with the Rac1 inhibitor NSC23766, as was cytokine release from OG-permeabilized platelets reconstituted with N17Rac1. Importantly, WASp phosphorylation was unaffected by manipulation of Rac1 signaling, in agreement with a report from Tomasevic et al. (46), who showed that Rac1 has minimal WASp modulating activity. Collectively, these data allow us to conclude that WASp and Rac1 independently regulate platelet secretion and exert opposing effects. By contrast, OG-permeabilized platelets reconstituted with dominant negative N17Cdc42 had increased cytokine secretion but no WASp phosphorylation following PAR1 ligation, indicating that Cdc42 is the upstream effector for platelet WASp. Pleines et al. (47) recently reported increased α- and dense-granule secretion from thrombin-stimulated Cdc42-null platelets. These observations fit with the concept that binding of Cdc42 to WASp is required for WASp phosphorylation and activation (19, 37, 45). Because WASp is the downstream target of Cdc42, WASp-deficient platelets should exhibit increased secretion. We therefore suggest a model in which ligation of the PAR receptor on platelets recruits WASp as well as leads to the GTP charging of Cdc42. One salient feature of our model is that phospho-WASp targets TGF-β1-containing granules (Fig. 6), inhibiting their centralization and fusion with the OCS.
FIGURE 6.
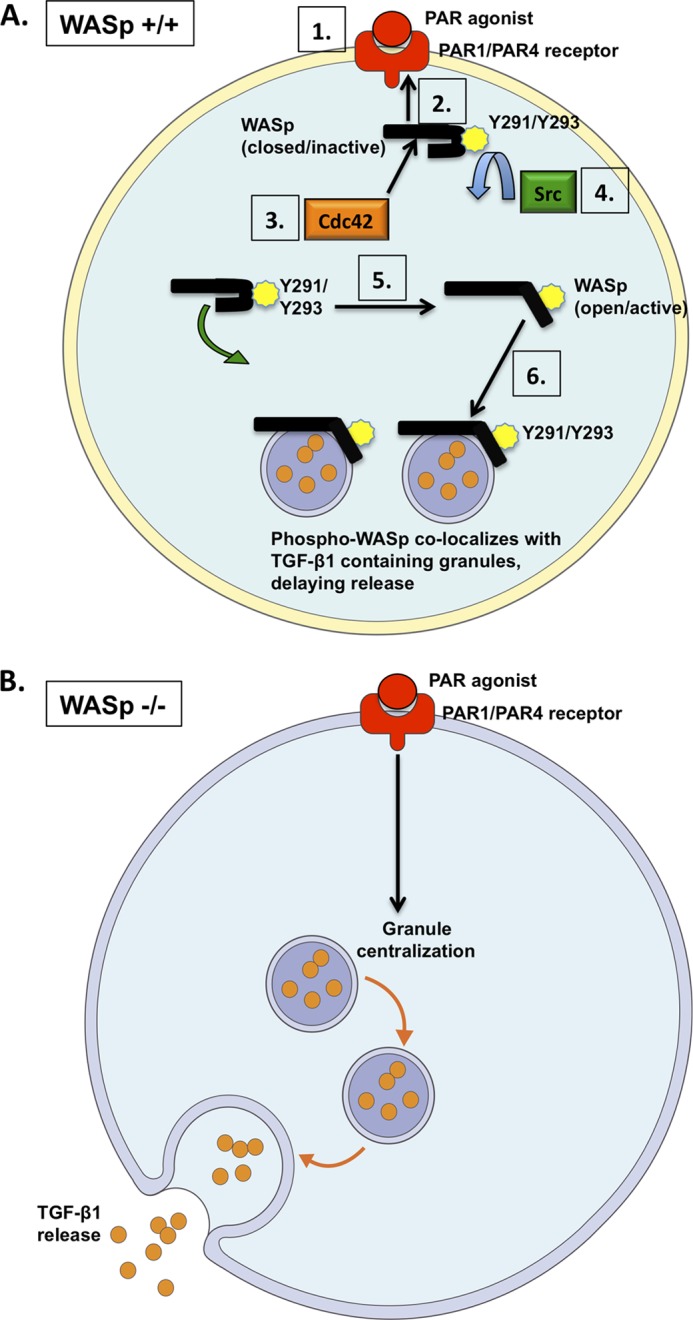
Model for WASp-mediated release of TGF-β1 in platelets. A, WASp is recruited to the PAR1/PAR4 receptor following agonist stimulation (steps 1 and 2). WASp is depicted in its closed, inactive conformation. Binding of Cdc42 to WASp (step 3) promotes the phosphorylation of WASp at Tyr-291/Tyr-293, which is mediated by Src family kinases (step 4). Phosphorylation transforms WASp into its open, active conformation (step 5). Phospho-WASp interacts with TGF-β1-containing α granules to control their spatial and temporal release (step 6). B, in the absence of WASp (or following WASp inactivation), TGF-β1-containing granules are freely trafficked within the platelet cytosol (marked by premature granule centralization and increased cytokine release).
TGF-β1 is a potent cytokine with significant effects on T-lymphocyte-mediated immunity (4). Lymphopenia and defective T-cell function are important features of WAS (50), and TGF-β1 is known to inhibit T-cell proliferation (4). This encourages speculation that increased TGF-β1 secretion from WASp-null platelets may contribute to the increased plasma levels of TGF-β1 and partly explains the atypical T-cell behavior observed in WAS, especially because platelet-derived cytokines directly modulate T-cell function (51). Paradoxically, WAS patients also suffer from autoimmune reactions (52) that are ascribed to dysfunctional regulatory T-lymphocytes, or Tregs (53). Tregs are a subset of T-cells that suppress autoimmune reactions via a phenomenon known as T-cell tolerance (4). The induction of Tregs from their precursor cells is TGF-β1-dependent, and Treg dysfunction may result from disrupted TGF-β1 signaling (4). Conceivably, an increase in TGF-β1 secretion by WASp-null platelets may represent a compensatory reaction to this deficiency.
Future studies may identify a specific function for WASp in mediating signals among different immune cells, while providing additional insight into the role of platelets in the host response to pathogens and contributing to a better understanding of WAS. In conclusion, our results identify WASp as an important regulator of TGF-β1 secretion downstream of Cdc42 and SFKs, corroborating the hypothesis that WASp has a specialized role in platelets, independent of actin assembly.
Acknowledgments
We thank Fumi Nakamura for assistance with protein purification and Scott Snapper for providing the DNA primers used for WASp genotyping.
This work was supported, in whole or in part, by National Institutes of Health Grants HL059561-11 (to J. H. H. and H. F.) and HL107146 (to K. M. H.). This work was also supported by a Canadian Institutes of Health Research Clinician-Scientist Award (to H. K.).
- WAS
- Wiskott-Aldrich syndrome
- SFK
- Src family kinase
- PAR4-AP
- PAR-4 receptor agonist peptide
- TRAP
- PAR-1 receptor-activating peptide
- OG
- octyl glucopyranoside
- FITC
- fluorescein isothiocyanate
- TRITC
- tetramethylrhodamine isothiocyanate
- CA
- central acidic domain
- VCA
- verprolin homology central acidic domain.
REFERENCES
- 1. Stegner D., Nieswandt B. (2011) Platelet receptor signaling in thrombus formation. J. Mol. Med. 89, 109–121 [DOI] [PubMed] [Google Scholar]
- 2. Semple J. W., Italiano J. E., Jr., Freedman J. (2011) Platelets and the immune continuum. Nat. Rev. Immunol. 11, 264–274 [DOI] [PubMed] [Google Scholar]
- 3. Assoian R. K., Komoriya A., Meyers C. A., Miller D. M., Sporn M. B. (1983) Transforming growth factor-β in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 258, 7155–7160 [PubMed] [Google Scholar]
- 4. Li M. O., Wan Y. Y., Sanjabi S., Robertson A. K., Flavell R. A. (2006) Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 24, 99–146 [DOI] [PubMed] [Google Scholar]
- 5. Battinelli E. M., Markens B. A., Italiano J. E., Jr. (2011) Release of angiogenesis regulatory proteins from platelet α granules. Modulation of physiologic and pathologic angiogenesis. Blood 118, 1359–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jonnalagadda D., Izu L. T., Whiteheart S. W. (2012) Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood 120, 5209–5216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hartwig J. H., Barkalow K., Azim A., Italiano J. (1999) The elegant platelet. Signals controlling actin assembly. Thromb. Haemost. 82, 392–398 [PubMed] [Google Scholar]
- 8. Vostal J. G., Shulman N. R. (1993) Vinculin is a major platelet protein that undergoes Ca2+-dependent tyrosine phosphorylation. Biochem. J. 294, 675–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vidal C., Geny B., Melle J., Jandrot-Perrus M., Fontenay-Roupie M. (2002) Cdc42/Rac1-dependent activation of the p21-activated kinase (PAK) regulates human platelet lamellipodia spreading. Implication of the cortical-actin binding protein cortactin. Blood 100, 4462–4469 [DOI] [PubMed] [Google Scholar]
- 10. Feng S., Reséndiz J. C., Lu X., Kroll M. H. (2003) Filamin A binding to the cytoplasmic tail of glycoprotein Ibα regulates von Willebrand factor-induced platelet activation. Blood 102, 2122–2129 [DOI] [PubMed] [Google Scholar]
- 11. Petrich B. G., Fogelstrand P., Partridge A. W., Yousefi N., Ablooglu A. J., Shattil S. J., Ginsberg M. H. (2007) The antithrombotic potential of selective blockade of talin-dependent integrin αIIbβ3 (platelet GPIIb-IIIa) activation. J. Clin. Invest. 117, 2250–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barkalow K., Witke W., Kwiatkowski D. J., Hartwig J. H. (1996) Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. J. Cell Biol. 134, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Falet H., Chang G., Brohard-Bohn B., Rendu F., Hartwig J. H. (2005) Integrin αIIbβ3 signals lead cofilin to accelerate platelet actin dynamics. Am. J. Physiol. Cell Physiol. 289, C819–C825 [DOI] [PubMed] [Google Scholar]
- 14. Falet H., Hoffmeister K. M., Neujahr R., Italiano J. E., Jr., Stossel T. P., Southwick F. S., Hartwig J. H. (2002) Importance of free actin filament barbed ends for Arp2/3 complex function in platelets and fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 99, 16782–16787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Calaminus S. D., McCarty O. J., Auger J. M., Pearce A. C., Insall R. H., Watson S. P., Machesky L. M. (2007) A major role for Scar/WAVE-1 downstream of GPVI in platelets. J. Thromb. Haemost. 5, 535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Derry J. M., Ochs H. D., Francke U. (1994) Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell 79, 922. [PubMed] [Google Scholar]
- 17. Thrasher A. J., Burns S. O. (2010) WASP. A key immunological multitasker. Nat. Rev. Immunol. 10, 182–192 [DOI] [PubMed] [Google Scholar]
- 18. Snapper S. B., Meelu P., Nguyen D., Stockton B. M., Bozza P., Alt F. W., Rosen F. S., von Andrian U. H., Klein C. (2005) WASP deficiency leads to global defects of directed leukocyte migration in vitro and in vivo. J. Leukocyte Biol. 77, 993–998 [DOI] [PubMed] [Google Scholar]
- 19. Park H., Cox D. (2009) Cdc42 regulates Fc γ receptor-mediated phagocytosis through the activation and phosphorylation of Wiskott-Aldrich syndrome protein (WASP) and neural-WASP. Mol. Biol. Cell 20, 4500–4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westerberg L. S., de la Fuente M. A., Wermeling F., Ochs H. D., Karlsson M. C., Snapper S. B., Notarangelo L. D. (2008) WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function. Blood 112, 4139–4147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Badour K., Zhang J., Shi F., McGavin M. K., Rampersad V., Hardy L. A., Field D., Siminovitch K. A. (2003) The Wiskott-Aldrich syndrome protein acts downstream of CD2 and the CD2AP and PSTPIP1 adaptors to promote formation of the immunological synapse. Immunity 18, 141–154 [DOI] [PubMed] [Google Scholar]
- 22. Trifari S., Sitia G., Aiuti A., Scaramuzza S., Marangoni F., Guidotti L. G., Martino S., Saracco P., Notarangelo L. D., Roncarolo M. G., Dupré L. (2006) Defective Th1 cytokine gene transcription in CD4+ and CD8+ T cells from Wiskott-Aldrich syndrome patients. J. Immunol. 177, 7451–7461 [DOI] [PubMed] [Google Scholar]
- 23. Falet H., Hoffmeister K. M., Neujahr R., Hartwig J. H. (2002) Normal Arp2/3 complex activation in platelets lacking WASp. Blood 100, 2113–2122 [PubMed] [Google Scholar]
- 24. Falet H., Marchetti M. P., Hoffmeister K. M., Massaad M. J., Geha R. S., Hartwig J. H. (2009) Platelet-associated IgAs and impaired GPVI responses in platelets lacking WIP. Blood 114, 4729–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Azuma T., Witke W., Stossel T. P., Hartwig J. H., Kwiatkowski D. J. (1998) Gelsolin is a downstream effector of rac for fibroblast motility. EMBO J. 17, 1362–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Snapper S. B., Rosen F. S., Mizoguchi E., Cohen P., Khan W., Liu C. H., Hagemann T. L., Kwan S. P., Ferrini R., Davidson L., Bhan A. K., Alt F. W. (1998) Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 9, 81–91 [DOI] [PubMed] [Google Scholar]
- 27. Witke W., Sharpe A. H., Hartwig J. H., Azuma T., Stossel T. P., Kwiatkowski D. J. (1995) Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell 81, 41–51 [DOI] [PubMed] [Google Scholar]
- 28. Hartwig J. H., DeSisto M. (1991) The cytoskeleton of the resting human blood platelet. Structure of the membrane skeleton and its attachment to actin filaments. J. Cell Biol. 112, 407–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoffmeister K. M., Felbinger T. W., Falet H., Denis C. V., Bergmeier W., Mayadas T. N., von Andrian U. H., Wagner D. D., Stossel T. P., Hartwig J. H. (2003) The clearance mechanism of chilled blood platelets. Cell 112, 87–97 [DOI] [PubMed] [Google Scholar]
- 30. Hartwig J. H., Bokoch G. M., Carpenter C. L., Janmey P. A., Taylor L. A., Toker A., Stossel T. P. (1995) Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 82, 643–653 [DOI] [PubMed] [Google Scholar]
- 31. Hoffmeister K. M., Falet H., Toker A., Barkalow K. L., Stossel T. P., Hartwig J. H. (2001) Mechanisms of cold-induced platelet actin assembly. J. Biol. Chem. 276, 24751–24759 [DOI] [PubMed] [Google Scholar]
- 32. Faruqi T. R., Weiss E. J., Shapiro M. J., Huang W., Coughlin S. R. (2000) Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J. Biol. Chem. 275, 19728–19734 [DOI] [PubMed] [Google Scholar]
- 33. Flaumenhaft R. (2003) Molecular basis of platelet granule secretion. Arterioscler. Thromb. Vasc. Biol. 23, 1152–1160 [DOI] [PubMed] [Google Scholar]
- 34. Larsen E., Celi A., Gilbert G. E., Furie B. C., Erban J. K., Bonfanti R., Wagner D. D., Furie B. (1989) PADGEM protein. A receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell 59, 305–312 [DOI] [PubMed] [Google Scholar]
- 35. Falet H., Barkalow K. L., Pivniouk V. I., Barnes M. J., Geha R. S., Hartwig J. H. (2000) Roles of SLP-76, phosphoinositide 3-kinase, and gelsolin in the platelet shape changes initiated by the collagen receptor GPVI/FcR γ-chain complex. Blood 96, 3786–3792 [PubMed] [Google Scholar]
- 36. Flaumenhaft R., Dilks J. R., Rozenvayn N., Monahan-Earley R. A., Feng D., Dvorak A. M. (2005) The actin cytoskeleton differentially regulates platelet α-granule and dense-granule secretion. Blood 105, 3879–3887 [DOI] [PubMed] [Google Scholar]
- 37. Torres E., Rosen M. K. (2003) Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol. Cell 11, 1215–1227 [DOI] [PubMed] [Google Scholar]
- 38. Gross B. S., Wilde J. I., Quek L., Chapel H., Nelson D. L., Watson S. P. (1999) Regulation and function of WASp in platelets by the collagen receptor, glycoprotein VI. Blood 94, 4166–4176 [PubMed] [Google Scholar]
- 39. Oda A., Ikeda Y., Ochs H. D., Druker B. J., Ozaki K., Handa M., Ariga T., Sakiyama Y., Witte O. N., Wahl M. I. (2000) Rapid tyrosine phosphorylation and activation of Bruton's tyrosine/Tec kinases in platelets induced by collagen binding or CD32 cross-linking. Blood 95, 1663–1670 [PubMed] [Google Scholar]
- 40. Lutskiy M. I., Shcherbina A., Bachli E. T., Cooley J., Remold-O'Donnell E. (2007) WASP localizes to the membrane skeleton of platelets. Br. J. Haematol. 139, 98–105 [DOI] [PubMed] [Google Scholar]
- 41. Kim S., Kunapuli S. P. (2011) Negative regulation of Gq-mediated pathways in platelets by G12/13 pathways through Fyn kinase. J. Biol. Chem. 286, 24170–24179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Azim A. C., Barkalow K., Chou J., Hartwig J. H. (2000) Activation of the small GTPases, rac and cdc42, after ligation of the platelet PAR-1 receptor. Blood 95, 959–964 [PubMed] [Google Scholar]
- 43. Rutledge T. W., Whiteheart S. W. (2004) Studies of secretion using permeabilized platelets. Methods Mol. Biol. 272, 109–120 [DOI] [PubMed] [Google Scholar]
- 44. Grainger D. J., Wakefield L., Bethell H. W., Farndale R. W., Metcalfe J. C. (1995) Release and activation of platelet latent TGF-β in blood clots during dissolution with plasmin. Nat. Med. 1, 932–937 [DOI] [PubMed] [Google Scholar]
- 45. Dovas A., Cox D. (2010) Regulation of WASp by phosphorylation. Activation or other functions? Commun. Integr. Biol. 3, 101–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomasevic N., Jia Z., Russell A., Fujii T., Hartman J. J., Clancy S., Wang M., Beraud C., Wood K. W., Sakowicz R. (2007) Differential regulation of WASP and N-WASP by Cdc42, Rac1, Nck, and PI(4,5)P2. Biochemistry 46, 3494–3502 [DOI] [PubMed] [Google Scholar]
- 47. Pleines I., Eckly A., Elvers M., Hagedorn I., Eliautou S., Bender M., Wu X., Lanza F., Gachet C., Brakebusch C., Nieswandt B. (2010) Multiple alterations of platelet functions dominated by increased secretion in mice lacking Cdc42 in platelets. Blood 115, 3364–3373 [DOI] [PubMed] [Google Scholar]
- 48. Pandey D., Goyal P., Dwivedi S., Siess W. (2009) Unraveling a novel Rac1-mediated signaling pathway that regulates cofilin dephosphorylation and secretion in thrombin-stimulated platelets. Blood 114, 415–424 [DOI] [PubMed] [Google Scholar]
- 49. Akbar H., Kim J., Funk K., Cancelas J. A., Shang X., Chen L., Johnson J. F., Williams D. A., Zheng Y. (2007) Genetic and pharmacologic evidence that Rac1 GTPase is involved in regulation of platelet secretion and aggregation. J. Thromb. Haemost. 5, 1747–1755 [DOI] [PubMed] [Google Scholar]
- 50. Cleland S. Y., Siegel R. M. (2011) Wiskott-Aldrich syndrome at the nexus of autoimmune and primary immunodeficiency diseases. FEBS Lett. 585, 3710–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gerdes N., Zhu L., Ersoy M., Hermansson A., Hjemdahl P., Hu H., Hansson G. K., Li N. (2011) Platelets regulate CD4+ T-cell differentiation via multiple chemokines in humans. Thromb. Haemost. 106, 353–362 [DOI] [PubMed] [Google Scholar]
- 52. Catucci M., Castiello M. C., Pala F., Bosticardo M., Villa A. (2012) autoimmunity in Wiskott-Aldrich syndrome. An unsolved enigma. Front. Immunol. 3, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Humblet-Baron S., Sather B., Anover S., Becker-Herman S., Kasprowicz D. J., Khim S., Nguyen T., Hudkins-Loya K., Alpers C. E., Ziegler S. F., Ochs H., Torgerson T., Campbell D. J., Rawlings D. J. (2007) Wiskott-Aldrich syndrome protein is required for regulatory T cell homeostasis. J. Clin. Invest. 117, 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]