

Background: The κ-opioid receptor can be activated by structurally diverse agonists.
Results: Four structurally diverse agonists differentially bound to and activated wild type and mutant κ-opioid receptors.
Conclusion: The structural features of the agonists dictate how they interact with and stabilize Gi-signaling receptor conformations.
Significance: The results provide insights into the structural basis of opioid receptor ligand recognition and activation.
Keywords: G Protein-coupled Receptors (GPCR), Mutagenesis Site-specific, Opiate Opioid, Pharmacology, Receptor Structure-Function
Abstract
The crystal structures of opioid receptors provide a novel platform for inquiry into opioid receptor function. The molecular determinants for activation of the κ-opioid receptor (KOR) were studied using a combination of agonist docking, functional assays, and site-directed mutagenesis. Eighteen positions in the putative agonist binding site of KOR were selected and evaluated for their effects on receptor binding and activation by ligands representing four distinct chemotypes: the peptide dynorphin A(1–17), the arylacetamide U-69593, and the non-charged ligands salvinorin A and the octahydroisoquinolinone carboxamide 1xx. Minimally biased docking of the tested ligands into the antagonist-bound KOR structure generated distinct binding modes, which were then evaluated biochemically and pharmacologically. Our analysis identified two types of mutations: those that affect receptor function primarily via ligand binding and those that primarily affect function. The shared and differential mechanisms of agonist binding and activation in KOR are further discussed. Usually, mutations affecting function more than binding were located at the periphery of the binding site and did not interact strongly with the various ligands. Analysis of the crystal structure along with the present results provide fundamental insights into the activation mechanism of the KOR and suggest that “functional” residues, along with water molecules detected in the crystal structure, may be directly involved in transduction of the agonist binding event into structural changes at the conserved rotamer switches, thus leading to receptor activation.
Introduction
The three classic opioid receptors termed μ (MOR),2 δ (DOR), and κ (KOR) and the closely related nociceptin receptor share 60–80% of their primary amino acid sequence (1) and are the molecular targets for a large number of prescribed and abused analgesic and psychoactive drugs. The structures of all four opioid receptors in complex with antagonists have recently been solved, allowing invaluable insights into the specificity determinants (2–5). Also, crystal structures of several class A GPCRs in activated or partially activated states have revealed conformational changes associated with the intracellular portion of the transmembrane helices (particularly TM5, TM6, and TM7) involving conserved amino acid motifs in the intracellular TM helical bundle (6). These helical rearrangements are modulated by a set of ligand-receptor interactions in the orthosteric binding site located in the extracellular region of the helical bundle. These ligand-dependent changes are usually much smaller in scale and involve a variety of non-conserved residues in different GPCRs, a consequence of the highly diverse nature of the corresponding GPCR ligands (6–8). Determining which binding site residues are responsible for ligand recognition and activation is currently an area of intense research. We have only recently begun to understand the complex ways in which GPCRs, their ligands, and their intracellular binding partners are able to differentially interact with one another (9). Perhaps due to relatively small conformational changes in the GPCR binding site upon activation, it has been shown that an inactive crystal structure can occasionally be of value in predicting agonist binding poses (10–13), revealing initial “recognition motifs” that may be common to both agonists and antagonists (6).
Previous studies (5, 14–18) have provided a wealth of information regarding the binding affinities and efficacies of a variety of ligand chemotypes to KOR mutants. This type of work, predominantly using antagonists (19), assisted in the description of a generic opioid binding site as well as specificity sites that are unique for each opioid receptor type. The generic opioid binding site is similar in all opioid receptors and is known as the “message” region. The term “address” was used historically to describe the specificity determinants of the different opioid receptors (these terms illustrate the idea that a general opioid message is being delivered to the specific address of KOR). However, the definition of the address determinants is by no means invariant. For example, the KOR crystal structure, as well as mutagenesis data, showed that the κ-selective compound JDTic does not bind to any of the previously described address residues, indicating an expanded address determinant for KOR. Although agonist binding and activation of KOR in different mutant receptors have been previously reported with a focus on different regions (5, 16, 20, 21), all of the available data cannot be easily reconciled or compared because of differences in assay conditions, radioligands employed, and other laboratory-specific variables. Here, to minimize such variability, we performed a parallel study examining the effects of mutations in 18 positions surrounding the KOR binding pocket on radioligand binding and receptor activation by representatives of four chemically distinct groups of KOR-selective agonists. Dynorphin A, an endogenous KOR ligand, is a peptide containing several basic amino acids. It is generally thought that these basic residues interact specifically with acidic residues unique to the KOR in the address region of the receptor (22, 23). The other ligands are small molecules: U-69593, an oxaspiro member of the arylacetamide class; salvinorin A, a highly potent and selective, non-nitrogenous hallucinogenic neoclerodane diterpenoid (24); and compound 1xx, a recently discovered non-basic full agonist octahydroisoquinolinone carboxamide with nanomolar potency as well as 700- and 2000-fold selectivity for KOR over MOR and DOR, respectively (25). Models of the different ligands bound to the KOR crystal structure were generated by minimally biased docking and assessed in the context of our biochemical studies. Interestingly, the mutagenesis data identified two types of mutated positions: 1) positions that affect receptor function through ligand binding and 2) positions that affect function more than binding. Mutations in some of these residues had a different effect for different ligands, whereas others had a similar effect on all of the tested compounds. The consequences of these observations are discussed in the structural framework of the docking models, which provide fundamentally new insights into the chemotype-specific modes of action of KOR agonists.
EXPERIMENTAL PROCEDURES
Radioligand Binding Assays
Radioligand binding assays were performed as previously detailed (20). In brief, crude cell membranes were prepared by lysing transfected HEK-T cells in binding buffer (50 mm Tris-Cl, 10 mm MgCl2, and 0.1 mm EDTA at pH 7.40) and centrifugation at 30,000 × g for 20 min. Membrane pellets were then flash-frozen in liquid nitrogen and stored at −80 °C until use in binding assays. Binding assays were conducted in total volumes of 0.25 ml in 96-well plates in a binding buffer containing 0.3–0.6 nm [3H]diprenorphine for a total of 60 min at room temperature. Assays were terminated by harvesting over 0.3% polyethyleneimine-treated, 96-well filter mats using a 96-well Filtermate harvester and three quick washes with ice-cold binding buffer. The filter mats were dried, and then scintillant was melted onto the filters, and the radioactivity retained on the filters was counted in a Wallac MicroBeta TriLux plate scintillation counter. Ki determinations were performed by using at least eight concentrations of unlabeled ligand spanning a 10,000-fold dose range. Diprenorphine Kd and receptor expression levels (Bmax) for the different mutants were determined using homologous competition binding. The raw data were analyzed by Prism 5.01 (GraphPad Software, Inc., San Diego, CA) to give Kd, Bmax, and Ki values reported as the means ± S.E.
Functional Assays
Point mutations were introduced into human KOR using the QuikChange site-directed mutagenesis II kit (Agilent), and all mutations were verified by Sanger automated sequencing. HEK293T cells were co-transfected with plasmids encoding the cAMP biosensor GloSensor-22F (Promega) and the different human KOR variants as described (26). After an 18-h incubation at 37 °C, the cells were seeded (at 20,000 cells/20 μl/well) into white, clear bottom, 384-well tissue culture plates precoated with poly-l-lysine (25 mg/liter; Sigma). Cells were plated in DMEM containing 1% dialyzed FBS. After a 24-h recovery, the medium was replaced with 20 μl of Drug Buffer (1× Hanks' balanced salt solution, 20 mm HEPES, pH 7.4), and the cells were treated with 10 μl of 3× test or reference drug prepared in Drug Buffer. After 20 min, cAMP production was stimulated and detected by treatment with 10 μl of 1.2 μm (4×) isoproterenol in 8% (4×) GloSensor reagent. Luminescence per well per second was read on a Wallac MicroBeta TriLux plate scintillation counter. Data were normalized to the isoproterenol response (0%), and the maximal salvinorin A-induced inhibition (100%) and regressed using the sigmoidal dose-response function built into GraphPad Prism 5.01.
Molecular Modeling
Modeling of the KOR-RB-64 complex has been described previously (5, 20). GOLDSuite 5.1 (Cambridge Crystallographic Data Centre, Cambridge, UK) was employed to perform automated docking tasks, and SYBYL-X version 1.3 (Tripos International, St. Louis, MO) was used to perform all additional modeling. Default parameters were used unless otherwise noted. All computing tasks were performed on Intel Xeon-based or AMD Opteron-based Linux workstations running CentOS 5.5 or Intel Xeon-based Mac Pro workstations running Mac OS X 10.6 (Snow Leopard).
The structure of the KOR was prepared for docking by extracting the “B” chain (protein only) from the Protein Data Bank file (entry 4DJH), adding hydrogen atoms, and extracting the JDTic ligand. Five water molecules (Wat1307, Wat1311, Wat1313, Wat1314, and Wat1316) located in the B chain binding site were also extracted and saved individually for use in the docking exercises. The structures of the compounds to be docked were sketched in SYBYL and energy-minimized using the Tripos Force Field (Gasteiger-Hückel charges, distance-dependent dielectric constant = 4.0; non-bonded interaction cut-off = 8 Å; termination criterion = energy gradient < 0.05 kcal/(mol × Å) for 100,000 iterations). To make the modeling computationally tractable using automated docking routines, only the first eight N-terminal residues of dynorphin A were included; this truncated dynorphin retains low nanomolar potency at KOR (26). For dynorphin A(1–8), the ligand was initially prepared by sketching the sequence in a fully extended β-strand conformation using the Protein Builder function of SYBYL with charged termini. Using the crystal structure of the KOR-JDTic complex as a guide, the two N-terminal residues of dynorphin A (i.e. Tyr1 and Gly2) were manually aligned to the peptide-like structure of JDTic, and an excellent agreement between the two structures was found. Specifically, the phenolic ring of dynorphin A Tyr1 was then spatially aligned to the phenolic portion of the tetrahydroisoquinoline fragment of JDTic (B chain), and dynorphin A backbone torsion angles were adjusted manually to match the JDTic “backbone” torsions for the two initial dynorphin A residues (Tyr1-Gly2). This conformation was used as a starting point for the docking of dynorphin A. The aligned Tyr1-Gly2 fragment was saved for later use as a docking constraint in GOLD 5.1.
GOLD 5.1 flexible docking was performed without constraints for all ligands except dynorphin A(1–8). GoldScore was used as the scoring/fitness function. A binding site definition consisting of a 15-Å radius sphere around the Asp138(3.32) Cβ atom was employed. Two docking runs were performed for each ligand. The first run involved keeping the side chain conformations as they were defined in the crystal structure. For the second run, selected side chains (Gln115(2.60), Asp138(3.32), Ile290(6.51), Ile294(6.55), Tyr312(7.35), Tyr313(7.36), and Ile316(7.39)) were allowed to be flexible during the docking run to reduce the bias imposed by the “imprint” or “ghost” left behind by the original JDTic ligand. Side chain flexibility for individual residues was modeled by incorporation of conformations (i.e. rotameric states) obtained from the Penultimate Rotamer Library (27) in addition to the conformation found in the crystal structure. Because water molecules are very important participants in the ligand binding process, five water molecules associated with the ligand (Wat1307, Wat1311, Wat1313, Wat1314, and Wat1316) in chain B were selected for inclusion in the docking experiments. The two water molecules linking JDTic to the imidazole ring of His291(6.52) (Wat1307 and Wat1311) were included as part of the binding site. In addition, the remaining water molecules (Wat1313, Wat1314, and Wat1316) were allowed to toggle on or off during the individual docking runs (i.e. these waters were not automatically present in the binding site but were included if their presence strengthened the interaction of the ligand with the receptor, as determined by the scoring function). Ten to thirty ligand poses were generated for each run. While docking dynorphin A(1–8), the rotatable bonds in the aligned portion of the peptide (Tyr1-Gly2) were kept rigid, and a scaffold constraint (penalty = 5.0) was used to maintain the original JDTic-aligned orientation of Tyr1-Gly2. GOLD was then allowed to freely find optimal solutions for the Gly3–Ile8 portion of dynorphin A. The final receptor-ligand complex for each ligand was chosen interactively by selecting the highest scoring pose that was consistent with experimentally derived information about the binding mode of the ligand. The chosen solution(s) was then merged back into the KOR, along with any necessary water molecules. Manual modification of some KOR side chain and ligand torsion angles was performed after completion of the automated docking routines to further optimize receptor-ligand interactions. These modifications involved adjustment of the rotameric state of amino acid side chains in the binding site to an alternative low energy rotamer that enhanced the association of the receptor and the ligand. A typical example would be to “flip” the side chain amide of a glutamine residue to allow formation of a hydrogen bond with the ligand. Finally, the complexes were energy-minimized in SYBYL using the previously stated parameters. The stereochemical quality of the final models was assessed using PROCHECK. As a control, docking experiments were performed using JDTic (the ligand co-crystallized with the KOR) to assess the effectiveness of the docking algorithm. Under all docking conditions used in this study, the top-scoring solution reproduced the observed binding mode to within less than 1.0 Å root mean square deviation and also reproduced the ligand-associated water molecules found in the crystal structure.
RESULTS
To study the agonist binding determinants of KOR, 18 positions around the orthosteric ligand binding site of KOR were mutated to alanine and, in some cases, other amino acids. The mutant receptors were then evaluated for radioligand binding and functional Gi activity using a genetically encoded cAMP biosensor as detailed previously (5). The compounds tested in these assays represent four chemically distinct groups of KOR-selective agonists of widely differing structural types and classes (Fig. 1): the peptide dynorphin A(1–17), the arylacetamide U-69593, and the non-charged ligands salvinorin A (24) and the octahydroisoquinolinone carboxamide 1xx (25). We first describe the effects that the different mutations have on the affinities and potencies of the tested ligands and then describe structural models of the bound ligands and propose structural explanations for some of these effects.
FIGURE 1.
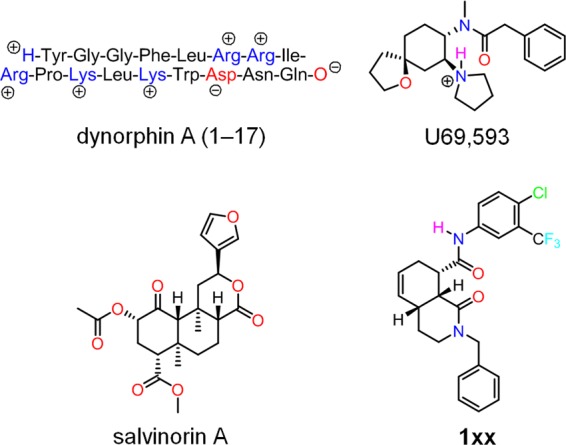
KOR-selective agonists used in this study.
The Effect of Mutations on the Binding and Gi Signaling by the Tested Ligands
The affinities and potencies of the four drugs were tested in wild type (WT) KOR and 26 single point mutants (Tables 1 and 2 and Fig. 2). Expression levels and diprenorphine affinity of the various mutants (Table 1, Kd and Bmax values) were comparable with those of WT in most cases, except for the two lowest expressing mutants (L212A and I135A). The effects of these extremely low expression levels on the apparent potency of the receptor were taken into consideration, and we avoid drawing conclusions on the effects of these mutations on activity relative to WT. However, their relative effect on function (differential effects on the different ligands) and their effects on ligand binding are still relevant. The data in Fig. 2 present the effects of the mutations relative to the WT KOR. The affinities and potencies of the drugs in WT KOR are comparable with previously reported values (21, 25). All of the tested mutations affected ligand binding and/or receptor activation to some degree. In our experiments, we could see a differential effect on binding and activation by the various compounds, with some of the mutations having a similar effect on all or some of the ligands, whereas others had effects specific for only one or two ligands.
TABLE 1.
Summary of competition binding in KOR mutants
Binding affinities (pKi ± S.E.) of the tested ligands for the different mutants were measured using [3H]diprenorphine competition. Bmax values (pmol/mg ± S.E.) and diprenorphine affinities (pKd ± S.E.) were determined by a homologous competition assay (n = 2–6).
| KOR variant | Diprenorphine |
pKi |
||||
|---|---|---|---|---|---|---|
| Bmax | Kd | Salvinorin A | U-69593 | Dynorphin A(1–17) | 1xx | |
| pmol/mg | ||||||
| WT | 4.79 ± 0.88 | 8.97 ± 0.16 | 7.9 ± 0.09 | 7.65 ± 0.2 | 8.1 ± 0.08 | 6.65 ± 0.1 |
| Y66A (1.38) | 5.07 ± 1.05 | 8.85 ± 0.06 | 7.57 ± 0.09 | 6.56 ± 0.11 | 7.11 ± 0.1 | 6.26 ± 0.03 |
| Q115A (2.60) | 3.88 ± 0.93 | 8.88 ± 0.03 | 6.23 ± 0.29 | 6.94 ± 0.02 | 6.91 ± 0.12 | 6.24 ± 0.21 |
| Y119A (2.64) | 6.96 ± 0.23 | 8.56 ± 0.02 | 6.58 ± 0.06 | 6.55 ± 0.04 | 6.69 ± 0.18 | 5.26 ± 0.12 |
| Y119F (2.64) | 5.36 ± 0.39 | 8.73 ± 0.08 | 7.61 ± 0.09 | 7.48 ± 0.18 | 7.8 ± 0.3 | 6.28 ± 0.06 |
| I135A (3.29) | 0.33 ± 0.09 | 9.02 ± 0.33 | 8.08 ± 0.1 | 7.71 ± 0.06 | 8.21 ± 0.2 | 6.66 ± 0.08 |
| D138A (3.32) | 5.94 ± 1.03 | 8.31 ± 0.08 | 8.45 ± 0.11 | 5.54 ± 0.04 | 5.28 ± 0.01 | 7.52 ± 0.06 |
| D138N (3.32) | 3.29 ± 0.47 | 8.69 ± 0.18 | 9.23 ± 0.01 | 5.36 ± 0 | 5.79 ± 0.06 | 8.4 ± 0.14 |
| Y139A (3.33) | 2.32 ± 0.32 | 8.44 ± 0.06 | 7.59 ± 0 | 7.67 ± 0.06 | 7.46 ± 0.17 | 6.45 ± 0.06 |
| Y139F (3.33) | 2.21 ± 0.13 | 8.86 ± 0.04 | 7.72 ± 0.29 | 7.57 ± 0.04 | 7.54 ± 0.17 | 6.52 ± 0.05 |
| M142A (3.36) | 1.16 ± 0.21 | 8.33 ± 0.07 | 7.24 ± 0.03 | 7.66 ± 0.05 | 7.79 ± 0.12 | 7.69 ± 0.11 |
| K200A (ECL2) | 1.56 ± 0.16 | 8.76 ± 0.08 | 8.03 ± 0.21 | 7.95 ± 0.14 | 8.32 ± 0.14 | 6.75 ± 0.03 |
| L212A (ECL2) | 0.18 ± 0.06 | 8.84 ± 0.35 | 8.08 ± 0.1 | 8.48 ± 0.17 | 8.27 ± 0.2 | 6.99 ± 0.16 |
| M226A (5.38) | 3.17 ± 0.36 | 8.73 ± 0.11 | 8.14 ± 0.07 | 7.98 ± 0.01 | 8.15 ± 0.23 | 6.74 ± 0.01 |
| K227A (5.39) | 2.62 ± 0.39 | 9.32 ± 0.3 | 8.36 ± 0.07 | 8.61 ± 0.1 | 8.66 ± 0.23 | 7.29 ± 0.04 |
| H291A (6.52) | 0.58 ± 0.05 | 8.25 ± 0.11 | 7.73 ± 0.15 | 7.22 ± 0.13 | 7.04 ± 0.15 | 6.41 ± 0.05 |
| H291F (6.52) | 0.59 ± 0.12 | 8.43 ± 0 | 8.11 ± 0.05 | 8.12 ± 0.03 | 7.76 ± 0.05 | 7.19 ± 0.07 |
| I294A (6.55) | 2.41 ± 0.26 | 8.6 ± 0.25 | 8.41 ± 0.14 | 7.6 ± 0.14 | 6.84 ± 0.02 | 6.32 ± 0.03 |
| E297A (6.58) | 3.84 ± 0.28 | 9.01 ± 0.19 | 8.14 ± 0.08 | 7.94 ± 0.1 | 7.32 ± 0.05 | 6.96 ± 0.05 |
| Y312A (7.35) | 2.21 ± 0.44 | 9.15 ± 0 | 7.7 ± 0.09 | 6.93 ± 0.08 | 7.96 ± 0.23 | 6.41 ± 0.19 |
| Y312F (7.35) | 3.05 ± 0.24 | 9 ± 0.17 | 7.73 ± 0.23 | 7.13 ± 0.17 | 7.71 ± 0.26 | 6.47 ± 0.12 |
| Y313A (7.36) | 4.95 ± 0.33 | 8.28 ± 0.69 | 5.85 ± 0.09 | 7.39 ± 0.1 | 7.35 ± 0.05 | 5.65 ± 0.29 |
| Y313F (7.36) | 3.42 ± 0.54 | 8.84 ± 0.47 | 7.51 ± 0.04 | 7.23 ± 0.16 | 7.9 ± 0.18 | 6.29 ± 0.22 |
| I316A (7.39) | 3.7 ± 0.29 | 8.63 ± 0.34 | 4.84 ± 0.08 | 6.82 ± 0.07 | 6.8 ± 0.1 | 6.05 ± 0.03 |
| Y320A (7.43) | 1.89 ± 0.46 | 8.5 ± 0.06 | 6.81 ± 0.03 | 6.54 ± 0.05 | 8.18 ± 0.12 | 5.38 ± 0.25 |
| Y320F (7.43) | 1.31 ± 0.23 | 8.85 ± 0.21 | 7.1 ± 0.02 | 6.73 ± 0.12 | 7.75 ± 0.1 | 6.33 ± 0.14 |
TABLE 2.
Summary of functional assays in the tested mutants
A cAMP inhibition assay was used to measure the potencies (pEC50 ± S.E.) of the tested ligands at the different mutants (n = 3–5). ND, not determined.
| KOR variant | Salvinorin A | U-96593 | Dynorphin A(1–17) | 1xx |
|---|---|---|---|---|
| WT | 9.87 ± 0.05 | 8.49 ± 0.08 | 8.32 ± 0.06 | 7.29 ± 0.09 |
| Y66A (1.38) | 9.01 ± 0.05 | 7.75 ± 0.05 | 7.98 ± 0.06 | 6.81 ± 0.04 |
| Q115A (2.60) | 8.58 ± 0.05 | 8.5 ± 0.04 | 7.19 ± 0.06 | 7.14 ± 0.05 |
| Y119A (2.64) | 8.41 ± 0.04 | 8.04 ± 0.05 | 7.26 ± 0.06 | 6.23 ± 0.04 |
| Y119F (2.64) | 9.28 ± 0.03 | 8.09 ± 0.06 | 8.42 ± 0.04 | 6.86 ± 0.04 |
| I135A (3.29) | 8.06 ± 0.04 | 7.14 ± 0.04 | 7.67 ± 0.04 | 5.78 ± 0.05 |
| D138A (3.32) | 9.6 ± 0.13 | ND | ND | 7.44 ± 0.09 |
| D138N (3.32) | 10.6 ± 0.03 | 5.16 ± 0.25 | 5.72 ± 0.05 | 8.04 ± 0.05 |
| Y139A (3.33) | 8.14 ± 0.05 | 7.55 ± 0.07 | 6.93 ± 0.04 | 6.4 ± 0.07 |
| Y139F (3.33) | 9.72 ± 0.04 | 8.48 ± 0.04 | 7.65 ± 0.06 | 6.86 ± 0.05 |
| M142A (3.36) | 7.77 ± 0.04 | 7.71 ± 0.04 | 6.97 ± 0.05 | 7.55 ± 0.04 |
| K200A (ECL2) | 10.03 ± 0.03 | 8.9 ± 0.03 | 8.8 ± 0.03 | 7.1 ± 0.03 |
| L212A (ECL2) | 8.21 ± 0.04 | 7.78 ± 0.04 | 7.1 ± 0.04 | 5.88 ± 0.04 |
| M226A (5.38) | 10.43 ± 0.05 | 9.73 ± 0.05 | 8.86 ± 0.05 | 7.37 ± 0.05 |
| K227A (5.39) | 9.29 ± 0.03 | 8.38 ± 0.03 | 7.93 ± 0.06 | 6.65 ± 0.07 |
| H291A (6.52) | 8.14 ± 0.06 | 6.87 ± 0.06 | 6.58 ± 0.07 | 6.05 ± 0.08 |
| H291F (6.52) | 8.79 ± 0.04 | 8.37 ± 0.04 | 7.09 ± 0.04 | 6.64 ± 0.04 |
| I294A (6.55) | 10.06 ± 0.06 | 8.33 ± 0.08 | 7.14 ± 0.05 | 6.46 ± 0.07 |
| E297A (6.58) | 9.93 ± 0.03 | 8.93 ± 0.04 | 7.36 ± 0.04 | 7.19 ± 0.04 |
| Y312A (7.35) | 8.21 ± 0.04 | 7.19 ± 0.03 | 7.33 ± 0.04 | 6.44 ± 0.05 |
| Y312F (7.35) | 9.56 ± 0.03 | 8.25 ± 0.04 | 8.24 ± 0.04 | 7.15 ± 0.05 |
| Y313A (7.36) | 8.05 ± 0.04 | 8.38 ± 0.05 | 7.52 ± 0.04 | 6.49 ± 0.06 |
| Y313F (7.36) | 9.55 ± 0.04 | 8.32 ± 0.03 | 8.23 ± 0.06 | 7.24 ± 0.07 |
| I316A (7.39) | 6.6 ± 0.11 | 7.72 ± 0.05 | 6.9 ± 0.05 | 6.4 ± 0.04 |
| Y320A (7.43) | 6.45 ± 0.05 | 6.08 ± 0.1 | 7 ± 0.08 | ND |
| Y320F (7.43) | 7.67 ± 0.05 | 6.81 ± 0.05 | 7.12 ± 0.04 | 6.6 ± 0.07 |
FIGURE 2.

Effect of the different mutations on binding affinity and potencies of the tested ligands. Histogram bar heights represent the difference between the KOR mutant and WT KOR potency (ΔpIC50) and affinity (ΔpKi) of the tested ligands salvinorin A (red), 1xx (red with diagonal hash marks), U-69593 (light blue), and dynorphin A (dark blue). Asterisks indicate low expressing mutants; the effect on function is at least partially due to low expression levels.
Polar and Charged Residues
Asp138(3.32), whose acidic side chain is responsible for the formation of a salt bridge with many amine-containing ligands, was mutated to alanine and to asparagine. Both mutations severely impaired the binding and potency of the basic ligands, which is consistent with the necessity for ionic bond formation at Asp138(3.32) for basic ligands. At the same time, our results unexpectedly revealed that these mutations increased the binding affinity of both non-charged ligands. The alanine mutation had a very small effect on the potencies of non-charged ligands, whereas the asparagine mutation had a strong positive effect on the potencies of both non-charged ligands. The greater effect of the asparagine mutant on the potency of the non-basic ligands compared with the alanine mutation suggests a possible hydrogen bond interaction that is distinct from those possible with more classical ligands (see discussion below). The mutation Q115A (TM2) had a variable effect; whereas the binding of salvinorin A and dynorphin A was significantly decreased, the binding of 1xx and U-69593 was altered to a smaller degree. The potencies of salvinorin A and dynorphin A decreased substantially, but those of 1xx and U-69593 were unaffected.
An alanine mutation in Lys227(5.39), at the entrance to the binding site, had a uniformly small to moderate positive effect on ligand binding while having a small negative effect on receptor activation. Based on the crystal structure, Lys227(5.39) forms a salt bridge with Glu297(6.58) at the extracellular end of TM6. TM6 is known to be important for GPCR activation, and disruption of the Lys227(5.39)-Glu297(6.58) interaction may hamper the ability of the KOR to adopt a Gi-interacting conformation. E297A, one helical turn above Ile294(6.55) in TM6, affected only the affinity and potency of dynorphin A and not the other three compounds. Glu297(6.58) is one of the acidic residues found at the opening of the binding site with which the basic residues in dynorphin A have been proposed to interact and that also serves as an “address” locus for classical morphinan-based ligands (28).
Mutations in His291(6.52) lowered receptor expression (Bmax) by about 8-fold (Table 1). An alanine mutation at this position decreased receptor affinity for dynorphin A by more than 1 order of magnitude; however, for the rest of the tested ligands, both alanine and phenylalanine mutations had only small effects on ligand binding. The effect on receptor activation, however, is uniformly profound for the alanine mutation (more than 1.5 orders of magnitude). The phenylalanine mutation decreased the potencies of salvinorin A and dynorphin A by an order of magnitude and had a smaller effect on 1xx and especially U-69593, which retained an almost WT-like potency. A lysine mutation at this position resulted in a complete lack of binding by any ligand, which could have been due either to low expression or to disruption of the binding site (data not shown).
Tyrosine residues have an aromatic moiety and a polar hydroxyl group, both of which may have an important role in ligand binding and receptor activation. Therefore, in several cases, these residues were mutated to an alanine, removing aromatic and polar interactions, or to phenylalanine, removing only the polar interactions. Mutations at position Tyr119(2.64) presented an interesting profile. The effect of the Y119A mutation on the binding of all ligands was larger than 1 order of magnitude. The same mutant affected receptor activation of the non-charged ligands by 1 order of magnitude or more, whereas the basic ligands (dynorphin A and U-69593) were affected by less than 5-fold. The phenylalanine mutation impaired the binding of all ligands to a much lesser degree, whereas its effect on activity was small and negative (decreased potency) for three of the four ligands but small and positive (increased potency) for dynorphin A. Both alanine and phenylalanine mutations at Tyr139(3.33) had a very small effect on ligand binding, but whereas the phenylalanine mutation had almost no effect on potencies, the alanine mutant exhibited a 0.75–1.5-order of magnitude decrease in the potency of each of the ligands. This suggests that decreased steric and/or hydrophobic interactions in the Y139A mutant compared with the Y139F mutant hamper the receptor's ability to adopt an active Gi-recognizing conformation.
The mutagenesis of residues in TM7 revealed an interesting pattern. Alanine and phenylalanine mutations at Tyr312(7.35) had only a slight effect on the binding of U-69593; however, the alanine mutation had a strong deleterious effect on the potencies of all tested compounds, which was rescued in the phenylalanine mutation. This result clearly points to an essential role of Tyr312(7.35), and specifically its aromatic ring, in receptor activation. Tyr313(7.36) had similar effects on both binding and function, and in both tests, the phenylalanine rescued the phenotype of the alanine mutation. The Y313A mutation was especially detrimental to salvinorin A binding and its ability to activate KOR. The Y320A mutation affected the binding of all tested ligands except dynorphin A, with a similar but more pronounced effect on function. Although the phenylalanine mutation managed to rescue most of the binding affinities, the potencies of all tested ligands were reduced, especially those of salvinorin A and U-69593.
Although Tyr66(1.38) does not form a direct contact with the ligand in the KOR crystal structure, an alanine mutation at this position had a robust effect on the binding of U-69593 and dynorphin A and only a small effect on the non-basic compounds salvinorin A and 1xx. Interestingly, the effect on activation by all compounds was similar and small (ΔpEC50 < 1.0).
Non-polar Residues
Ile316(7.39) is oriented directly into the binding site, and an alanine mutation in this position impaired both binding and function for all tested ligands. The I135A mutation on TM3 barely affected the binding of any ligand. Although the comparison with WT for this mutant is problematic because of its low expression levels, we observe that this mutation affected the activation by uncharged ligands to a higher degree than the basic ligands, suggesting that Ile135(3.29) may be important for receptor activation by non-basic ligands, perhaps by partially compensating for the absence of a salt bridge normally formed with the ligand at Asp138(3.32) one helical turn below Ile135(3.29). The M142A mutation had no effect (U-69593) or a small negative effect on the binding of all ligands except 1xx; the affinity of this compound increased by an order of magnitude, but its potency was not substantially altered. The potencies of the remaining compounds were substantially decreased, with that of salvinorin A being the most severely impaired. The alanine mutation at Met226(5.38) did not have a substantial effect on the affinity of any tested ligand and had a small to moderate positive effect on the potency of all of the tested ligands except 1xx, which was not affected. Met226(5.38) is not directly in the binding site cavity; however, it is part of the cluster of hydrophobic residues located at the junction of TM4, ECL2, and TM5. The decreased size of the alanine relative to the methionine side chain may serve to destabilize inactive conformation(s) of the receptor. I294A, one helix turn above His291(6.52), affected the binding and activity of dynorphin A and 1xx but not salvinorin A and U-69593. The affinity of dynorphin A was reduced by more than 1 order of magnitude, and the potencies of both 1xx and dynorphin A were reduced similarly.
Mutations in ECL2
Lys200(ECL2) and Leu212(ECL2) were mutated with an attempt to understand the involvement of ECL2 in binding and function. The K200A mutation did not substantially affect the binding of any of the tested ligands, and only a slight increase was observed in the potency for dynorphin A; this may be attributable to the removal of potentially unfavorable interactions with basic residues in the full-length dynorphin A(1–17).
The L212A mutation had a strong negative effect on the expression of the receptor; however, the binding affinities were not dramatically affected, and the potency shift of about 1 order of magnitude can be attributed to the low expression levels. Leu212(ECL2) is part of a conserved hydrophobic cluster located at the top of TM4/TM5 that possibly serves to stabilize the receptor in an inactive conformation (for discussions, see below).
Prediction of a Putative Binding Mode for Dynorphin A(1–8)
Dynorphin A utilized in this study is a KOR-preferring 17-residue peptide that contains the same Tyr-Gly-Gly-Phe (YGGF) motif at the N terminus as is present in the μ- and δ-selective endorphins and enkephalins. Because the tyrosine residue is essential for activity, it is often speculated that the phenolic ring of morphinan opioids and dynorphin A serve similar functions (for a review of the dynorphin structure-activity relationship, see Ref. 29). Alignment of the occupied binding sites of the KOR, MOR, and DOR using recently published crystal structures (Protein Data Bank codes 4DJH, 4DKL, and 4EJ4, respectively) revealed that the phenolic rings of the opioids occupy a position similar to that of the phenolic portion of the tetrahydroisoquinoline ring of JDTic and most likely the N-terminal aromatic residue of the opioid receptor-selective neuropeptides (2). Using similar logic, the amino group of the terminal tyrosine may mimic the basic amine of morphinans. Until the crystal structures of the opioid receptors became available, there appeared to be no concrete evidence to support these assumptions, which, nonetheless have served as a convenient working hypothesis for decades (30). The positively charged C-terminal region of dynorphin A has been suggested to interact with acidic residues in the extracellular region of the receptor, specifically with ECL2 (31). This notion strengthened the idea of a general ligand-receptor complex topography in which the N-terminal amino group of dynorphin A forms an ionic interaction with the conserved Asp138(3.32), whereas the C-terminal portion extends toward the extracellular loops and perhaps beyond into the aqueous phase.
The truncated dynorphin, dynorphin A(1–8), +H3N-Tyr1-Gly2-Gly3-Phe4-Leu5-Arg6-Arg7-Ile8-COO−, was used in the docking studies to simplify the calculations. The truncated analog retains the potency and selectivity (32) of the parent peptide while containing the message (residues 1–5) and a portion of the highly positively charged address C terminus (residues 6–8) (22, 29).
In the predicted KOR-dynorphin A(1–8) complex (Fig. 3), chosen on the basis of docking score and agreement with experimental evidence, the YGGF message sequence remained in the same location as JDTic and interacted extensively with message residues in the lower portion of the binding site (Fig. 3). To our knowledge, there is no conclusive evidence that the C-terminal address sequence of dynorphin A shows any preference for regular secondary structure (in contrast to UFP-101, an orphanin FQ analog (4)). The dynorphin A(1–8) C-terminal address residues Leu5-Arg6-Arg7-Ile8 docked in an extended conformation, where the predicted binding pose created a network of interactions with non-conserved or partially conserved address residues at each position, including the important specificity-conferring basic residues Arg6 and Arg7.
FIGURE 3.
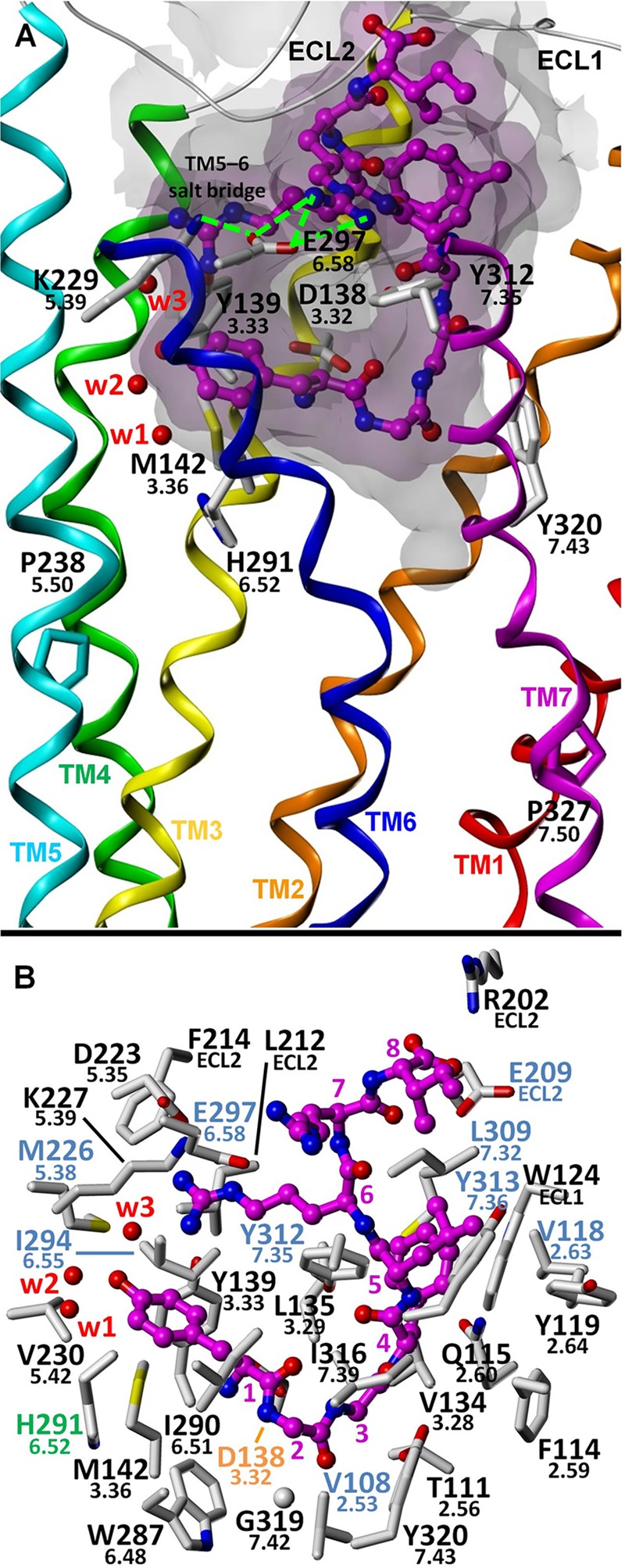
Proposed binding mode for dynorphin A(1–8). A, putative interaction of Arg7 of dynorphin A with Glu297(6.58) probably disturbs the stability of a salt bridge formed between Lys227(5.39) and Glu297(6.58) (potential hydrogen bonds are indicated with green lines). This salt bridge may serve to attenuate G protein-mediated signaling by limiting the ability of TM6 to adopt an active-like conformation, an effect that is probably analogous to the formation of the Ser222(5.43)–Asn344(6.55) hydrogen bond (and other nonpolar interactions) upon binding of the β-arrestin-selective agonist ergotamine at the 5-HT2B receptor. B, putative binding mode showing KOR amino acid residues within 4 Å of the ligand. Water molecules W1–W4 correspond to Wat1311, -1307, -1316, and -1314, respectively.
The docked dynorphin A model suggests a salt bridge between the N-terminal tyrosine (Tyr1) amino group and the Asp138(3.32) carboxylate (Fig. 3). The D138A and D138N mutations both essentially eliminate the potency and affinity of dynorphin A. Additional specific polar interactions were also noted. However, although in our model the phenolic -OH group of Tyr320(7.43) acts as a hydrogen bond donor to the dynorphin A Gly3 backbone carbonyl oxygen atom, its removal has only a small effect on binding, whereas the effect on function is small relative to the effect of this mutation on the other tested ligands. The amide side chain oxygen atom of Gln115(2.60) forms a hydrogen bond with the backbone NH group of dynorphin A Leu5, consistent with the large detrimental effect of the Q115A mutation (more than 10-fold) on binding and function. Glu297(6.58) is unique to the KOR and is largely responsible for the selectivity of dibasic ligands, such as norbinaltorphimine (33) and 5′-guanidinonaltrindole (34), over MOR and DOR. The model shows an ionic interaction between the address guanidinium group of dynorphin A Arg7 and the carboxylate of Glu297(6.58), consistent with a substantially detrimental effect of the E297A mutation and its specificity for dynorphin A (Fig. 3).
Although hydrophobic interactions are difficult to identify using mutagenesis studies, they are likely to be important for the affinity of dynorphin A for the KOR. In our model, Phe4 of the YGGF message domain is located in a pocket delineated by hydrophobic residues that are mostly conserved among KOR, MOR, and DOR; these include Wat124(ECL1), Val134(3.28), Ile135(3.29), and the disulfide linkage residue Cys210(ECL2).
Prediction of a Putative Binding Mode for U-69593
The arylacetamide class of small molecule KOR-selective opioids has been extensively studied, and there are several well known members, including U-69593, which is commonly employed as a radiolabel in KOR competitive binding assays.
The predicted binding mode for U-69593 is shown in Fig. 4. As with other basic amine-containing ligands, a salt bridge is evident between the ammonium ion and Asp138(3.32). Mutations at other nearby residues, including M142A and I294A, are well tolerated. The oxaspiro ring is located in the same small pocket as the N-cyclopropylmethyl group of the morphinan ligands and is surrounded by Val108(2.53), Trp287(6.48), and Tyr320(7.43). Mutation of Tyr320(7.43) to either phenylalanine or (especially) alanine has a profound detrimental effect on the potency of U-69593. However, the effect of these mutations on binding is less profound, an observation consistent with the high affinity of U-50488, which lacks the oxaspiro ring of U-69593. These data, along with the model, suggest that the hydrogen bond formed between Tyr320(7.43) and Thr111(2.56) is somehow important for the proper function of the receptor in this case. Interestingly, it also appears thermodynamically favorable for a crystallographic water molecule to bridge the oxaspiro oxygen atom and a side chain carboxylate atom from Asp138(3.32).
FIGURE 4.
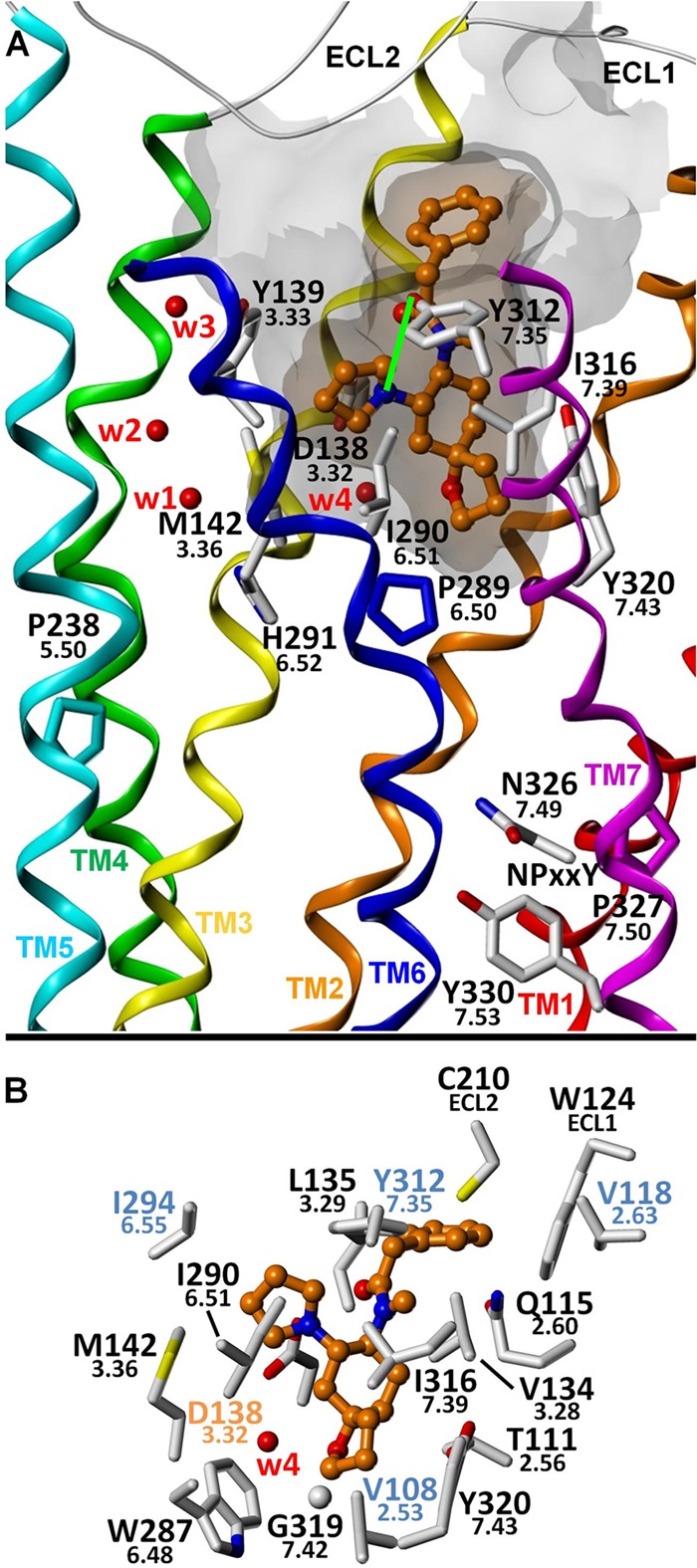
Proposed binding mode for U-69593. A, putative interaction of U-69593 with Ile316(7.39) and Ile290(6.51), binding site residues at positions that belong to a conserved network of key GPCR non-covalent interactions. Mutation of either Ile316 or Tyr320(7.43) one turn above significantly reduces the potency of all tested agonists, consistent with the importance of Ile316 for proper GPCR structure and function. Structurally, a likely explanation for this effect is that these mutations significantly hinder the ability of TM7 and its NPXXY motif to adopt a G protein-recognizing conformation. A hydrogen bond (shown as a green line) may be formed between Tyr312(7.35) and the pyrrolidine ring nitrogen atom in an alternate ring puckering conformation. B, putative binding mode showing KOR amino acid residues within 4 Å of the ligand. Water molecules W1–W4 correspond to Wat1311, -1307, -1316, and -1314, respectively.
Prediction of a Putative Binding Mode for 1xx
Some members of the recently discovered octahydroisoquinolinone class are specific KOR agonists with affinities in the low to mid nanomolar range (5–200 nm) and are non-basic (25); one of the best binding members (1xx) was chosen for study in the current work. The putative binding mode was identified for 1xx from the best scoring docked solution; the results for 1xx are presented in Fig. 5.
FIGURE 5.
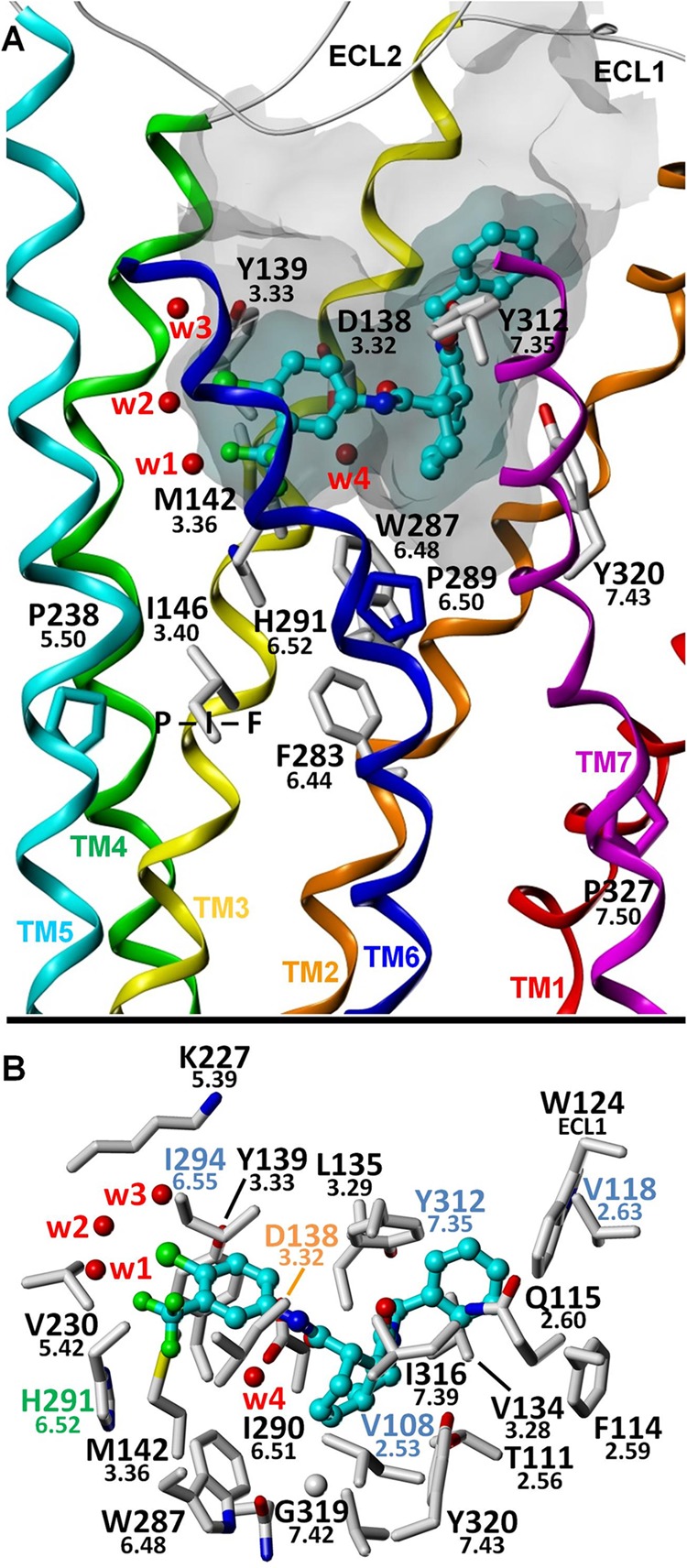
Proposed binding mode for 1xx. A, putative interaction of 1xx with Met142(3.36) at the bottom of the intrahelical ligand binding pocket. Mutation of Met142 to alanine adversely affects the potency of all tested agonists except 1xx, and the affinity of 1xx, unlike that of the other agonists, was substantially increased in the M142A mutant. Because Met142 is directly adjacent to and interacts extensively with the His291(6.52) and Trp287(6.48), its mutation to alanine disrupts the ability of the KOR to function. However, the putative binding mode of 1xx suggests that its bulky 4-chloro-3-trifluoromethyl group, located directly adjacent to Met142, could maintain the signaling ability of the M142A mutant by preventing collapse in the region caused by the missing methionine atoms. B, putative binding mode showing KOR amino acid residues within 4 Å of the ligand. Water molecules W1–W4 correspond to Wat1311, -1307, -1316, and -1314, respectively.
Examination of Table 1 and Fig. 2 shows that three mutations substantially increase the binding affinity of 1xx: D138A, D138N, and M142A. In the binding mode presented here, the Asp138(3.32) carboxylate group is near the octahydroisoquinolinone core, but it does not create a favorable interaction with it. Our model predicts the existence of an extended hydrogen bond network between Asp138(3.32), a bound water molecule, and the amide moiety of 1xx. However, this interaction is probably suboptimal, because by mutating Asp138(3.32) to the smaller and nonpolar alanine, the unfavorable desolvation cost associated with non-basic ligand binding to the charged Asp138(3.32) may be removed (35), thus increasing binding affinity. Because mutation of Asp138(3.32) to asparagine resulted in even greater affinity for 1xx, we propose that a favorable hydrogen bond may then be directly formed between Asn138 and the exocyclic amide carbonyl oxygen atom (i.e. without the intermediacy of a water molecule). The 4-chloro-3-trifluoromethylphenyl group of 1xx is in close proximity to the Met142(3.36) side chain. The M142A mutation resulted in a significantly higher binding affinity, possibly due to removal of unfavorable stereoelectronic interactions of the sulfur and halogen atoms in the WT receptor. Alternatively, direct or indirect effects may change the shape of the binding cavity to allow 1xx to bind more tightly.
There are three mutations that decrease the affinity of 1xx by an order of magnitude or more: Y119A, Y313A, and Y320A. As mentioned previously, Tyr119(2.64) is not directly in the binding site but is part of an aromatic cluster of residues that probably affects ligand binding indirectly. In the KOR crystal structure, Tyr119(2.64) interacts directly with Tyr313(7.36) via edge-face aromatic interactions. When either residue is mutated to alanine, the binding affinity drops markedly; however, the corresponding phenylalanine mutants retain almost all of the binding affinity of the WT KOR. These results suggest that Tyr119(2.64) and Tyr313(7.36) act together to indirectly maintain the shape of the binding cavity. In the proposed interaction mode for 1xx, Tyr119(2.64) and Tyr313(7.36) are relatively far from the ligand. The Y320A mutation nearly abolishes 1xx activity as well as being very detrimental to its binding, whereas the phenylalanine mutation at this position restores almost all of its binding affinity and functionality. This implies that the aromatic ring of Tyr320(7.43) is important both for binding and activation, probably though hydrophobic interactions with the ligand. This is consistent with the proposed model, in which Tyr320(7.43) directly faces the octahydroisoquinolinone core.
Several other mutations decrease the binding affinity of 1xx but to a lesser degree. Ile316(7.39), adjacent to the isoquinolinone core in the proposed model, lowers the affinity by about half a log unit when mutated to alanine. Ile294(6.55), unique to the human KOR, is located near the 4-chloro-3-trifluoromethylphenyl “arm” of 1xx. This is also where the Tyr1 residue of dynorphin A is proposed to bind. The N-benzyl “arm” of 1xx is oriented toward the same hydrophobic pocket as the dynorphin A Phe4 side chain and may also engage in π-stacking or NH-π interactions with the Gln115(2.60) side chain, which when mutated to alanine was found to reduce the affinity by about half a log unit. His291(6.52) is located very near to the 1xx trifluoromethyl group. Interestingly, the H291F mutant was found to have an increased affinity for 1xx by about half of a log unit. This may be due to the increased hydrophobic character of the phenyl compared with the imidazole group.
Prediction of a Putative Binding Mode for Salvinorin A
GOLD docking of salvinorin A (Fig. 1) produced two likely initial interaction modes based on the JDTic-KOR structure. However, one of these poses was more consistent with the mutagenesis data, suggesting that it is closer to the actual binding mode. Examination of the proposed model (Fig. 6) reveals intimate interactions with Ile316(7.39) and Gln115(2.60). The tricyclic core of salvinorin A binds in the same location as the phenylpiperidine fragment of JDTic and is flanked primarily by these residues, which is consistent with the effects of mutations at these positions. Mutation of Ile316(7.39) to alanine has a highly deleterious effect on binding affinity and potency. Q115A and V118K were previously shown to have dramatically reduced affinity for salvinorin A compared with WT, and in this work, we show that both the binding affinity and potency of salvinorin A for Q115A are substantially reduced. The 2-acetoxy group is oriented toward TM5, TM6, and the associated “aromatic cluster” (36, 37) and is directly adjacent to the highly conserved Asp138(3.32). As with 1xx, the D138A mutation has increased affinity for salvinorin A compared with WT, and the model suggests that this is due to the removal of unfavorable desolvation cost of the charged Asp138 residue. Additionally, both affinity and potency are further increased for the D138N mutant; the model suggests that this is due to hydrogen bond formation between a 2-acetoxy oxygen atom acceptor and an Asn138(3.32) side chain -NH donor. The Y320A mutation reduces the affinity by about 10-fold, and Y320F reduces it by 5-fold, suggesting that both the aromatic ring and the hydroxyl group of Tyr320(7.43) are important for optimal binding of salvinorin A. The 4-position methyl ester is located near the same small pocket that is bounded in part by the conserved residues Trp287(6.53) and Tyr320(7.43) and which is occupied by the isopropyl substituent in JDTic and by the N-cyclopropylmethyl substituent of morphinan-based KOR ligands, such as norbinaltorphimine and 5′-guanidinonaltrindole. In the proposed model, the methyl ester group is π-stacked with Tyr320(7.43), and a hydrogen bond may be formed between its phenolic -OH group and the ester group oxygen atoms.
FIGURE 6.
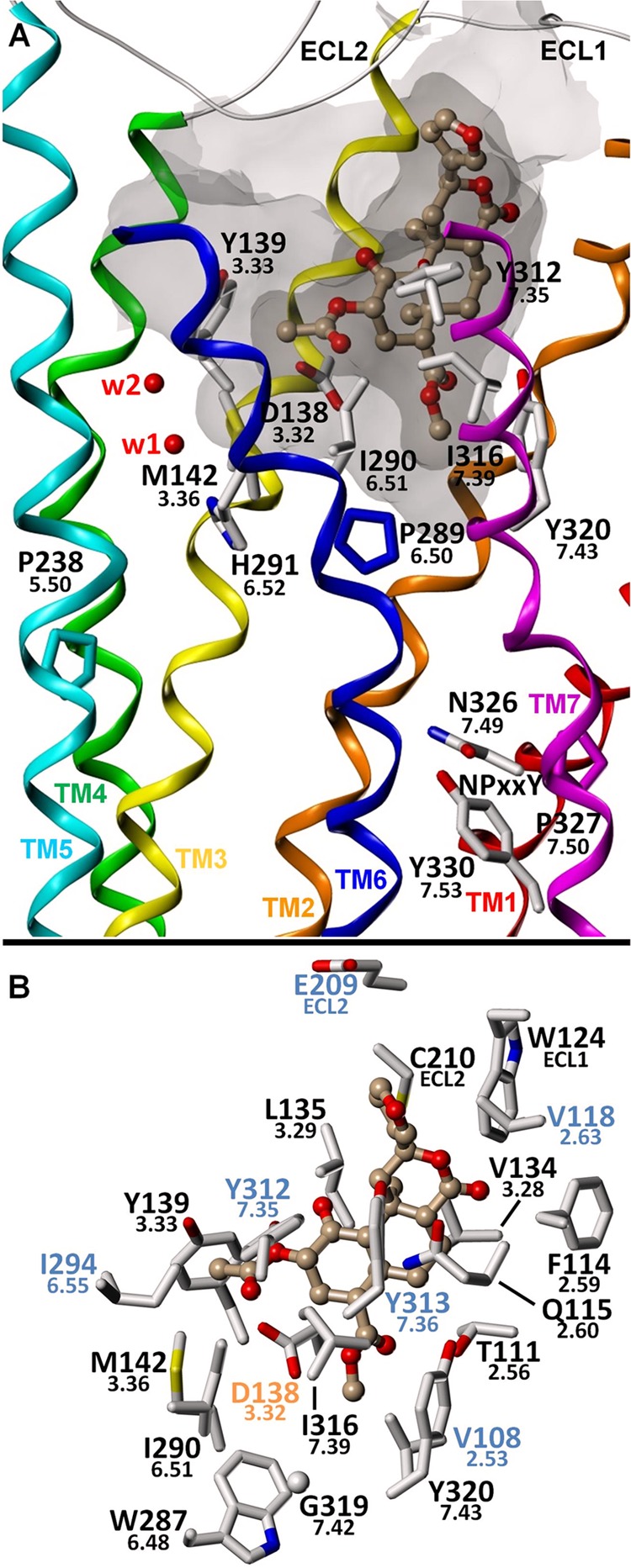
Proposed binding mode for salvinorin A. A, putative interaction of salvinorin A with, among other residues, Ile316(7.39) and Ile290(6.51) in the KOR orthosteric binding site. Like U-69593, the close association of the ligand with TM7 probably interferes with the ability of TM7 to adopt a G protein-recognizing conformation. B, putative binding mode showing KOR amino acid residues within 4 Å of the ligand. Water molecules W1–W4 correspond to Wat1311, -1307, -1316, and -1314, respectively.
Both the Y119A and Y313A mutations severely reduced the binding of salvinorin A, the effects of which were reversed in the Y119F and Y313F mutations. As mentioned previously, Tyr119(2.64) is not directly accessible from the binding site, and Tyr313(7.36) is located on its edge; the aromatic side chains of these residues probably serve to stabilize an agonist-binding conformation of the receptor. The binding of salvinorin A was found to be particularly sensitive to the Y313A mutation. This may be due to both the lack of a strong ionic receptor-ligand interaction and its location or orientation(s) within the binding cavity, because non-basic ligands, such as salvinorin A and 1xx, would be more sensitive to small variations in the structure of the receptor binding site.
DISCUSSION
In this work, we provide new insights into the selective recognition and activation mechanisms of distinct KOR agonist chemotypes. Our data are consistent with models predicting that GPCR agonists with distinct chemotypes will have differential and unique modes of binding and, consequently, receptor activation. These results are likely to be of broad general significance to the field of GPCR research, because they clearly indicate that structurally distinct agonists interact with both shared and unique residues and that unitary models of agonist binding and activation are not tenable.
Not surprisingly, the affinities and potencies of the basic amine-containing compounds dynorphin A and U-69593 are drastically reduced by D138A and D138N mutations (30). Significantly, and in contrast to basic residue-containing agonists, the affinities and potencies of the non-basic compounds 1xx and salvinorin A were unaffected by D138A (16), whereas in both cases, D138N caused a large increase in both affinity and potency. The observed change in binding affinity follows a parallel trend; mutation to alanine modestly, and to asparagine strongly, increases the affinity of 1xx and salvinorin A, whereas the affinity of dynorphin A and U-69593 is reduced by at least 1000-fold. The progressively stronger interactions of D138A and D138N for the non-basic ligands can be attributed to reduced unfavorable desolvation cost and an increase in favorable hydrogen bond interactions with other portions of the non-basic ligand, respectively. These data imply that the high affinity and potency of salvinorin A and congeners is mostly due to hydrophobic interactions as well as other polar interactions. In biogenic amine receptors, mutation of Asp(3.32) typically abolishes both ligand affinity and agonist activity (38–40), consistent with its role as the ligand amino group binding site.
Other mutations also displayed differential effects on the affinities and potencies of the various ligands. For instance, 1) M142A had deleterious effects on all ligands except 1xx; 2) E297A affected dynorphin binding more than any of the other ligands; and 3) Y312A affected U-69593 and salvinorin A more than the other two ligands, while 4) Y320A affected all small molecule ligands to a much greater degree than the peptide dynorphin. Modeling of the M142A mutation creates a new cavity in the “wall” of the binding site that may allow 1xx to dock more tightly and thereby stabilize an active state. The docking positions of both salvinorin A and U-69593 are closer to the extracellular portion of the binding site than the other ligands, and their stability in the binding cavity depends to a higher degree on the interactions with the tyrosine at position 312. Dynorphin interacts with a large network of residues; therefore, its binding is less affected by some of the individual mutations, such as Tyr320(7.43). On the other hand, the “classic” address domain mutation E297A (5, 19) affects dynorphin binding to a higher degree than any of the other tested ligands, probably due to the important interaction of this residue with the sixth arginine of dynorphin.
Differential Effects of Mutations on Binding and Function
An analysis of the relationship between the effects of the various mutations on ligand binding and function can provide novel insights into both shared and distinctive features of receptor activation by the different ligand scaffolds. However, this analysis must be done carefully and take into consideration that although receptor expression levels do not affect binding affinity (41), they can affect the apparent potency of the receptor in functional assays. Therefore, when we analyzed the relationship between the effect of mutations on binding and function (Fig. 7), we excluded from the analysis mutants with extremely low expression levels (Leu212(ECL2) and Ile135(3.29)) and considered only differences in effects that are greater than 10-fold. In most cases, the effect of the mutations on binding was roughly equivalent to their effect on function (within 1 order of magnitude), suggesting that the majority of the effect on function is due to the mutation's effect on ligand binding. However, in some instances, the mutations had a larger effect on function than on binding (by at least 1 order of magnitude). Mutations in Tyr139(3.33), Met142(3.36), Tyr320(7.43), Tyr312(7.35), and Lys227(5.39) affected receptor function by a mechanism additional to, or different from, their direct effect on ligand binding. One possibility for such a mechanism is their potential involvement in the transduction of the activation signal through the receptor affecting some key residues (rotamer switches) directly or indirectly.
FIGURE 7.

The relationship between the effect of mutations on binding and function of the different ligands. The effect of mutations on binding affinity (ΔpKi) is compared with their effect on function (ΔpIC50) for the tested ligands salvinorin A, 1xx, dynorphin A, and U-69593. The middle diagonal line indicates identical effects on binding and function. The outside lines are 1 log unit above and below this “line of identical effect.” Gray points indicate mutations that affected function more than binding. Black points outside the 1 log difference line belong to the low expressing mutations L212A and I135A.
Several conserved residues have been postulated to be involved in the transduction of agonist binding into signaling. These conserved residues are part of larger motifs and are thought to be “rotamer switches”; changes in these residues are thought to be important for the propagation of signal. Some of the important switches are the DRY motif in the intracellular end of TM3, the NPXXY motif at the intracellular end of TM7, and the P-I-F motif connecting TM3, TM5, and TM6 (Fig. 8) (6, 42, 43). An analysis of the structural changes between the active and inactive states of three different class A GPCRs revealed that rotameric changes in these motifs correlated with the larger scale movements of helices, predominately TM5, TM6, and TM7, upon activation (6).
FIGURE 8.

Proposed activation pathways in the κ-opioid receptor. A, an overall view of the JDTic-bound KOR crystal structure with the important rotamer switch motifs (green residues, DRY; orange residues, NPXXY; purple residues, P-I-F motif). B, focus on the interface between TM3, TM5, and TM6, showing the proposed structural relationship between some of the tested positions and the P-I-F motif. C, the predicted changes in the rotameric state of Ile(3.40) and Phe(6.44) (orange) upon activation (based on the analysis of the active state P-I-F motif in several GPCR structures) introduce a collision with the water molecule trapped between Ile146 and His291 in the KOR crystal structure, suggesting that this water molecule is displaced upon KOR activation. D, a surface representation of the binding site of KOR. Mutagenesis of Tyr139 can create a cleft in the wall of the binding site. Salvinorin A and 1xx are represented by thin white and yellow sticks, respectively.
An examination of Figs. 2 and 7 reveals several mutations that affect function differently than binding. Mutation of residues Tyr139(3.33), Met142(3.36), Tyr320(7.43), Tyr312(7.35), and Lys227(5.39) did not have a marked effect on expression levels relative to wild type. Tyr320(7.43) and Lys227(5.39) are involved in intrahelical interactions in the antagonist bound state (5), and their effect on function reflects the importance of these interactions for the transduction of signal. Mutation of Lys227(5.39) to an alanine had a positive effect on binding of all ligands but a small negative effect on their ability to induce signal. This residue interacts with Glu297(6.58) in a salt bridge (Fig. 8) and thus was thought to be directly linked to the proposed movement of TM6 during receptor activation (6, 42). However, a mutation in Glu297 did not have such an effect, and an examination of the crystal structure reveals that, in the antagonist-bound state, Lys227 interacted not only with Glu297 but also with a water molecule that connects it to Tyr139 (Fig. 8), another position that affected function more than binding. Taken together, these data suggest that when Glu297 is mutated, the structured water molecule may be able to stabilize the lysine in the correct position and allow correct activation of the receptor.
Tyr320(7.43) is a conserved residue in many GPCRs, and it interacts with the TM3 Asp(3.32) in biogenic amine receptors as well as in other opioid receptors (2–4, 43–45). The interaction between TM3 and TM7 has been shown to be important for receptor activation, although the details of this interaction change between different receptors (6). In the JDTic-bound KOR structure, Tyr320(7.43) forms a hydrogen bond with Thr111(2.56) (5). This is a characteristic of the KOR-JDTic complex, and it is possible that, in other KOR complexes, this tyrosine behaves similarly to other receptors and interacts with Asp(3.32), an interaction that can affect the movement of TM7 during activation as well as the DRY motif at the intracellular end of TM3.
A Cluster of Residues in the Interface of TM3, TM5, and TM6 Has a Distinct Functional Role in KOR Activation
Figs. 2 and 7 show that mutations in Tyr139(3.33), Met142(3.36), Lys227(5.39), and His291(6.52) affected receptor function more than binding in most cases. The modest effect that the Y139A mutation had on binding affinity suggests that it does not disrupt the binding site directly but affects function in a binding-independent and ligand-specific manner. An alanine mutation in Tyr139(3.33) can disrupt the binding site, and it is conceivable that some chemical moieties (like the methyl group at position 2 of salvinorin A) may insert into this cavity and interfere with the signal propagation via the TM3 rotamer switch (Fig. 8D). Although mutations in His291(6.52) decreased receptor expression levels markedly, they had a remarkable effect on function relative to binding. This effect, along with its location at the interface of TM3, TM5, and TM6, suggests that it may have a role in transducing the ligand binding event to a signal. Further examination of this region in the JDTic-bound crystal structure of KOR reveals that Tyr139(3.33), Met142(3.36), Lys227(5.39), and His291(6.52) form a water-mediated network connecting the edge of the binding site to the critical P-I-F motif (Fig. 8). This may suggest a role in the transduction of the binding event into a signaling event. These “functional” residues are close enough to the binding site to be affected by the binding event itself, and thus, subtle changes in their conformation or polarity could result in changes in the positions of water molecules in the receptor (Fig. 8C). In the inactive state, Tyr139 supports the conformation of Met142, thereby stabilizing the conformation of His291. A water molecule is trapped between the imidazole ring of His291(6.52) and Ile146(3.40), stabilizing the “inactive” rotameric state of the P-I-F motif. The rotameric state of Ile(3.40) in the activated state of serotonin receptors and adrenergic receptors (6, 42, 43) is fairly conserved. Replacing the inactive rotamer of Ile146 in the crystal structure with the “active” rotamer would cause a clash with the water molecule (Fig. 8C) and remove the water from its position. This analysis provides further support to the role of Tyr139(3.33), Met142(3.36), and His291(6.52) in the activation of the receptor, perhaps via water molecules.
Y312A is an interesting mutation; the potencies of all of the tested agonists were similarly impaired (0.8–1.5 orders of magnitude), but only the affinity for U-69593 was substantially decreased. This decrease in affinity is maintained with the Y312F mutation at the same position, although the receptor activity is restored to nearly WT levels. Taken together, these data suggest a direct interaction between U-69593 and the phenolic group of Tyr312(7.35). However, this interaction is not critical for the role of the aromatic moiety in receptor activation. In our model, the phenolic group is close to the heterocyclic amine in a manner that can promote a hydrogen bond between them (Fig. 4).
Collectively, our findings suggest specific roles of different residues in the binding and activation of the κ-opioid receptor by agonist molecules of distinct chemotypes. The ligands tested here represent four divergent chemotypes, and although they all share a similar docked position in the binding site, we were able to predict some ligand-specific interactions as well as interactions that are shared among all of the tested ligands. The non-basic ligands have been shown to bind to KOR despite a suboptimal contact with Asp138(3.32), although this interaction can be dramatically improved by changing this acidic side chain to asparagine. We have also suggested that residues in TM3, -5, -6, and -7 may be directly involved in receptor activation in a manner that does not affect binding.
Tremendous strides in understanding the active states of GPCRs (5, 6, 9, 43, 45–47) have been reported recently, and in this work, we explored the activities of chemically distinct agonist molecules that can use both shared and different molecular determinants in the propagation of signals in the receptor. Using the antagonist-bound structure of KOR and minimally biased docking, we generated models to provide a structural context for the initial interaction of these determinants. Ultimately, however, agonist-bound crystal structures, perhaps in complex with effectors, may be required to definitively delineate signaling mechanisms and GPCR activation determinants. Nonetheless, our findings highlight the complexity of both the recognition and activation mechanism(s) for a single GPCR and provide new insights into how distinct chemotypes at a single GPCR dictate their functions.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grant R01 DA017204 (to B. L. R.); the NIH, NIMH, Psychoactive Drug Screening Program (University of North Carolina at Chapel Hill) (to B. L. R.); and NIH PSI:Biology Grant U54 GM094618 (to V. K. and R. C. S.).
- MOR
- μ-opioid receptor
- DOR
- δ-opioid receptor
- KOR
- κ-opioid receptor
- ECL
- extracellular loop
- GPCR
- G protein-coupled receptor
- TM
- transmembrane.
REFERENCES
- 1. Stevens C. W., Brasel C. M., Mohan S. (2007) Cloning and bioinformatics of amphibian μ, δ, κ, and nociceptin opioid receptors expressed in brain tissue. Evidence for opioid receptor divergence in mammals. Neurosci. Lett. 419, 189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Granier S., Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Weis W. I., Kobilka B. K. (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485, 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., Granier S. (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson A. A., Liu W., Chun E., Katritch V., Wu H., Vardy E., Huang X.-P., Trapella C., Guerrini R., Calo G., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 485, 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X.-P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katritch V., Cherezov V., Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katritch V., Cherezov V., Stevens R. C. (2012) Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 33, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Audet M., Bouvier M. (2012) Restructuring G-protein-coupled receptor activation. Cell 151, 14–23 [DOI] [PubMed] [Google Scholar]
- 9. Vardy E., Roth B. L. (2013) Conformational ensembles in GPCR activation. Cell 152, 385–386 [DOI] [PubMed] [Google Scholar]
- 10. Negri A., Rives M.-L., Caspers M. J., Prisinzano T. E., Javitch J. A., Filizola M. (2013) Discovery of a novel selective κ-opioid receptor agonist using crystal structure-based virtual screening. J. Chem. Inf. Model. 53, 521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katritch V., Abagyan R. (2011) GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol. Sci. 32, 637–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katritch V., Reynolds K. A., Cherezov V., Hanson M. A., Roth C. B., Yeager M., Abagyan R. (2009) Analysis of full and partial agonists binding to β2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J. Mol. Recognit. 22, 307–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weiss D. R., Ahn S., Sassano M. F., Kleist A., Zhu X., Strachan R., Roth B. L., Lefkowitz R. J., Shoichet B. K. (2013) Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem. Biol. 8, 1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferguson D. M., Kramer S., Metzger T. G., Law P. Y., Portoghese P. S. (2000) Isosteric replacement of acidic with neutral residues in extracellular loop-2 of the κ-opioid receptor does not affect dynorphin A(1–13) affinity and function. J. Med. Chem. 43, 1251–1252 [DOI] [PubMed] [Google Scholar]
- 15. Hjorth S. A., Thirstrup K., Schwartz T. W. (1996) Radioligand-dependent discrepancy in agonist affinities enhanced by mutations in the κ-opioid receptor. Mol. Pharmacol. 50, 977–984 [PubMed] [Google Scholar]
- 16. Kane B. E., McCurdy C. R., Ferguson D. M. (2008) Toward a structure-based model of salvinorin A recognition of the κ-opioid receptor. J. Med. Chem. 51, 1824–1830 [DOI] [PubMed] [Google Scholar]
- 17. Metzger T. G., Paterlini M. G., Ferguson D. M., Portoghese P. S. (2001) Investigation of the selectivity of oxymorphone- and naltrexone-derived ligands via site-directed mutagenesis of opioid receptors. Exploring the “address” recognition locus. J. Med. Chem. 44, 857–862 [DOI] [PubMed] [Google Scholar]
- 18. Owens C. E., Akil H. (2002) Determinants of ligand selectivity at the κ-receptor based on the structure of the orphanin FQ receptor. J. Pharmacol. Exp. Ther. 300, 992–999 [DOI] [PubMed] [Google Scholar]
- 19. Portoghese P. S. (1989) Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 10, 230–235 [DOI] [PubMed] [Google Scholar]
- 20. Yan F., Bikbulatov R. V., Mocanu V., Dicheva N., Parker C. E., Wetsel W. C., Mosier P. D., Westkaemper R. B., Allen J. A., Zjawiony J. K., Roth B. L. (2009) Structure-based design, synthesis, and biochemical and pharmacological characterization of novel salvinorin A analogues as active state probes of the κ-opioid receptor. Biochemistry 48, 6898–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yan F., Mosier P. D., Westkaemper R. B., Stewart J., Zjawiony J. K., Vortherms T. A., Sheffler D. J., Roth B. L. (2005) Identification of the molecular mechanisms by which the diterpenoid salvinorin A binds to κ-opioid receptors. Biochemistry 44, 8643–8651 [DOI] [PubMed] [Google Scholar]
- 22. Chavkin C., Goldstein A. (1981) Specific receptor for the opioid peptide dynorphin. Structure-activity relationships. Proc. Natl. Acad. Sci. U.S.A. 78, 6543–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patkar K. A., Murray T. F., Aldrich J. V. (2009) The effects of C-terminal modifications on the opioid activity of [N-benzylTyr1]dynorphin A-(1–11) analogues. J. Med. Chem. 52, 6814–6821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roth B. L., Baner K., Westkaemper R., Siebert D., Rice K. C., Steinberg S., Ernsberger P., Rothman R. B. (2002) Salvinorin A. A potent naturally occurring nonnitrogenous κ opioid selective agonist. Proc. Natl. Acad. Sci. U.S.A. 99, 11934–11939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Frankowski K. J., Ghosh P., Setola V., Tran T. B., Roth B. L., Aubé J. (2010) N-Alkyl-octahydroisoquinolin-1-one-8-carboxamides. Selective and nonbasic κ-opioid receptor ligands. ACS Med. Chem. Lett. 1, 189–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Allen J. A., Yost J. M., Setola V., Chen X., Sassano M. F., Chen M., Peterson S., Yadav P. N., Huang X.-P., Feng B., Jensen N. H., Che X., Bai X., Frye S. V., Wetsel W. C., Caron M. G., Javitch J. A., Roth B. L., Jin J. (2011) Discovery of β-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U.S.A. 108, 18488–18493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lovell S. C., Word J. M., Richardson J. S., Richardson D. C. (2000) The penultimate rotamer library. Proteins 40, 389–408 [PubMed] [Google Scholar]
- 28. Filizola M., Devi L. A. (2012) How opioid drugs bind to receptors. Nature 485, 314–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Naqvi T., Haq W., Mathur K. B. (1998) Structure-activity relationship studies of dynorphin A and related peptides. Peptides 19, 1277–1292 [DOI] [PubMed] [Google Scholar]
- 30. Kong H., Raynor K., Reisine T. (1994) Amino acids in the cloned mouse κ receptor that are necessary for high affinity agonist binding but not antagonist binding. Regul. Pept. 54, 155–156 [Google Scholar]
- 31. Wang J. B., Johnson P. S., Wu J. M., Wang W. F., Uhl G. R. (1994) Human κ opiate receptor second extracellular loop elevates dynorphin's affinity for human μ/κ chimeras. J. Biol. Chem. 269, 25966–25969 [PubMed] [Google Scholar]
- 32. Mansour A., Hoversten M. T., Taylor L. P., Watson S. J., Akil H. (1995) The cloned μ, δ and κ receptors and their endogenous ligands. Evidence for two opioid peptide recognition cores. Brain Res. 700, 89–98 [DOI] [PubMed] [Google Scholar]
- 33. Larson D. L., Jones R. M., Hjorth S. A., Schwartz T. W., Portoghese P. S. (2000) Binding of norbinaltorphimine (nor-BNI) congeners to wild type and mutant μ and κ opioid receptors. Molecular recognition loci for the pharmacophore and address components of κ antagonists. J. Med. Chem. 43, 1573–1576 [DOI] [PubMed] [Google Scholar]
- 34. Sharma S. K., Jones R. M., Metzger T. G., Ferguson D. M., Portoghese P. S. (2001) Transformation of a κ-opioid receptor antagonist to a κ-agonist by transfer of a guanidinium group from the 5′- to 6′-position of naltrindole. J. Med. Chem. 44, 2073–2079 [DOI] [PubMed] [Google Scholar]
- 35. Honig B., Nicholls A. (1995) Classical electrostatics in biology and chemistry. Science 268, 1144–1149 [DOI] [PubMed] [Google Scholar]
- 36. Javitch J. A., Ballesteros J. A., Weinstein H., Chen J. (1998) A cluster of aromatic residues in the sixth membrane-spanning segment of the dopamine D2 receptor is accessible in the binding-site crevice. Biochemistry 37, 998–1006 [DOI] [PubMed] [Google Scholar]
- 37. Westkaemper R. B., Runyon S. P., Bondarev M. L., Savage J. E., Roth B. L., Glennon R. A. (1999) 9-(Aminomethyl)-9,10-dihydroanthracene is a novel and unlikely 5-HT2A receptor antagonist. Eur. J. Pharmacol. 380, R5–R7 [DOI] [PubMed] [Google Scholar]
- 38. Kristiansen K., Kroeze W. K., Willins D. L., Gelber E. I., Savage J. E. (2000) A highly conserved aspartic acid (Asp 155) anchors the terminal amine moiety of tryptamines and is involved in membrane targeting of the 5-HT2A serotonin receptor but does not participate in activation via a “salt-bridge disruption” mechanism. J. Pharmacol. Exp. Ther. 293, 735–746 [PubMed] [Google Scholar]
- 39. Ohta K., Hayashi H., Mizuguchi H., Kagamiyama H., Fujimoto K., Fukui H. (1994) Site-directed mutagenesis of the histamine H1 receptor. Roles of aspartic acid107, asparagine198 and threonine194. Biochem. Biophys. Res. Commun. 203, 1096–1101 [DOI] [PubMed] [Google Scholar]
- 40. Strader C. D., Sigal I. S., Candelore M. R., Rands E., Hill W. S., Dixon R. A. (1988) Conserved aspartic acid residues 79 and 113 of the β-adrenergic receptor have different roles in receptor function. J. Biol. Chem. 263, 10267–10271 [PubMed] [Google Scholar]
- 41. Kenakin T. (2009) A Pharmacology Primer: Theory, Application, and Methods, 3rd Ed., Elsevier Academic Press, Burlington, MA [Google Scholar]
- 42. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wacker D., Wang C., Katritch V., Han G. W., Huang X.-P., Vardy E., McCorvy J. D., Jiang Y., Chu M., Siu F. Y., Liu W., Xu H. E., Cherezov V., Roth B. L., Stevens R. C. (2013) Structural features for functional selectivity at serotonin receptors. Science 340, 615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chien E. Y., Liu W., Zhao Q., Katritch V., Han G. W., Hanson M. A., Shi L., Newman A. H., Javitch J. A., Cherezov V., Stevens R. C. (2010) Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang C., Jiang Y., Ma J., Wu H., Wacker D., Katritch V., Han G. W., Liu W., Huang X.-P., Vardy E., McCorvy J. D., Gao X., Zhou X. E., Melcher K., Zhang C., Bai F., Yang H., Yang L., Jiang H., Roth B. L., Cherezov V., Stevens R. C., Xu H. E. (2013) Structural basis for molecular recognition at serotonin receptors. Science 340, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu J. J., Horst R., Katritch V., Stevens R. C., Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nygaard R., Zou Y., Dror R. O., Mildorf T. J., Arlow D. H., Manglik A., Pan A. C., Liu C. W., Fung J. J., Bokoch M. P., Thian F. S., Kobilka T. S., Shaw D. E., Mueller L., Prosser R. S., Kobilka B. K. (2013) The dynamic process of β2-adrenergic receptor activation. Cell 152, 532–542 [DOI] [PMC free article] [PubMed] [Google Scholar]