

Background: Dopaminergic neurons are susceptible to dopamine-induced toxicity.
Results: Dopamine induces the cytosolic sulfotransferase SULT1A3 via a dopamine D1-NMDA receptor-coupled mechanism.
Conclusion: Induction of SULT1A3 significantly protects cells from dopamine neurotoxicity.
Significance: The dysregulation of SULT1A3 expression may be a risk factor for neurodegenerative diseases involving dopamine.
Keywords: Calcineurin; Dopamine Receptors; ERK; Glutamate Receptors Ionotropic (AMPA, NMDA); Metabolism; Sulfotransferase
Abstract
Dopamine neurotoxicity is associated with several neurodegenerative diseases, and neurons utilize several mechanisms, including uptake and metabolism, to protect them from injury. Metabolism of dopamine involves three enzymes: monoamine oxidase, catechol O-methyltransferase, and sulfotransferase. In primates but not lower order animals, a sulfotransferase (SULT1A3) is present that can rapidly metabolize dopamine to dopamine sulfate. Here, we show that SULT1A3 and a closely related protein SULT1A1 are highly inducible by dopamine. This involves activation of the D1 and NMDA receptors. Both ERK1/2 phosphorylation and calcineurin activation are required for induction. Pharmacological agents that inhibited induction or siRNA targeting SULT1A3 significantly increased the susceptibility of cells to dopamine toxicity. Taken together, these results show that dopamine can induce its own metabolism and protect neuron-like cells from damage, suggesting that SULT1A3 activity may be a risk factor for dopamine-dependent neurodegenerative diseases.
Introduction
Dopamine is a neurotransmitter that exerts its biological effects primarily through a family of closely related G protein-coupled receptors located in both central and peripheral tissues (1). In humans, the dopaminergic system is involved in reward-based learning, addiction, and motivation (2). In addition, dopamine exhibits marked effects on the cardiovascular system (3). Abnormalities in dopamine signaling have been implicated in several neurological and psychiatric disorders including Parkinson disease, schizophrenia, Huntington disease, Alzheimer disease, attention deficit disorder, and addictive behavior (1, 4–6). Some of these diseases arise from dopamine-induced neurotoxicity that can result from elevated dopamine levels (7). The toxic actions of dopamine appear to involve several distinct mechanisms including autoxidation, followed by oxidative stress, and D1 receptor activation (8). In dopaminergic neurons, dopamine is stored in acidic vesicles, which protects the neurotransmitter from oxidation (9). However, increased cytosolic dopamine as a result of leakage into the cytosol or uptake from the extracellular space can lead to neuronal cell death (7).
Dopaminergic neurons utilize a combination of transporters and metabolic enzymes to attenuate intracellular dopamine concentrations, thereby minimizing toxicity. Transporters on the plasma membrane (dopamine active transporter) efficiently accumulate extracellular dopamine from the synapse following its release. This, in turn, is transported from the cytosol into intracellular vesicles by a second transporter (vesicular monoamine transporter 2) where the dopamine is stored for future use. There are three enzymes that metabolize dopamine to less toxic products (10). Two of these enzymes, monoamine oxidase and catechol O-methyltransferase, are found both inside and outside of the cell. They exhibit enzyme kinetics toward dopamine (Km ∼400 μm) that suggest that they function primarily at high substrate concentrations (11). The third enzyme, sulfotransferase 1A3 (SULT1A3),2 which is found in the cytoplasm of neuronal cells, sulfonates dopamine to the inactive dopamine sulfate. This metabolite accounts for >90% of dopamine in the circulation and the cerebrospinal fluid (12). SULT1A3 exhibits a much lower Km toward dopamine (∼ 5 μm), suggesting that it may have a detoxification role at low dopamine concentrations such as that found within neuronal cells (13, 14). Recently, it was shown that these enzymes may act in concert to sequentially metabolize catecholamines (15).
The SULT1A3 gene was originally cloned by Zhu et al. (16) in 1993 and has since been mapped to the short arm of chromosome 16 along with two other sulfotransferases SULT1A1 and SULT1A2 (17). A large genomic duplication in this chromosomal region has resulted in two almost identical copies of SULT1A3 at 16p11.2 and 16p12.1 (18). The second copy has been assigned the gene symbol SULT1A4. The SULT1A3 and 1A4 genes encode identical proteins, and both appear to be constitutively expressed (18). SULT1A3 is present in several tissues including the gastrointestinal tract, platelets, lung, kidney, and brain (19). The gene is only found in primates with no known sequence or functional equivalent in rodents and other lower order species. Several researchers have speculated that evolutionary pressure resulting from a greater catecholamine demand in primates may be responsible for its emergence (19–22).
The role of SULT1A3 in the gut is well established. It is essential in the presystemic elimination of dietary dopamine and other catecholamines. Moreover, gut SULT1A3 appears to be responsible for the metabolism of circulating dopamine (23). The identification of SULT1A3 in the brain, including dopaminergic regions of the midbrain (24), raises the possibility that it has a role in eliminating dopamine in the brain as well. In experiments designed to investigate the interaction of the orphan sulfotransferase SULT4A1 with other sulfotransferases, we observed a significant induction in SULT1A3 in the presence of dopamine in human SK-N-MC and SH-SY5Y cells. These observations led us to study the underlying mechanism of induction and to ask whether SULT1A3 had any role in protecting cells from dopamine toxicity. In the present study, we describe the molecular pathways involved in dopamine-dependent up-regulation of SULT1A3.
EXPERIMENTAL PROCEDURES
Materials
Dopamine hydrochloride, D1 receptor antagonist SCH 23390 hydrochloride, ketamine hydrochloride, dibutyryl cyclic AMP sodium salt, BAPTA-AM, FK506, epinephrine and sodium pyruvate were purchased from Sigma-Aldrich. MEK1/2 inhibitor SL327 and cyclosporin A were purchased from Calbiochem. Cocaine was purchased from the National Measurement Institute (NSW, Australia). Double-stranded RNAi (27-mer) targeting human SULT1A3 (SR304654), human GABP-α (SR301693) and universal scrambled negative control siRNA duplex (SR30004) were purchased from OriGene (Rockville, MD).
Cell Culture and Drug Treatments
Human SK-N-MC cells were maintained in minimal essential medium supplemented with 10% FBS, 2 mm l-glutamine, 1 mm sodium pyruvate, and antibiotics in a humidified atmosphere of 95% air, 5% CO2 at 37 °C. SH-SY5Y cells were maintained in Advanced Dulbecco's modified Eagle's medium-F12 supplemented with 10% FBS and antibiotics. Neuro 2A cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS and antibiotics. For treatments, cells were grown in 6-well culture plates (seeding density 1 × 105 cells/well). Cells were treated with dopamine or norepinephrine (50 μm) or dibutyryl cAMP (500 μm) and were incubated for the specified times at 37 °C. Control cells were treated with an equal concentration of solvent. Cells were pretreated for 30 min with different inhibitors including SCH23390 (10 μm), SL327 (20 μm), ketamine (10 μm), cocaine (10 μm), cyclosporin A (1 μm), BAPTA-AM (10 μm), or FK506 (10 μm) prior to the addition of dopamine. SK-N-MC cells were treated with dopamine (1, 10, 100 μm) for up to 48 h for dose- and time-dependent studies. After the duration of the experiment, cells were processed for protein and mRNA analysis.
Luciferase Assay
SK-N-MC cells were transiently transfected with either the SULT1A3 promoter construct pGL3-1117 or pGL3-basic (empty vector) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 24 h of transfection, some cells were treated with 100 μm dopamine for a further 24 h. Cells were then lysed and luciferase activity measured using a firefly luciferase assay kit (Promega) according to the manufacturer's protocol.
siRNA Treatment
Transient transfections were performed using Lipofectamine RNAiMAX for siRNA according to the manufacturer's protocol (Invitrogen). SK-N-MC cells (5 × 105 cells/well) were transfected using the forward transfection method. Briefly, SULT1A3 or GABP-α-specific siRNA oligomers were diluted in OptiMEM reduced serum medium and mixed with Lipofectamine reagent at a 1:1 ratio. After incubation for 10 min at room temperature, siRNA complexes were added to the cells at a final concentration of 10 nm. Scrambled siRNA was used as a negative control. Samples were prepared at 24, 48, 72, and 96 h, and gene knockdown was either evaluated by quantitative real-time PCR or Western blot analysis.
C2/CREB Transfection
SK-N-MC cells were seeded in 24-well plates at 1 × 105 cells/well and transfected with 1 μg of FLAG-C2/CREB plasmid DNA (a kind gift from Dr. Gerald Thiel, University of the Saarland Medical Center, Homburg, Germany) (25) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. After 24 h of transfection, cell lysates were prepared and Western blotting performed as described below.
Transfection of Neuro 2A Cells with FLAG SULT1A3 Plasmid
The coding region of SULT1A3 was inserted in-frame into the mammalian expression vector p3×FLAG-CMV7.1 (Sigma-Aldrich). Neuro 2A cells were transiently transfected with either p3×FLAG-CMV7.1 (empty vector) or pFLAG-SULT1A3 using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Transfection efficiency determined by co-transfection with an enhanced GFP construct was 89 ± 3%.
Assessment of Cell Toxicity
MTS assays were performed using the CellTiter 96 Aqueous One-solution assay kit (Promega) according to the manufacturer's protocol. SK-N-MC cells were seeded in 96-multiwell plates at 5 × 103 cells/well and treated as described. Cells were then washed with PBS, and the medium was replaced with 100 μl of medium without phenol red and 20 μl of MTS reagent. After a 1-h incubation at 37 °C, the absorbance at 490 nm was measured by a microplate reader (Polarstar Omega, BMG Labtech).
Protein Preparation and Immunoblot Analysis
Cell lysates were prepared by lysing cells on ice with radioimmuneprecipitation assay buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate with protease inhibitor mixture), and protein concentrations were determined by the Bradford method. Briefly, homogenate protein (20 μg) was separated by SDS-PAGE and then transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dried milk in Tris-buffered saline with Tween 20 (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, and 0.05% Tween 20) followed by an overnight incubation at 4 °C with primary antibody. After extensive washing, the blots were incubated for 1 h with the appropriate horseradish peroxidase-linked secondary antibody and developed using Western Lightning Plus-ECL (NEL105001EA; PerkinElmer Life Sciences). Densitometric analyses of the blots were performed on a Kodak image station 4000MM (Molecular Imaging Systems; Carestream Health, Rochester, NY).
For Western blotting, the following antibodies were used: polyclonal anti-human SULT1A1/3 (gift from Professor Mike Coughtrie, University of Dundee, Ninewells Hospital and Medical School, Dundee, Scotland; this antibody recognized both SULT1A1 and 1A3, which migrate differently in SDS-PAGE (26)), polyclonal total and phospho-p44/42 MAPK (ERK1/2) antibodies and α-tubulin (Cell Signaling Technology), HRP-conjugated anti-sheep IgG (A3415; Sigma-Aldrich), anti-mouse and anti-rabbit IgG (Jackson ImmunoResearch), and anti-FLAG M2 HRP (Sigma-Aldrich).
Quantitative Real-time PCR
Total RNA was isolated using a RNeasy mini kit (Qiagen) and reverse transcribed using oligo(dT)15 primer (Promega) and Superscript II reverse transcriptase (Invitrogen). Expression levels of mRNA were determined using an iCycler IQ real-time PCR detection system (Bio-Rad). First-strand cDNA was amplified using specific primers for SULT1A1 (forward primer, 5′-GTTGGCTCTGCAGGGTTTCTAGGA-3′ and reverse primer, 5′-CCCAAACCCCCTGCTGGCCAGCACCC-3′), SULT1A3 (forward primer, 5′-GGAACCCTCAGGGCTGGAG-3′ and reverse primer, 5′-CGTCCTTTGGGTTTCGGG-3′), GABP-α (qstar primer pair, HP205793), and β-actin (forward primer, 5′-CCTCGCCTTTGCCGATCC-3′ and reverse primer, 5′-GGATCTTCATGAGGTAGTCAGTC-3′). Reactions contained SensiMix Plus SYBR and fluorescein (Bioline, NSW, Australia) and 6 pmol of each primer in a total volume of 20 μl. Samples were amplified using the following conditions: initial denaturation at 95 °C for 15 min followed by 30 cycles of 95 °C for 20 s, 58 °C for 20 s for SULT1A1 and SULT1A3 or 60 °C for GABP-α and 72 °C for 20 s. Melt curve analysis confirmed specificity of the PCR, whereas agarose gel electrophoresis confirmed the correct size of the PCR product. Relative expression levels of mRNA were determined by the comparative CT method. CT values for the mRNA expression were normalized to CT values for β-actin and then expressed relative to control.
Data Analysis and Statistics
Data are expressed as mean ± S.E. Statistical comparisons between different treatments were assessed by Student's t tests and one-way analysis of variance assuming significance at a p value of 0.05 or less.
RESULTS
Dopamine Induces SULT1A1 and SULT1A3 in Neuronal-like Cells
Human SK-N-MC neuroepithelioma cells constitutively express both SULT1A1 and 1A3 (Fig. 1A). When these cells were treated with 100 μm dopamine for 48 h, a dose-dependent increase in SULT1A1/3 proteins was seen (Fig. 1A). As little as 10 μm dopamine resulted in a 2–3-fold induction of the enzymes. The increase in protein was accompanied by an increase in mRNA for both genes (Fig. 1B). Induction was not confined to SK-N-MC cells because the human SH-SY5Y neuroblastoma cells, which constitutively express only SULT1A3, also showed a 3-fold increase in the sulfotransferase protein following dopamine treatment (Fig. 1C). A time study in SK-N-MC cells showed no change in SULT1A1/3 expression within the first 4 h of treatment but a significant increase by 8 h (Fig. 1D). Interestingly, parallel changes in SULT1A1 and SULT1A3 were always observed in this study, suggesting that their cellular regulation is closely associated.
FIGURE 1.

Induction of SULT1A1 and SULT1A3 by dopamine. A, concentration-dependent induction of SULT1A1/3. SK-N-MC cells were treated with dopamine (DA) for 48 h following which SULT1A1/3 protein was determined by Western blotting. B, quantitation of SULT1A1 and 1A3 mRNA by qPCR. C, concentration-dependent induction of SULT1A3 in SH-SY5Y cells treated with dopamine for 48 h. D, time-dependent induction of SULT1A1/3 by 100 μm dopamine. E, concentration-dependent increase in ERK1/2 phosphorylation following dopamine treatment for 48 h. F, time-dependent increase in ERK1/2 phosphorylation in the presence of 100 μm dopamine. All results are the mean ± S.E. (error bars), n = 3, normalized to tubulin as loading control and are expressed as percentage of control (no dopamine for A, C, and E; zero time for D and F). Asterisks indicate data significantly different (p < 0.05) from control.
Dopamine treatment increased phosphorylation of the downstream target of dopamine receptor signaling, ERK1/2 (Fig. 1E). ERK1/2 phosphorylation increased in a dose-dependent manner similar to that for the sulfotransferases. However, the up-regulation of phospho-ERK1/2 was evident as early as 2 h after treatment (Fig. 1F), indicating that it preceded SULT1A1/3 induction.
SULT1A1/3 Induction Requires Dopamine D1 Receptor Activation and ERK1/2 Phosphorylation
SK-N-MC cells express dopamine D1 receptors with little or no D2, D3, D4, or D5 receptors (27). To test whether dopamine receptor activation was involved in SULT1A1/3 induction, cells were pretreated with the D1 receptor antagonist SCH23390 and then exposed to dopamine. Fig. 2A shows that SULT1A1/3 induction was significantly attenuated. By contrast, the dopamine transport inhibitor cocaine had no effect (Fig. 2B). These results suggested that induction of the sulfotransferases was mediated by D1 receptor activation.
FIGURE 2.
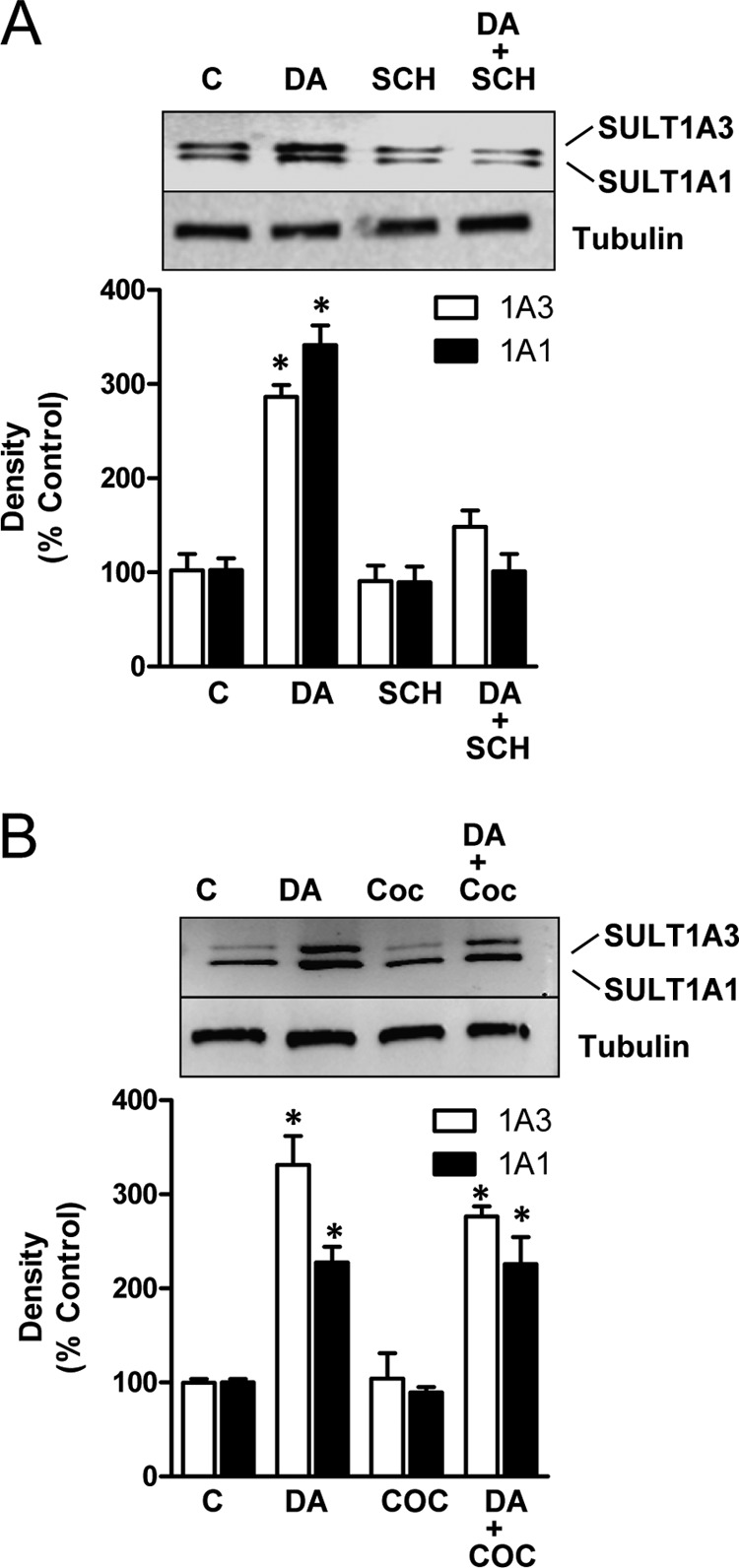
Inhibition of SULT1A1/3 induction. A, SK-N-MC cells were co-treated with 10 μm D1 receptor antagonist SCH23390 (SCH) and 100 μm dopamine (DA) for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. B, cells were co-treated with 10 μm dopamine active transporter inhibitor cocaine (COC) and 100 μm dopamine for 24 h. All results are the mean ± S.E. (error bars), n = 3, normalized to tubulin as loading control and are expressed as percentage of control (C). Asterisks indicate data significantly different (p < 0.05) from control by Student's t test.
We next asked whether phosphorylation of ERK1/2 was necessary for SULT1A1/3 induction. As expected, pretreatment with the MEK inhibitor SL327 blocked the increase in p44 and p42 (ERK1/2) phosphorylation by dopamine (Fig. 3A). In addition, it also blocked induction of the sulfotransferases (Fig. 3B). By contrast, both norepinephrine and dibutyryl cAMP increased ERK1/2 phosphorylation to the same extent as dopamine (Fig. 3C). However, neither of these treatments led to induction of SULT1A1/3 (Fig. 3D). Thus, ERK1/2 phosphorylation appears to be necessary, but not sufficient, for induction of both sulfotransferases. The lack of effect by dibutyryl cAMP suggests that signaling of the D1 receptor through cAMP is not the intracellular pathway that leads to SULT1A1/3 induction.
FIGURE 3.

Induction mechanism of SULT1A1/3. A, SK-N-MC cells were co-treated with 20 μm MEK inhibitor SL327 (SL) and 100 μm dopamine (DA) for 24 h. Phospho-ERK (p-ERK) and total ERK1/2 (Total-ERK) protein levels were quantified by Western blotting. B, cells were co-treated with 20 μm MEK inhibitor SL327 and 100 μm dopamine for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. C, effect of norepinephrine and dibutyryl cAMP on ERK1/2 phosphorylation is shown. Cells were treated with 100 μm dopamine, 50 μm norepinephrine (NA), or 500 μm dibutyryl cAMP for 24 h. Phospho-ERK and total ERK1/2 protein levels were quantified by Western blotting. D, cells were treated with 100 μm dopamine, 50 μm norepinephrine, or 500 μm dibutyryl cAMP for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. All results are the mean ± S.E. (error bars), n = 3, normalized to tubulin as loading control and are expressed as percentage of control (C). Asterisks indicate data significantly different (p < 0.05) from control by Student's t test.
SULT1A1/3 Induction Involves NMDA Receptors
D1 receptor activation can modulate NMDA receptor function through several different mechanisms including direct physical interaction (28, 29), enhanced cell surface receptor density (30), regulation of NMDA receptor phosphorylation (31), and inhibition of Ca2+/calmodulin inactivation of the NMDA channel (32). The dopamine D1 and NMDA receptors signal along pathways that converge at ERK1/2 (33, 34). NMDA receptor signaling results in an increase in intracellular calcium with subsequent activation of calmodulin and calcineurin (35). Calcineurin, a calcium-dependent phosphatase, can dephosphorylate a range of intracellular proteins and transcription factors including NFAT, MEF2A, Crz1, Drp1, Tau, and Rack1 (36). To test the possible involvement of NMDA receptors in SULT1A1/3 induction, SK-N-MC cells were pretreated with the NMDA receptor antagonist ketamine. Blockade of NMDA receptors completely inhibited any dopamine-dependent increase in sulfotransferase expression (Fig. 4A). Moreover, an increase in intracellular calcium was required because the calcium chelator BAPTA also blocked SULT1A1/3 induction (Fig. 4B). Finally, to determine whether calcineurin was required for induction, cells were pretreated with two calcineurin antagonists, cyclosporin and FK506. Cyclosporin binds to cyclophilin whereas FK506 binds to FKBP. Both of these complexes block the function of calcineurin (37). Pretreatment of SK-N-MC cells with either inhibitor significantly decreased dopamine-dependent up-regulation of SULT1A1/3 (Fig. 4, C and D). Taken together, these results show that dopamine induction of SULT1A1/3 requires NMDA receptor signaling, increased intracellular calcium, and calcineurin activation.
FIGURE 4.

Effect of NMDA signaling on SULT1A1/3 induction. A, SK-N-MC cells were co-treated with 10 μm NMDA receptor antagonist ketamine (Ket) and 100 μm dopamine (DA) for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. B, cells were co-treated with 10 μm NMDA receptor antagonist ketamine and 100 μm dopamine for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. C, cells were co-treated with 1 μm calcineurin inhibitor cyclosporin A (Cyc) and 100 μm dopamine for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. D, cells were co-treated with 10 μm calcineurin inhibitor FK506 and 100 μm dopamine for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. All results are the mean ± S.E. (error bars), n = 3, normalized to tubulin as loading control and are expressed as percentage of control (C). Asterisks indicate data significantly different (p < 0.05) from control by Student's t test.
The Ets Family Member GABP, but Not CREB, Is Involved in SULT1A1/3 Induction
To assess whether the induction of the sulfotransferases involved transcriptional up-regulation, we used a luciferase reporter construct containing a 1117-bp fragment of the SULT1A3 gene promoter, as reported elsewhere (38). SK-N-MC cells transfected with this construct showed a significant increase in luciferase activity following dopamine treatment compared with control (Fig. 5A). This result indicates that induction of SULT1A3 is, at least in part, due to increased transcription.
FIGURE 5.

Effect of transcription factors on SULT1A1/3 induction. A, the effect of dopamine on the SULT1A3 promoter was assessed using a luciferase reporter system containing 1117 bp upstream of the SULT1A3 exon 1. Transfected SK-N-MC cells were treated with dopamine or vehicle for 24 h before luciferase activity was determined. B, RT-PCR demonstrates inhibition of GABP-α mRNA by siRNA compared with a scrambled control sequence. Three independent experiments are shown. C, effect of GABP-α knockdown on SULT1A1/3 induction is shown. SK-N-MC cells were co-treated with 10 nm siRNA in the absence (C) or presence of 100 μm dopamine (DA) for 24 h. SULT1A1/3 protein levels were quantified by Western blotting. D, concentration-dependent phosphorylation of CREB (p-CREB) by dopamine was assessed for 24 h. E, effect of constitutively active CREB (C2/CREB) on SULT1A1/3 expression is shown. Cells were transfected with FLAG-tagged C2/CREB, and SULT1A1/3 protein levels were quantified by Western blotting 24 h later. The bottom blot is anti-FLAG antibody. Triplicate results are shown. All results are the mean ± S.E. (error bars), n = 3, normalized to tubulin as loading control and are expressed as percentage of control. Asterisks indicate data significantly different (p < 0.05) from control by Student's t test.
Promoter studies have shown that both SULT1A1 and 1A3 are regulated by the transcription factor Sp1 in HepG2 hepatoma cells (38). However, important differences were found between the two genes. SULT1A1 showed synergistic activation in the presence of Sp1 and the Ets family transcription factor GABP. By contrast, SULT1A3 did not show this synergy due to base changes in the region of the promoter that bound these two transcription factors. GABP is a dimer comprised of GABP-α and GABP-β, and it cooperates with NFAT, a downstream target of calcineurin, to regulate synaptic expression of utrophin (39). Because GABP only up-regulates SULT1A1, we used siRNA targeting the GABP-α subunit to deplete GABP-α mRNA and then examined dopamine induction of both sulfotransferases. Fig. 5B demonstrates the effectiveness of the siRNA. Moreover, the siRNA, but not a scrambled control, blocked induction of both SULT1A1 and 1A3 by dopamine, suggesting a direct or indirect role for the GABP transcription factor (Fig. 5C).
We have shown previously that expression of the brain-specific orphan sulfotransferase SULT4A1 is regulated by CREB (40), which can be up-regulated by dopamine via PKA-dependent phosphorylation (1). Treatment of SK-N-MC cells with dopamine showed an increase in CREB phosphorylation, but only at 100 μm drug (Fig. 5D). Because SULT1A1/3 was induced by as little as 10 μm dopamine (Fig. 1A), these results do not support a role for CREB. This is consistent with the lack of effect of dibutyryl cAMP, which also increases CREB phosphorylation (41). To further determine the role of CREB, we overexpressed a constitutively active CREB construct, C2/CREB, and quantified sulfotransferase expression (Fig. 5E). Although no increase in SULT1A1 was observed, there was a small but significant increase in SULT1A3. Thus, CREB may have a role in SULT1A3 expression at the higher dopamine concentrations. However, the results suggest that the adenylate cyclase-cAMP-PKA-CREB pathway has little effect on sulfotransferase expression in the model used in the present study.
Induction of SULT1A1/3 Protects Neuronal-like Cells from Dopamine Toxicity
In the presence of serum, dopamine exhibits much less toxicity in human neuronal cells (42, 43) compared with nonhuman cells, such as chick (44) or mouse (45) neurons. We hypothesized that this difference may be due to SULT1A3 induction during exposure to the catecholamine. To test this, cells were pretreated with several pharmacological agents that prevented SULT1A1/3 induction. The dopamine D1 receptor antagonist SL327, NMDA receptor antagonist ketamine, and calcineurin inhibitor cyclosporin A did not affect cell viability alone. However, in combination with 100 μm dopamine, significant loss of viability was seen (Fig. 6A). A full dose-response curve was constructed using SL327, which demonstrated a shift to the left and a decrease in the IC50 from ∼1 mm to 160 ± 23 μm (Fig. 6B).
FIGURE 6.

Effect of SULT1A1/3 induction on dopamine toxicity. A, SK-N-MC cells co-treated with various inhibitors and 100 μm dopamine (DA) for 24 h. Cell viability was then determined. B, dose-response curve for the 20 μm MEK inhibitor SL327. SK-N-MC cells were treated with inhibitor and increasing concentrations of dopamine for 24 h following which cell viability was determined. C, effect of various inhibitors on induction of SULT1A1/3 by 10 μm dopamine. SK-N-MC cells were co-treated with various inhibitors and 10 μm dopamine for 24 h. Representative Western blots are shown from three independent experiments. C, control; DA, dopamine; SCH, SCH23390; SL, SL327; Ket, ketamine. All results are the mean ± S.E. (error bars), n = 3, normalized to no dopamine treatment (control). Asterisks indicate data significantly different (p < 0.05) from control by Student's t test.
The enhanced dopamine toxicity following inhibition of SULT1A3 induction with the various pharmacological inhibitors (Fig. 6A) raised the possibility that their inhibition of SULT1A3 expression may have been due to cytotoxicity instead of interference with the induction pathway(s). To test this, we repeated several of the induction experiments using 10 μm instead of 100 μm dopamine as this concentration caused near maximal sulfotransferase induction (Fig. 1A) without toxicity (Fig. 6A). We saw the same effects for each inhibitor at this lower concentration, confirming that their ability to block SULT1A1/3 up-regulation was not due to dopamine toxicity (Fig. 6C).
Although these results strongly suggested that inhibition of SULT1A1/3 induction enhanced dopamine toxicity, they do not directly demonstrate a role for SULT1A3. Therefore, we selectively knocked down the sulfotransferase using siRNA. Three different sequences were used. Sequence siRNA-A and siRNA-B equally knocked down both sulfotransferases (Fig. 7A). By contrast, sequence siRNA-C preferentially knocked down SULT1A3. Cells pretreated with either siRNA-B (Fig. 7B) or siRNA-C (Fig. 7C) showed enhanced dopamine toxicity. For siRNA-B, the IC50 was 210 μm, and for siRNA-C, it was 125 μm, compared with a scrambled control where the IC50 was 930 μm. Next, we repeated the toxicity study in SH-SY5Y cells, which only express SULT1A3 (Fig. 1C). Again, knockdown of the sulfotransferase with siRNA significantly enhanced dopamine toxicity (Fig. 7D). Finally, we performed the reverse experiment using murine Neuro 2A cells, which are devoid of SULT1A3. Transfection resulted in SULT1A3 expression for at least 96 h in these cells (Fig. 7E). Dopamine IC50 was 75 ± 8 μm in control cells (empty vector). This increased to 415 ± 69 μm when SULT1A3 was introduced (Fig. 7F). These results support the siRNA data and show a central role for SULT1A3 in protecting cells from dopamine toxicity.
FIGURE 7.

Effect of down-regulation and up-regulation of SULT1A3 on dopamine toxicity. A, effect of three siRNA sequences on the expression of SULT1A1/3 protein. SK-N-MC cells were treated with 10 nm siRNA for 72 h, and SULT1A1/3 protein levels were measured by Western blotting. B, dose-response curve of the effect of siRNA-B on dopamine toxicity. SK-N-MC cells were treated with siRNA for 48 h and then increasing concentrations of dopamine for 24 h following which cell viability was determined. C, dose-response curve of the effect of siRNA-C on dopamine toxicity. SK-N-MC cells were treated with siRNA for 48 h and then increasing concentrations of dopamine for 24 h following which cell viability was determined. D, dose-response curve of the effect of siRNA-C on dopamine toxicity in SH-SY5Y cells. Cells were treated with siRNA for 48 h and then increasing concentrations of dopamine for 24 h following which cell viability was determined. E, expression of SULT1A3 in murine Neuro 2A cells was determined. Cells were transfected with empty vector or a FLAG-SULT1A3 construct, and cell lysates were collected up to 96 h for Western blot analysis. F, dose-response curve of the effect of SULT1A3 expression on dopamine toxicity in Neuro 2A cells. Cells were transfected for 48 h and then treated with increasing concentrations of dopamine for 24 h following which cell viability was determined. Error bars, S.E.
DISCUSSION
In the present study, we have shown that dopamine induces both SULT1A1 and 1A3 in human neuronal cell lines by a mechanism involving both dopamine D1 and NMDA receptors. Moreover, this induction appears to protect cells from dopamine-induced cytotoxicity. Studies with siRNA that selectively targeted SULT1A3 showed that this sulfotransferase was responsible for the decreased toxicity in both SK-N-MC and SH-SY5Y cells.
The increased expression of SULT1A1/3 following dopamine treatment appeared to require gene activation because there was an increase in mRNA for both genes. Moreover, down-regulation of the transcription factor GABP, which previously has been shown to regulate SULT1A1 expression (38), prevented induction. Nevertheless, these data do not prove increased transcription of the SULT1A1/3 genes by dopamine as mRNA stability and/or protein stability could account for the results reported here. Regardless, the activation of D1 receptor and phosphorylation of ERK1/2 were necessary. However, the latter by itself was not sufficient to increase SULT1A1/3 because neither norepinephrine nor dibutyryl cAMP increased either sulfotransferase despite increasing phospho-p44/p42. These results suggested that at least two converging pathways were involved in SULT1A1/3 induction. Using a range of pharmacological agents, we showed that induction involves coupling of the D1 and NMDA receptors (28, 29, 31, 32) and activation of calcineurin. Calcineurin, in turn, signals through several target proteins including the transcription factor NFAT. In rat islet cells, insulin induction by glucose requires the formation of NFAT-Maf and NFAT-C/EBP complexes, with ERK1/2 phosphorylation modulating the partners of the calcineurin-dependent NFAT (46). This co-dependent signaling has also been reported in cardiomyocytes (47), embryonic stem cells (48), and myoblasts (49).
SULT1A3 joins a very long list of drug-metabolizing enzymes that are induced by their own substrates (50, 51). This induction is usually dose-dependent, rapid, and reversible (50). In this study, we observed an increase in SULT1A1/3 as early as 8 h after treatment, and this was sustained for at least 48 h. These sulfotransferases are the first phase I or phase II enzymes shown to be induced by a dopamine D1-NMDA receptor-coupled mechanism. However, further human studies are required to confirm induction in vivo. The lack of an animal model for SULT1A3 will make these studies more challenging.
A unique feature of the SULT1A3 gene is its presence only in primates. Several researchers have speculated that evolutionary pressure resulting from greater catecholamine demand in humans may be responsible for its emergence. Because SULT1A3 resides in a chromosomal region of low copy repeats (16p11.2), there is a possibility that multiple copies of the gene are present in humans, and like the CYP2D6 gene, copy number varies among individuals. Consistent with this notion is the recent discovery of an identical gene (SULT1A4) located at 16p12.1. To date, the SULT1A4 gene has only been found in humans, and evolutionary analysis indicates that it arose due to postspeciation duplication. Given the results from the present study that indicate sulfonation is important in dopamine toxicity, gene copy number may be a significant risk factor in catecholamine-induced neurodegenerative disease. High copy number may be protective whereas low copy number, gene deletion, or a loss in inducibility may result in an increased susceptibility. A comprehensive study in appropriate cohorts is required to address this possibility.
This work was supported by National Health and Medical Research Council of Australia Grant GNT1005899.
- SULT
- sulfotransferase
- C2/CREB
- constitutively active CREB
- CREB
- cAMP-responsive element-binding protein
- GABP
- GA-binding protein
- NFAT
- nuclear factor of activated T cells.
REFERENCES
- 1. Beaulieu J. M., Gainetdinov R. R. (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217 [DOI] [PubMed] [Google Scholar]
- 2. Bromberg-Martin E. S., Matsumoto M., Hikosaka O. (2010) Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68, 815–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tayebati S. K., Lokhandwala M. F., Amenta F. (2011) Dopamine and vascular dynamics control: present status and future perspectives. Curr. Neurovasc. Res. 8, 246–257 [DOI] [PubMed] [Google Scholar]
- 4. Blum K., Chen A. L., Giordano J., Borsten J., Chen T. J., Hauser M., Simpatico T., Femino J., Braverman E. R., Barh D. (2012) The addictive brain: all roads lead to dopamine. J. Psychoactive Drugs 44, 134–143 [DOI] [PubMed] [Google Scholar]
- 5. Ungless M. A., Grace A. A. (2012) Are you or aren't you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 35, 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu J., Xiao H., Sun H., Zou L., Zhu L. Q. (2012) Role of dopamine receptors in ADHD: a systematic meta-analysis. Mol. Neurobiol. 45, 605–620 [DOI] [PubMed] [Google Scholar]
- 7. Chen L., Ding Y., Cagniard B., Van Laar A. D., Mortimer A., Chi W., Hastings T. G., Kang U. J., Zhuang X. (2008) Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J. Neurosci. 28, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J., Rusnak M., Luedtke R. R., Sidhu A. (2004) D1 dopamine receptor mediates dopamine-induced cytotoxicity via the ERK signal cascade. J. Biol. Chem. 279, 39317–39330 [DOI] [PubMed] [Google Scholar]
- 9. Lotharius J., Brundin P. (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 3, 932–942 [DOI] [PubMed] [Google Scholar]
- 10. Avramovich-Tirosh Y., Amit T., Bar-Am O., Zheng H., Fridkin M., Youdim M. B. (2007) Therapeutic targets and potential of the novel brain-permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer's disease. J. Neurochem. 100, 490–502 [DOI] [PubMed] [Google Scholar]
- 11. Yan M., Webster L. T., Jr., Blumer J. L. (2002) Kinetic interactions of dopamine and dobutamine with human catechol-O-methyltransferase and monoamine oxidase in vitro. J. Pharmacol. Exp. Ther. 301, 315–321 [DOI] [PubMed] [Google Scholar]
- 12. Cedarbaum J. M., Olanow C. W. (1991) Dopamine sulfate in ventricular cerebrospinal fluid and motor function in Parkinson's disease. Neurology 41, 1567–1570 [DOI] [PubMed] [Google Scholar]
- 13. Cappai R., Leck S. L., Tew D. J., Williamson N. A., Smith D. P., Galatis D., Sharples R. A., Curtain C. C., Ali F. E., Cherny R. A., Culvenor J. G., Bottomley S. P., Masters C. L., Barnham K. J., Hill A. F. (2005) Dopamine promotes α-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 19, 1377–1379 [DOI] [PubMed] [Google Scholar]
- 14. Sai Y., Wu Q., Le W., Ye F., Li Y., Dong Z. (2008) Rotenone-induced PC12 cell toxicity is caused by oxidative stress resulting from altered dopamine metabolism. Toxicol. in Vitro 22, 1461–1468 [DOI] [PubMed] [Google Scholar]
- 15. Kurogi K., Alazizi A., Liu M. Y., Sakakibara Y., Suiko M., Sugahara T., Liu M. C. (2012) Concerted actions of the catechol O-methyltransferase and the cytosolic sulfotransferase SULT1A3 in the metabolism of catecholic drugs. Biochem. Pharmacol. 84, 1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu X., Veronese M. E., Bernard C. C., Sansom L. N., McManus M. E. (1993) Identification of two human brain aryl sulfotransferase cDNAs. Biochem. Biophys. Res. Commun. 195, 120–127 [DOI] [PubMed] [Google Scholar]
- 17. Thomae B. A., Rifki O. F., Theobald M. A., Eckloff B. W., Wieben E. D., Weinshilboum R. M. (2003) Human catecholamine sulfotransferase (SULT1A3) pharmacogenetics: functional genetic polymorphism. J. Neurochem. 87, 809–819 [DOI] [PubMed] [Google Scholar]
- 18. Hildebrandt M. A., Salavaggione O. E., Martin Y. N., Flynn H. C., Jalal S., Wieben E. D., Weinshilboum R. M. (2004) Human SULT1A3 pharmacogenetics: gene duplication and functional genomic studies. Biochem. Biophys. Res. Commun. 321, 870–878 [DOI] [PubMed] [Google Scholar]
- 19. Riches Z., Stanley E. L., Bloomer J. C., Coughtrie M. W. (2009) Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie.” Drug Metab. Dispos. 37, 2255–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dajani R., Cleasby A., Neu M., Wonacott A. J., Jhoti H., Hood A. M., Modi S., Hersey A., Taskinen J., Cooke R. M., Manchee G. R., Coughtrie M. W. (1999) X-ray crystal structure of human dopamine sulfotransferase, SULT1A3: molecular modeling and quantitative structure-activity relationship analysis demonstrate a molecular basis for sulfotransferase substrate specificity. J. Biol. Chem. 274, 37862–37868 [DOI] [PubMed] [Google Scholar]
- 21. Dajani R., Hood A. M., Coughtrie M. W. (1998) A single amino acid, Glu-146, governs the substrate specificity of a human dopamine sulfotransferase, SULT1A3. Mol. Pharmacol. 54, 942–948 [DOI] [PubMed] [Google Scholar]
- 22. Gamage N., Barnett A., Hempel N., Duggleby R. G., Windmill K. F., Martin J. L., McManus M. E. (2006) Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 90, 5–22 [DOI] [PubMed] [Google Scholar]
- 23. Goldstein D. S., Swoboda K. J., Miles J. M., Coppack S. W., Aneman A., Holmes C., Lamensdorf I., Eisenhofer G. (1999) Sources and physiological significance of plasma dopamine sulfate. J. Clin. Endocrinol. Metab. 84, 2523–2531 [DOI] [PubMed] [Google Scholar]
- 24. Salman E. D., Kadlubar S. A., Falany C. N. (2009) Expression and localization of cytosolic sulfotransferase (SULT) 1A1 and SULT1A3 in normal human brain. Drug Metab. Dispos. 37, 706–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thiel G., Al Sarraj J., Vinson C., Stefano L., Bach K. (2005) Role of basic region leucine zipper transcription factors cyclic AMP response element-binding protein (CREB), CREB2, activating transcription factor 2 and CAAT/enhancer-binding protein α in cyclic AMP response element-mediated transcription. J. Neurochem. 92, 321–336 [DOI] [PubMed] [Google Scholar]
- 26. Richard K., Hume R., Kaptein E., Stanley E. L., Visser T. J., Coughtrie M. W. (2001) Sulfation of thyroid hormone and dopamine during human development: ontogeny of phenol sulfotransferases and arylsulfatase in liver, lung, and brain. J. Clin. Endocrinol. Metab. 86, 2734–2742 [DOI] [PubMed] [Google Scholar]
- 27. Jassen A. K., Yang H., Miller G. M., Calder E., Madras B. K. (2006) Receptor regulation of gene expression of axon guidance molecules: implications for adaptation. Mol. Pharmacol. 70, 71–77 [DOI] [PubMed] [Google Scholar]
- 28. Lee F. J., Xue S., Pei L., Vukusic B., Chéry N., Wang Y., Wang Y. T., Niznik H. B., Yu X. M., Liu F. (2002) Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 111, 219–230 [DOI] [PubMed] [Google Scholar]
- 29. Fiorentini C., Gardoni F., Spano P., Di Luca M., Missale C. (2003) Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-d-aspartate receptors. J. Biol. Chem. 278, 20196–20202 [DOI] [PubMed] [Google Scholar]
- 30. Hallett P. J., Spoelgen R., Hyman B. T., Standaert D. G., Dunah A. W. (2006) Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J. Neurosci. 26, 4690–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Snyder G. L., Fienberg A. A., Huganir R. L., Greengard P. (1998) A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J. Neurosci. 18, 10297–10303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen G., Greengard P., Yan Z. (2004) Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 101, 2596–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valjent E., Pascoli V., Svenningsson P., Paul S., Enslen H., Corvol J. C., Stipanovich A., Caboche J., Lombroso P. J., Nairn A. C., Greengard P., Hervé D., Girault J. A. (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kaphzan H., O'Riordan K. J., Mangan K. P., Levenson J. M., Rosenblum K. (2006) NMDA and dopamine converge on the NMDA-receptor to induce ERK activation and synaptic depression in mature hippocampus. PLoS One 1, e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marshall J., Dolan B. M., Garcia E. P., Sathe S., Tang X., Mao Z., Blair L. A. (2003) Calcium channel and NMDA receptor activities differentially regulate nuclear C/EBPβ levels to control neuronal survival. Neuron 39, 625–639 [DOI] [PubMed] [Google Scholar]
- 36. Li H., Rao A., Hogan P. G. (2011) Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 21, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rusnak F., Mertz P. (2000) Calcineurin: form and function. Physiol. Rev. 80, 1483–1521 [DOI] [PubMed] [Google Scholar]
- 38. Hempel N., Wang H., LeCluyse E. L., McManus M. E., Negishi M. (2004) The human sulfotransferase SULT1A1 gene is regulated in a synergistic manner by Sp1 and GA binding protein. Mol. Pharmacol. 66, 1690–1701 [DOI] [PubMed] [Google Scholar]
- 39. Angus L. M., Chakkalakal J. V., Méjat A., Eibl J. K., Bélanger G., Megeney L. A., Chin E. R., Schaeffer L., Michel R. N., Jasmin B. J. (2005) Calcineurin-NFAT signaling, together with GABP and peroxisome PGC-1α, drives utrophin gene expression at the neuromuscular junction. Am. J. Physiol. Cell Physiol. 289, C908–917 [DOI] [PubMed] [Google Scholar]
- 40. Butcher N. J., Mitchell D. J., Burow R., Minchin R. F. (2010) Regulation of mouse brain-selective sulfotransferase sult4a1 by cAMP response element-binding protein and activating transcription factor-2. Mol. Pharmacol. 78, 503–510 [DOI] [PubMed] [Google Scholar]
- 41. Mayr B., Montminy M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 42. Jeon S. M., Cheon S. M., Bae H. R., Kim J. W., Kim S. U. (2010) Selective susceptibility of human dopaminergic neural stem cells to dopamine-induced apoptosis. Exp. Neurobiol. 19, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lev N., Ickowicz D., Barhum Y., Lev S., Melamed E., Offen D. (2009) DJ-1 protects against dopamine toxicity. J. Neural Transm. 116, 151–160 [DOI] [PubMed] [Google Scholar]
- 44. Gandhi S., Vaarmann A., Yao Z., Duchen M. R., Wood N. W., Abramov A. Y. (2012) Dopamine-induced neurodegeneration in a PINK1 model of Parkinson's disease. PLoS One 7, e37564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ziv I., Melamed E., Nardi N., Luria D., Achiron A., Offen D., Barzilai A. (1994) Dopamine induces apoptosis-like cell death in cultured chick sympathetic neurons: a possible novel pathogenetic mechanism in Parkinson's disease. Neurosci. Lett. 170, 136–140 [DOI] [PubMed] [Google Scholar]
- 46. Lawrence M. C., McGlynn K., Park B. H., Cobb M. H. (2005) ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J. Biol. Chem. 280, 26751–26759 [DOI] [PubMed] [Google Scholar]
- 47. Sanna B., Bueno O. F., Dai Y. S., Wilkins B. J., Molkentin J. D. (2005) Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol. Cell. Biol. 25, 865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li X., Zhu L., Yang A., Lin J., Tang F., Jin S., Wei Z., Li J., Jin Y. (2011) Calcineurin-NFAT signaling critically regulates early lineage specification in mouse embryonic stem cells and embryos. Cell Stem Cell 8, 46–58 [DOI] [PubMed] [Google Scholar]
- 49. Oh M., Dey A., Gerard R. D., Hill J. A., Rothermel B. A. (2010) The CCAAT/enhancer binding protein β(C/EBPβ) cooperates with NFAT to control expression of the calcineurin regulatory protein RCAN1–4. J. Biol. Chem. 285, 16623–16631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Handschin C., Meyer U. A. (2003) Induction of drug metabolism: the role of nuclear receptors. Pharmacol. Rev. 55, 649–673 [DOI] [PubMed] [Google Scholar]
- 51. Xu C., Li C. Y., Kong A. N. (2005) Induction of phase I, II, and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 28, 249–268 [DOI] [PubMed] [Google Scholar]