

Background: Bacterial pathogens use virulence proteins to inhibit the host innate immune system.
Results: The Escherichia coli O157:H7 NleH1 protein interacts with the host CRKL protein.
Conclusion: CRKL may recruit NleH1 to a host kinase on which NleH1 performs its inhibitory function.
Significance: These data clarify a mechanism by which E. coli inhibits innate immunity.
Keywords: Escherichia coli, Innate Immunity, NF-κB, Type III Secretion System, Virulence factors, CRKL, IKKβ, NleH1
Abstract
Enterohemorrhagic Escherichia coli and other attaching/effacing bacterial pathogens cause diarrhea in humans. These pathogens use a type III secretion system to inject virulence proteins (effectors) into host cells, some of which inhibit the innate immune system. The enterohemorrhagic E. coli NleH1 effector prevents the nuclear translocation of RPS3 (ribosomal protein S3) to inhibit its participation as a nuclear “specifier” of NF-κB binding to target gene promoters. NleH1 binds to RPS3 and inhibits its phosphorylation on Ser-209 by IκB kinase-β (IKKβ). However, the precise mechanism of this inhibition is unclear. NleH1 possesses a Ser/Thr protein kinase activity that is essential both for its ability to inhibit the RPS3/NF-κB pathway and for full virulence of the attaching/effacing mouse pathogen Citrobacter rodentium. However, neither RPS3 nor IKKβ is a substrate of NleH1 kinase activity. We therefore screened ∼9,000 human proteins to identify NleH1 kinase substrates and identified CRKL (v-Crk sarcoma virus CT10 oncogene-like protein), a substrate of the BCR/ABL kinase. Knockdown of CRKL abundance prevented NleH1 from inhibiting RPS3 nuclear translocation and NF-κB activity. CRKL residues Tyr-198 and Tyr-207 were required for interaction with NleH1. Lys-159, the kinase-active site of NleH1, was necessary for its interaction with CRKL. We also identified CRKL as an IKKβ interaction partner, mediated by CRKL Tyr-198. We propose that the CRKL interaction with IKKβ recruits NleH1 to the IKKβ complex, where NleH1 then inhibits the RPS3/NF-κB pathway.
Introduction
Many Gram-negative bacterial pathogens inject virulence proteins (effectors) into host cells through a type III secretion system (T3SS)2 (1). These effectors are thought to be critical for bacterial pathogenesis and transmission between hosts. Enterohemorrhagic Escherichia coli (EHEC) causes hemorrhagic colitis in humans and is the leading infectious cause of pediatric renal failure (2). This E. coli virotype is transmitted to humans through contaminated meat, water, and vegetables. EHEC encodes numerous T3SS effectors, the presence of which correlates with the ability of strains to cause severe disease and outbreaks of disease in humans (3).
A subset of these effectors function as inhibitors of the innate immune system of intestinal epithelial cells (4–7). For instance, NleB disrupts the recruitment of GAPDH (8) and TRADD (TNF receptor-associated death domain protein) (9) to TRAF2 (TNF receptor-associated factor 2) (8, 9). NleC is a zinc metalloprotease that cleaves the NF-κB p65 subunit to block IL-8 production during infection (10–13). NleD cleaves JNK to inhibit AP-1 pathway activation (10). NleE methylates TAB2/3 to inhibit NF-κB activity in response to TNF and IL-1β (5, 6, 14). In addition to its role in EHEC adhesion and “pedestal” formation, Tir (translocated intimin receptor) also inhibits NF-κB activation in response to TNF stimulation (7).
NF-κB is sequestered in the cytoplasm by inhibitory IκB proteins that mask NF-κB nuclear localization signals (15). Pathogen-associated molecular pattern recognition by Toll-like receptors activates IκB kinase-β (IKKβ), leading to phosphorylation of the IκBs, followed by their ubiquitination and degradation by the 26 S proteasome. After NF-κB translocation to the nucleus, this transcription factor binds κB sites within target gene promoters and regulates transcription by recruiting co-activators/co-repressors (16). RPS3 (ribosomal protein S3) has been recently implicated in host-pathogen interactions (17). After its phosphorylation on Ser-209 by IKKβ (18), RPS3 translocates to the nucleus and guides NF-κB to specific κB sites by increasing the affinity of the NF-κB p65 subunit for a subset of target gene promoters (16).
The NleH effectors are conserved among the attaching/effacing (A/E) pathogens EHEC and enteropathogenic E. coli and the mouse pathogen Citrobacter rodentium. EHEC encodes two forms of NleH, NleH1 and NleH2 (19), whereas C. rodentium encodes only one ortholog of NleH, which functions similarly to EHEC NleH1 (19, 20). In addition to binding to the Bax inhibitor-1 protein to block apoptosis during enteropathogenic E. coli infection (21, 22), NleH1 also binds to RPS3 and prevents its nuclear translocation by inhibiting IKKβ-mediated phosphorylation of RPS3 Ser-209 (18). NleH1 possesses a Ser/Thr protein kinase activity that is essential both for its ability to inhibit the RPS3/NF-κB pathway and for full virulence of C. rodentium (23). However, neither RPS3 nor IKKβ is a substrate of NleH1 kinase activity.
Here, we identified CRKL (v-Crk sarcoma virus CT10 oncogene-like protein) as a target of the NleH1 kinase. We determined both that CRKL interacts with IKKβ and that CRKL knockdown prevents NleH1 from inhibiting RPS3 nuclear translocation and NF-κB activity. We propose that the CRKL interaction with IKKβ recruits NleH1 to the IKKβ complex, where NleH1 then inhibits the RPS3/NF-κB pathway.
EXPERIMENTAL PROCEDURES
Plasmids, Chemicals, and Antibodies
The strains and plasmids used in this study are described in Table 1. All chemicals and antibodies were used according to the manufacturers' recommendations. Antibodies were obtained from the following sources: anti-poly(ADP-ribose) polymerase, BD Biosciences; anti-RPS3, Proteintech Group; anti-CRKL, Santa Cruz Biotechnology; and anti-β-actin, anti-FLAG, anti-HA, and anti-α-tubulin, Sigma. CRKL was amplified from HEK293T RNA using an RNeasy mini kit (Qiagen) and a ProtoScript II first strand cDNA synthesis kit (New England Biolabs), and the CRKL open reading frame was generated by PCR. To produce the CRKL(Y198F), CRKL(Y207F), and CRKL(Y198F,Y207F) mutants, p3×FLAG-CRKL was used as a PCR template, and a two-step PCR was used to generate appropriate PCR products. All mutants were verified by DNA sequencing.
TABLE 1.
Strains and plasmids used in this study
| Strain/plasmid | Description | Source/Ref. |
|---|---|---|
| Strains | ||
| E. coli BL21(DE3) | E. coli F− ompT hsdSB (rB−mB−) gal dcm (DE3) | Novagen |
| BL21(DE3)/CRKL-pET28a | His-CRKL | This study |
| BL21(DE3)/NleH1-pET28a | His-EHEC NleH1 | Ref. 4 |
| BL21(DE3)/NleH1(K159A)-pET28a | His-EHEC NleH1 (K159A) | Ref. 4 |
| BL21(DE3)/NleH1-pET42a | GST-EHEC NleH1 | Ref. 4 |
| Plasmids | ||
| κB(5×)-luciferase | Firefly luciferase driven by RPS3/κB site | Promega |
| pTKRL-luciferase | Renilla luciferase | Promega |
| pET28a | Bacterial hexahistidine fusion expression | Novagen |
| CRKL-pET28a | His-CRKL | This study |
| NleH1-pET28a | His-EHEC NleH1 | Ref. 4 |
| NleH1(K159A)-pET28a | His-EHEC NleH1 (K159A) | Ref. 4 |
| pET42a | Bacterial GST fusion expression | Novagen |
| NleH1-pET42a | GST-EHEC NleH1 | Ref. 4 |
| HA | HA fusion expression | Clontech |
| NleC-HA | HA fused to E. coli EDL933 NleC | This study |
| NleE-HA | HA fused to E. coli EDL933 NleE | This study |
| NleH1-HA | HA fused to E. coli EDL933 NleH1 | Ref. 4 |
| NleH1(K159A)-HA | HA fused to E. coli EDL933 NleH1 (K159A) | Ref. 4 |
| CRKL-HA | HA fused to CRKL | This study |
| 3×FLAG | FLAG expression | Sigma |
| 3×FLAG-CRKL | FLAG-CRKL | This study |
| 3×FLAG-CRKL(Y198F) | FLAG-CRKL (Y198F) | This study |
| 3×FLAG-CRKL(Y207F) | FLAG-CRKL (Y207F) | This study |
| 3×FLAG-CRKL(Y198F,Y207F) | FLAG-CRKL (Y198F,Y207F) | This study |
| 3×FLAG-RPS3 | FLAG-RPS3 | Ref. 16 |
| 3×FLAG-IKKβ | FLAG-IKKβ | Ref. 18 |
| HA-IKKβ | HA-IKKβ | This study |
| 3×FLAG-IKKβ(SSAA) | FLAG-IKKβ(SSAA) | Ref. 18 |
Cell Culture and Transient DNA Transfection
HeLa and HEK293T cells were maintained in DMEM. HCT-8 cells were maintained in RPMI 1640 medium. Media were supplemented with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate and with 10% FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2. For immunoblot analysis, cells were seeded into 6-well plates or 10-cm diameter dishes, and DNA was transfected into subconfluent cells using Lipofectamine 2000 reagent (Invitrogen) or PolyJet reagent (SignaGen Laboratories). For luciferase reporter assays, HEK293T and HCT-8 cells were seeded into 24-well plates and transfected using PolyJet or Lipofectamine 2000, respectively.
siRNA Transfection
Two different siRNAs targeting CRKL, as well as a negative control siRNA, were obtained from OriGene. Cells were seeded into 6-well plates and transfected with 30 nm siRNA (final concentration) using Lipofectamine 2000 reagent.
Protein Purification
CRKL was cloned into pET28a. WT NleH1 and NleH1(K159A) were cloned into pET28a and pET42a and expressed in E. coli BL21(DE3) cells. Bacterial cultures were grown to A600 = 0.5, and isopropyl β-d-thiogalactopyranoside was added to a final concentration of 1 mm. After 2 h of additional growth, cells were pelleted and lysed in either His lysis buffer (5 mm imidazole, 0.5 m NaCl, and 20 mm Tris-HCl, pH 7.9) or GST BugBuster protein extraction reagent (Novagen). After sonication and centrifugation, the supernatants were applied to either nickel-nitrilotriacetic acid-agarose (Qiagen) or GST bead slurries (Novagen) and incubated overnight at 4 °C. Slurries were washed several times, and proteins were eluted and analyzed by 12% SDS-PAGE.
Kinase Substrate Identification Assays
Recombinant NleH1 (5 nm) was supplied to Invitrogen for use in ProtoArray® kinase substrate identification assays and incubated in 100 mm MOPS, pH 7.2, 100 mm NaCl, 10 mg/ml BSA, 10 mm MnCl2, 0.1% Nonidet P-40, and 1 mm DTT supplemented with [γ-33P]ATP. Buffer containing [γ-33P]ATP but lacking NleH1 was used as a negative control. These solutions (120 μl) were applied to microarray slides that had been preblocked in PBS and 1% BSA. Arrays were covered with a glass coverslip and incubated at 30 °C for 1 h. After incubation, arrays were transferred to 50-ml conical tubes and washed four times with 40 ml of 0.5% SDS in water. Arrays were dried by placing them in a slide holder and spinning them in a tabletop centrifuge equipped with a microplate rotor at 1,000 rpm for 2 min. Arrays were placed in an x-ray film cassette and covered with clear plastic wrap and a phosphorimaging screen. Exposure of the arrays to the phosphorimaging screen was carried out for 20 h prior to scanning on a Cyclone PhosphorImager at a resolution of 600 dots/inch. The TIFF file produced from the PhosphorImager was processed using Adobe Photoshop. GenePix 6 software was used to overlay the mapping of human proteins in the array list file on each array image with a fixed feature size of 200 μm (diameter). Pixel intensities for each spot on the array were determined from the software after incorporating nearest neighbor analysis, background correction, and Z-score transformation.
In Vitro Kinase Assay
His-CRKL (100 ng/μl) was incubated with 100 ng/μl His-NleH1 (WT or K159A) in the presence of 0.5 μCi of [γ-32P]ATP (GE Healthcare) in kinase reaction buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 100 mm NaCl, 1 mm DTT, and 1 mm ATP) at 30 °C for 1 h. Reactions were terminated by adding 5× SDS sample buffer and boiled for 5 min. Samples were analyzed by SDS-PAGE and autoradiography.
Nuclear Fractionation
Cytosolic and nuclear protein extracts were obtained as described previously (4). Briefly, HeLa or HEK293T cells were transfected with the VN-HA or NleH1-HA plasmid and with CRKL siRNA. After 48 h, cells were stimulated with TNF (50 ng/ml, 30 min) and harvested. Cells were resuspended for 10 min on ice in 10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT, and 0.05% (v/v) Nonidet P-40. Lysates were centrifuged at 900 × g for 10 min at 4 °C, and supernatants were collected as cytosolic fractions. The pellets were resuspended in 5 mm HEPES, pH 7.9, 1.5 mm MgCl2, 300 mm NaCl, 0.2 mm EDTA, 0.5 mm DTT, and 26% (v/v) glycerol; homogenized; and incubated on ice for 30 min. Supernatants were collected as nuclear fractions after centrifugation at 22,000× g for 20 min at 4 °C. Data were analyzed by Western blotting for nuclear RPS3. Poly(ADP-ribose) polymerase and actin were used to normalize the protein concentrations of nuclear and cytoplasmic fractions, respectively.
Luciferase Reporter Assays
HEK293T and HCT-8 cells were cotransfected with a firefly luciferase construct driven by a consensus κB site and the Renilla luciferase pTKRL plasmid (Promega) at a ratio of 10:1, together with the VN-HA, NleC-HA, NleE-HA, and NleH1-HA expression plasmids and siRNAs. Transfected cells were cultured for 48 h and then stimulated with TNF (50 ng/ml, 30 min). Cells were lysed with passive lysis buffer, and lysates were analyzed using the Dual-Luciferase kit (Promega), with firefly fluorescence units normalized to Renilla fluorescence units. Luciferase reporter assays were performed in triplicate with at least three independently transfected cell populations.
Co-immunoprecipitation Assay
Transfected cells were scraped into PBS, pooled, centrifuged at 16,200 × g for 5 min, resuspended in PBS, and recentrifuged. Supernatants were removed, and cells were lysed in 20 mm Tris-HCl, pH 8.0, 2 mm EDTA, 137 mm NaCl, 1% (v/v) Nonidet P-40, and 10% (v/v) glycerol supplemented with Complete protease inhibitor mixture (Thermo Scientific). Samples were incubated on ice for 30 min, and cell lysates were collected by centrifugation at 7,800 × g for 10 min at 4 °C. Protein G-Dynabeads (Invitrogen) were first incubated with 2 μg of the appropriate antibody for 30 min at room temperature. After washing, ∼300 μg of protein from cell lysates was added to antibody-fused protein G-Dynabeads and adsorbed overnight at 4 °C. The mixture was pelleted and washed several times with cold PBS. The beads were resuspended in 5× SDS sample buffer, boiled for 5 min, and analyzed by immunoblotting.
Immunoblotting
Cells were washed with PBS, and cell extracts were prepared by adding radioimmune precipitation assay buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P-40, and Complete protease inhibitor mixture), incubating on ice for 30 min, and centrifuging. Equal amounts of protein from the supernatants were separated by SDS-PAGE, transferred to nitrocellulose membranes, blocked in Odyssey blocking buffer (LI-COR) for 1 h at room temperature, and probed overnight with the appropriate primary antibodies. After washing with PBS, the membranes were incubated with IRDye 680RD or 800CW secondary antibody (LI-COR) for 1 h. Blots were imaged using an Odyssey infrared imaging system (LI-COR).
RESULTS
NleH1 Phosphorylates CRKL in Vitro and Co-immunoprecipitates with CRKL in Mammalian Cell Culture
The T3SS effector protein NleH1, encoded by the A/E bacterial pathogens E. coli and C. rodentium, inhibits the phosphorylation of the NF-κB subunit RPS3 by IKKβ, thus limiting host NF-κB pathway activation and promoting bacterial colonization (4, 18). NleH1 is a Ser/Thr kinase, and Lys-159 (in E. coli O157:H7 EDL933) is critical for its kinase activity. However, neither RPS3 nor IKKβ is a substrate of NleH1 kinase activity (18).
To identify NleH1 kinase substrates and their potential contribution to NleH1-mediated inhibition of the NF-κB pathway, we used an in vitro kinase array (Invitrogen). Incubating purified recombinant NleH1 with an array of ∼9,000 human proteins allowed us to identify three candidate substrates (Fig. 1A): EPS8L2 (EGF receptor kinase pathway substrate 8-like protein 2), MAPRE1 (microtubule-associated protein RP/EB family member 1), and CRKL. EPS8L2 is responsible for functional redundancy in the receptor tyrosine kinase-activated signaling pathway, leading to actin remodeling (24). MAPRE1 functions in microtubule polymerization and spindle function by stabilizing microtubules and anchoring them at centrosomes (25). CRKL contains an SH2 domain and two SH3 domains and mediates the transduction of intracellular signals (26). CRKL is tyrosine-phosphorylated and interacts directly with the transforming protein BCR/ABL (27). Among these three candidate substrates of NleH1, we confirmed the specific phosphorylation of CRKL by NleH1 using purified recombinant proteins. WT NleH1, but not NleH1(K159A), phosphorylated CRKL in vitro (Fig. 1B). We failed to confirm the interactions between either EPS8L2 or MAPRE1 and NleH1 (data not shown). These potential kinase substrates of NleH1 were therefore not studied further.
FIGURE 1.
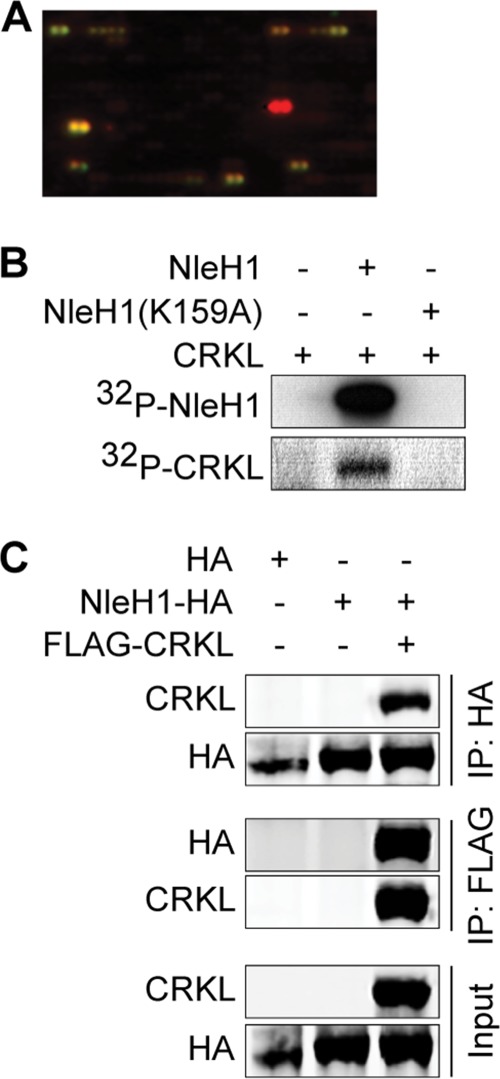
NleH1 phosphorylates CRKL in vitro and co-immunoprecipitates with CRKL from mammalian cells. A, identification of CRKL (yellow spot) as a substrate of NleH1 kinase activity using a kinase substrate array (Invitrogen). B, recombinant NleH1, but not NleH1(K159A), phosphorylated recombinant CRKL in vitro. C, HEK293T cells were cotransfected with FLAG-CRKL and NleH1-HA. After 48 h, cell lysates were immunoprecipitated (IP) with either anti-HA or anti-FLAG antibody, followed by immunoblotting with anti-FLAG or anti-HA antibody. The protein expression levels of NleH1 and CRKL in cell lysates are indicated in the input fraction.
To determine whether NleH1 interacts with CRKL in mammalian cells, we performed co-immunoprecipitation experiments. After transfecting both NleH1-HA and FLAG-CRKL, cell lysates were immunoprecipitated with either anti-HA or anti-FLAG antibody and subsequently immunoblotted. NleH1-HA, but not an HA epitope control, interacted with FLAG-CRKL under both co-immunoprecipitation conditions (Fig. 1C).
CRKL Is Required for NleH1 to Inhibit RPS3 Nuclear Translocation and NF-κB Activity
To determine whether the interaction between NleH1 and CRKL contributes to the ability of NleH1 to inhibit RPS3 nuclear translocation and its role in NF-κB-dependent transcription, we first established siRNA knockdown conditions to reduce the steady-state levels of CRKL in HEK293T cells (Fig. 2A). CRKL knockdown cells were cotransfected with either NleH1-HA or an HA epitope control plasmid. Cells were treated with TNF for 30 min (to induce RPS3 nuclear translocation) before harvesting cell lysates and fractionating them to separate cytoplasmic from nuclear contents. RPS3 nuclear translocation induced by TNF was then assessed by immunoblotting.
FIGURE 2.

CRKL is required for NleH1 to inhibit RPS3 nuclear translocation and NF-κB activity. A, CRKL knockdown with siRNAs. B, RPS3 nuclear translocation assay. HEK293T cells were cotransfected with NleH1-HA or an HA epitope control and with two independent CRKL siRNAs (siCRKL) or with a nonspecific siRNA (ns siRNA). After 48 h, cells were treated with TNF (50 ng/ml, 30 min), separated into nuclear and cytoplasmic fractions, and subjected to immunoblotting. PARP, poly(ADP-ribose) polymerase. C, quantification (n = 3) of the -fold change in nuclear RPS3 abundance after normalization to nuclear poly(ADP-ribose) polymerase abundance. Data are shown as means ± S.E. *, significant differences between the indicated pairwise comparisons (p < 0.05, t test). ns, not significant. D, NF-κB activity. HEK293T cells were cotransfected with NleH1, CRKL siRNA, a firefly luciferase construct driven by a consensus κB site, and a Renilla luciferase plasmid. Cells were stimulated with TNF (50 ng/ml, 30 min) after 48 h of transfection. NF-κB activity was determined by luciferase reporter assays. Data are shown as means ± S.E. of luciferase activity from three independent assays. *, significant differences between the indicated pairwise comparisons (p < 0.05, t test). The expression levels of CRKL, NleH-HA, and the HA control were determined by immunoblotting with anti-CRKL and anti-HA antibodies. E, NF-κB activity in HCT-8 cells. HCT-8 cells were cotransfected with CRKL siRNA and luciferase reporter plasmids in the presence or absence of NleC, NleE, or NleH1 and then treated with TNF (50 ng/ml, 30 min) 48 h after transfection. Data are shown as the means ± S.E. of luciferase activity from three independent assays. *, significant differences from unstimulated cells (p < 0.05, t test).
Treating WT cells with TNF induced an ∼9-fold increase in RPS3 translocation to the nucleus (Fig. 2, B and C). Transfecting NleH1-HA significantly inhibited RPS3 nuclear translocation. By contrast, in CRKL knockdown cells, NleH1-HA failed to inhibit RPS3 nuclear translocation. Knockdown of CRKL did not alter the ability of TNF to induce RPS3 nuclear translocation in cells lacking NleH1 (Fig. 2, B and C). Thus, WT levels of CRKL appear to be required for NleH1 to exert its inhibitory activity.
As RPS3 nuclear translocation is essential to NF-κB-dependent gene transcription, we subsequently assessed the impact of CRKL knockdown and NleH1-HA transfection on NF-κB-dependent luciferase reporter activity. Luciferase activity in WT cells transfected with NleH1-HA was significantly inhibited (Fig. 2D). By contrast, in cells with reduced CRKL abundance, NleH1-HA failed to inhibit NF-κB luciferase activity (Fig. 2D).
To determine whether CRKL knockdown would also affect the ability of NleH1 to inhibit NF-κB activation in intestinal epithelial cells, we cotransfected HCT-8 cells with CRKL siRNA and NleH1-HA and luciferase reporter plasmids. Similar to the results obtained using HEK293T cells, knockdown of CRKL in HCT-8 cells inhibited the ability of NleH1 to suppress NF-κB activation (Fig. 2E).
We also assessed whether CRKL knockdown would affect the ability of other E. coli effectors to inhibit NF-κB activation. However, neither NleC (10–13) nor NleE (5, 6, 14) was prevented from inhibiting NF-κB activation in CRKL knockdown cells (Fig. 2E), suggesting that the impact of CRKL knockdown is specific to NleH1.
Characterization of Amino Acids Mediating the NleH1-CRKL Interaction
Tyrosine phosphorylation of CRKL by BCR/ABL plays an important role in CRKL activation and signal transduction. CRKL residues Tyr-198 and Tyr-207 are phosphorylated in response to BCR/ABL transforming activity (27). To determine whether CRKL Tyr-198 and/or Tyr-207 is required for interaction with NleH1, we mutated these residues either singly or in tandem by site-directed mutagenesis. We then performed cotransfection and co-immunoprecipitation experiments. Mutating both Tyr-198 and Tyr-207 to phenylalanine residues abrogated the interaction between NleH1 and CRKL (Fig. 3A). By contrast, the single Y198F or Y207F mutation did not inhibit the NleH1-CRKL interaction.
FIGURE 3.

Characterization of amino acids mediating the NleH1-CRKL interaction. A, co-immunoprecipitation of CRKL(Y198F), CRKL(Y207F), and CRKL(Y198F,Y207F) after their cotransfection with NleH1. B, co-immunoprecipitation of WT and NleH1(K159A) with CRKL after cotransfection. IP, immunoprecipitation.
NleH1 Lys-159 is required for NleH1 kinase activity (Fig. 1B) (4, 18, 23). To determine whether NleH1 Lys-159 governs the interaction between NleH1 and CRKL, we compared the interaction of WT NleH1 versus NleH1(K159A) with CRKL. After performing cotransfection and co-immunoprecipitation assays, we determined that NleH1 Lys-159 is required for the NleH1-CRKL interaction (Fig. 3B).
CRKL Interacts with IKKβ, but Not with RPS3
IKKβ-mediated phosphorylation of RPS3 Ser-209 governs RPS3 nuclear import and subsequent NF-κB pathway activation (18). NleH1 kinase activity is required to inhibit IKKβ-mediated phosphorylation of RPS3 Ser-209, although NleH1 does not phosphorylate IKKβ (18). We considered whether CRKL might interact with IKKβ and/or RPS3 and thus recruit NleH1 during infection to inhibit the NF-κB pathway. To test this idea, we cotransfected HEK293T cells with CRKL-HA and FLAG-RPS3 or FLAG-IKKβ. After immunoprecipitating cell lysates with anti-FLAG antibody, we observed that CRKL interacted with IKKβ, but not with RPS3 (Fig. 4A). A reciprocal immunoprecipitation experiment with anti-HA antibody against CRKL also confirmed this result (Fig. 4B).
FIGURE 4.

CRKL interacts with IKKβ, but not with RPS3. A, HEK293T cells were cotransfected with CRKL-HA and FLAG-RPS3 or FLAG-IKKβ expression plasmid. Cells were harvested after 48 h of transfection. Samples were immunoprecipitated (IP) with anti-FLAG antibody and immunoblotted with anti-HA and anti-FLAG antibodies. B, co-immunoprecipitation using anti-HA antibody. C, HEK293T cells were cotransfected with HA-IKKβ and FLAG-CRKL (WT, Y198F, Y207F, or Y198F,Y207F) expression plasmids. Cells were harvested after 48 h of transfection and immunoprecipitated with either anti-HA or anti-FLAG antibody, followed by immunoblotting. D, HEK293T cells were cotransfected with CRKL-HA and FLAG-IKKβ (WT or kinase-dead SSAA mutant) expression plasmids. After 48 h, cell lysates were immunoprecipitated with either anti-HA or anti-FLAG antibody, followed by immunoblotting.
To assess the role of the CRKL Tyr-198 and Tyr-207 residues in mediating interaction with IKKβ, we performed cotransfection and reciprocal co-immunoprecipitation experiments with CRKL mutants. CRKL(Y198F) and CRKL(Y198F,Y207F) did not interact with IKKβ, whereas CRKL(Y207F) retained the ability to interact with IKKβ (Fig. 4C).
We also tested whether IKKβ kinase activity is required for IKKβ interaction with CRKL. To do this, we transfected a “kinase-dead” form of IKKβ (IKKβ(SSAA)) and performed co-immunoprecipitation experiments. CRKL interacted with both WT IKKβ and IKKβ(SSAA) (Fig. 4D), suggesting that IKKβ kinase activity is not required for the CRKL-IKKβ interaction.
DISCUSSION
Bacterial infection triggers an inflammatory response via host recognition of pathogen-associated molecular patterns, resulting in activation of the IKK complex. IKK phosphorylates IκBα, subsequently promoting its ubiquitination and degradation, releasing NF-κB for nuclear translocation. RPS3 is a non-Rel NF-κB subunit that significantly enhances p65 binding to DNA (16). The phosphorylation of RPS3 by IKKβ on Ser-209 is a critical determinant for the nuclear translocation of RPS3 before it participates in NF-κB-dependent transcriptional regulation (18). Many bacterial pathogens use a T3SS to inject effector proteins into host cells to subvert the innate immune response by inhibiting NF-κB activation (28).
The E. coli effector NleH1 is a Ser/Thr protein kinase that depends on Lys-159 for kinase activity (4). NleH1 inhibits the phosphorylation of RPS3 Ser-209 by IKKβ (18), but the mechanism is not understood. Although NleH1 kinase activity is required for its inhibitory activity, NleH1 does not directly phosphorylate either IKKβ or RPS3 (18). We speculated that NleH1 might phosphorylate one or more human proteins. We screened ∼9,000 human proteins to identify NleH1 kinase substrates and identified CRKL as the most promising candidate. We confirmed that recombinant CRKL is phosphorylated by WT NleH1, but not by NleH1(K159A). We also found that CRKL interacts with WT NleH1 but not with NleH1(K159A) in mammalian cells.
CRKL has been studied extensively for its role as a substrate of the BCR/ABL tyrosine kinase in chronic myelogenous leukemia (29). Other CRKL binding partners include the proto-oncoprotein CBL (30, 31), Cas (Crk-associated substrate), HEF1 (human enhancer of filamentation 1), and paxillin (32–34). Although the role of CRKL in bacterial pathogenesis is unknown, data suggest that its homolog, Crk, influences bacterial adherence and internalization. Abl tyrosine kinases are required for Shigella flexneri infection through a mechanism linked to Rho GTPase activation (Rac and Cdc42) (35). CRKL may also contribute to viral pathogenesis, as the SH3 domain-binding motif of A/NS1 (influenza A virus non-structural protein 1) is essential for binding to CRKL and can suppress the anti-viral JNK-ATF2 pathway (36).
In this study, we identified a role for CRKL in E. coli infection. We found that CRKL interacts with IKKβ and with NleH1. We determined that both Tyr-198 and Tyr-207 of CRKL are required for its interaction with NleH1 and that Tyr-198 is required for its interaction with IKKβ. CRKL is required for NleH1 to suppress the RPS3/NF-κB pathway, as CRKL knockdown significantly reduced the ability of NleH1 to inhibit RPS3 nuclear translocation and NF-κB activity normally stimulated by TNF. We propose that, during A/E pathogen infection, NleH1 is injected into host cells through the T3SS and then interacts with CRKL (Fig. 5). A model consistent with our data is that, in the absence of CRKL, NleH1 is not recruited to the IKKβ-RPS3 complex and fails to block RPS3 nuclear translocation. However, CRKL knockdown itself does not affect IKKβ, as RPS3 nuclear translocation and NF-κB activity were unchanged. This suggests that CRKL serves, in the context of E. coli infection, as an adapter protein to recruit NleH1 but does not appear to have a native role in regulating IKKβ activity.
FIGURE 5.
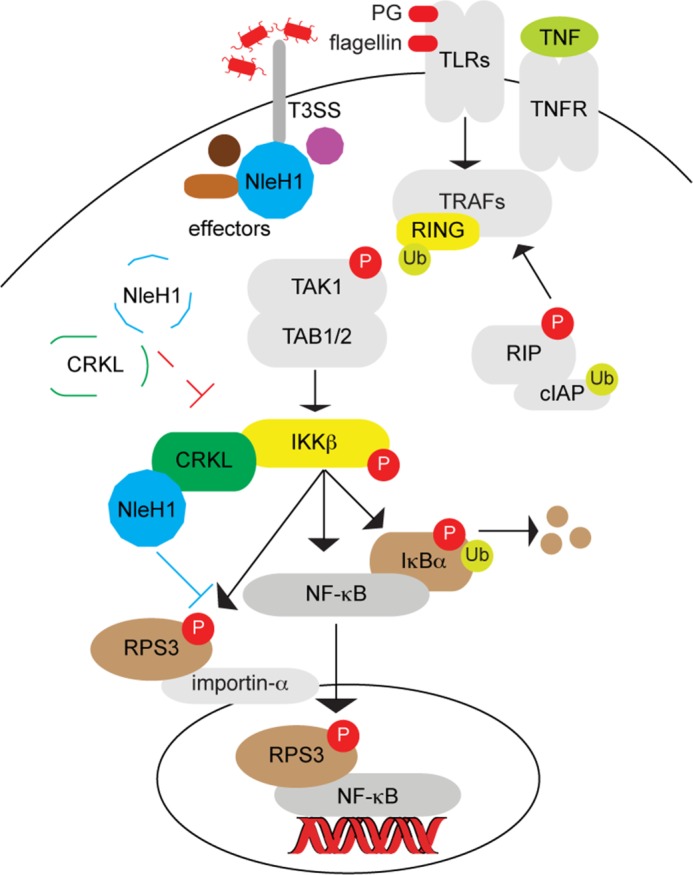
Model illustrating the potential role of CRKL in recruiting NleH1 to inhibit RPS3/NF-κB signaling during A/E pathogen infection. During A/E pathogen infection, NleH1 is injected into host cells through the T3SS and then interacts with CRKL. CRKL interacts with IKKβ and may recruit NleH1 to the RPS3-NF-κB complex, where NleH1 can then inhibit RPS3 phosphorylation. In the absence of CRKL (indicated by dashed lines), NleH1 is not recruited to the IKKβ-RPS3 complex and fails to block RPS3 nuclear translocation. PG, peptidoglycan; TLRs, Toll-like receptors; TNFR, TNF receptor; TRAF, TNF receptor-associated factor; Ub, ubiquitin; RIP, receptor-interacting protein; cIAP, cellular inhibitor of apoptosis protein.
The limitations of our study are that we did not identify the residue(s) on CRKL that are phosphorylated by NleH1 and did not determine the functional significance of such phosphorylation. Although we observed that NleH1 phosphorylates CRKL in vitro, it is possible that NleH1 is not an especially efficient kinase in vivo. Thus, the significance of the NleH1-CRKL interaction may be limited largely to the apparent requirement of NleH1 in CRKL expression for its targeting of the IKKβ-RPS3 complex.
This work was supported, in whole or in part, by National Institutes of Health Grant AI099002 from NIAID.
- T3SS
- type III secretion system
- EHEC
- enterohemorrhagic E. coli
- IKKβ
- IκB kinase-β
- A/E
- attaching/effacing.
REFERENCES
- 1. Cornelis G. R. (2010) The type III secretion injectisome, a complex nanomachine for intracellular 'toxin' delivery. Biol. Chem. 391, 745–751 [DOI] [PubMed] [Google Scholar]
- 2. Tzipori S., Sheoran A., Akiyoshi D., Donohue-Rolfe A., Trachtman H. (2004) Antibody therapy in the management of shiga toxin-induced hemolytic uremic syndrome. Clin. Microbiol. Rev. 17, 926–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coombes B. K., Wickham M. E., Mascarenhas M., Gruenheid S., Finlay B. B., Karmali M. A. (2008) Molecular analysis as an aid to assess the public health risk of non-O157 shiga toxin-producing Escherichia coli strains. Appl. Environ. Microbiol. 74, 2153–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gao X., Wan F., Mateo K., Callegari E., Wang D., Deng W., Puente J., Li F., Chaussee M. S., Finlay B. B., Lenardo M. J., Hardwidge P. R. (2009) Bacterial effector binding to ribosomal protein S3 subverts NF-κB function. PLoS Pathog. 5, e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nadler C., Baruch K., Kobi S., Mills E., Haviv G., Farago M., Alkalay I., Bartfeld S., Meyer T. F., Ben-Neriah Y., Rosenshine I. (2010) The type III secretion effector NleE inhibits NF-κB activation. PLoS Pathog. 6, e1000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Newton H. J., Pearson J. S., Badea L., Kelly M., Lucas M., Holloway G., Wagstaff K. M., Dunstone M. A., Sloan J., Whisstock J. C., Kaper J. B., Robins-Browne R. M., Jans D. A., Frankel G., Phillips A. D., Coulson B. S., Hartland E. L. (2010) The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-κB p65. PLoS Pathog. 6, e1000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ruchaud-Sparagano M. H., Mühlen S., Dean P., Kenny B. (2011) The enteropathogenic E. coli (EPEC) Tir effector inhibits NF-κB activity by targeting TNFα receptor-associated factors. PLoS Pathog. 7, e1002414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao X., Wang X., Pham T. H., Feuerbacher L. A., Lubos M. L., Huang M., Olsen R., Mushegian A., Slawson C., Hardwidge P. R. (2013) NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-κB activation. Cell Host Microbe 13, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li S., Zhang L., Yao Q., Li L., Dong N., Rong J., Gao W., Ding X., Sun L., Chen X., Chen S., Shao F. (2013) Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501, 242–246 [DOI] [PubMed] [Google Scholar]
- 10. Baruch K., Gur-Arie L., Nadler C., Koby S., Yerushalmi G., Ben-Neriah Y., Yogev O., Shaulian E., Guttman C., Zarivach R., Rosenshine I. (2011) Metalloprotease type III effectors that specifically cleave JNK and NF-κB. EMBO J. 30, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mühlen S., Ruchaud-Sparagano M. H., Kenny B. (2011) Proteasome-independent degradation of canonical NFκB complex components by the NleC protein of pathogenic Escherichia coli. J. Biol. Chem. 286, 5100–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sham H. P., Shames S. R., Croxen M. A., Ma C., Chan J. M., Khan M. A., Wickham M. E., Deng W., Finlay B. B., Vallance B. A. (2011) Attaching and effacing bacterial effector NleC suppresses epithelial inflammatory responses by inhibiting NF-κB and p38 mitogen-activated protein kinase activation. Infect. Immun. 79, 3552–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yen H., Ooka T., Iguchi A., Hayashi T., Sugimoto N., Tobe T. (2010) NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog. 6, e1001231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vossenkämper A., Marchès O., Fairclough P. D., Warnes G., Stagg A. J., Lindsay J. O., Evans P. C., Luong le A., Croft N. M., Naik S., Frankel G., MacDonald T. T. (2010) Inhibition of NF-κB signaling in human dendritic cells by the enteropathogenic Escherichia coli effector protein NleE. J. Immunol. 185, 4118–4127 [DOI] [PubMed] [Google Scholar]
- 15. Häcker H., Karin M. (2006) Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13. [DOI] [PubMed] [Google Scholar]
- 16. Wan F., Anderson D. E., Barnitz R. A., Snow A., Bidere N., Zheng L., Hegde V., Lam L. T., Staudt L. M., Levens D., Deutsch W. A., Lenardo M. J. (2007) Ribosomal protein S3: a KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 131, 927–939 [DOI] [PubMed] [Google Scholar]
- 17. Gao X., Hardwidge P. R. (2011) Ribosomal protein S3: a multifunctional target of attaching/effacing bacterial pathogens. Front. Microbiol. 2, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wan F., Weaver A., Gao X., Bern M., Hardwidge P. R., Lenardo M. J. (2011) IKKβ phosphorylation regulates RPS3 nuclear translocation and NF-κB function during infection with Escherichia coli strain O157:H7. Nat. Immunol. 12, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. García-Angulo V. A., Deng W., Thomas N. A., Finlay B. B., Puente J. L. (2008) Regulation of expression and secretion of NleH, a new non-locus of enterocyte effacement-encoded effector in Citrobacter rodentium. J. Bacteriol. 190, 2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tobe T., Beatson S. A., Taniguchi H., Abe H., Bailey C. M., Fivian A., Younis R., Matthews S., Marches O., Frankel G., Hayashi T., Pallen M. J. (2006) An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc. Natl. Acad. Sci. U.S.A. 103, 14941–14946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hemrajani C., Berger C. N., Robinson K. S., Marchès O., Mousnier A., Frankel G. (2010) NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc. Natl. Acad. Sci. U.S.A 107, 3129–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson K. S., Mousnier A., Hemrajani C., Fairweather N., Berger C. N., Frankel G. (2010) The enteropathogenic Escherichia coli effector NleH inhibits apoptosis induced by Clostridium difficile toxin B. Microbiology 156, 1815–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pham T. H., Gao X., Tsai K., Olsen R., Wan F., Hardwidge P. R. (2012) Functional differences and interactions between the Escherichia coli type III secretion system effectors NleH1 and NleH2. Infect. Immun. 80, 2133–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Offenhäuser N., Borgonovo A., Disanza A., Romano P., Ponzanelli I., Iannolo G., Di Fiore P. P., Scita G. (2004) The Eps8 family of proteins links growth factor stimulation to actin reorganization generating functional redundancy in the Ras/Rac pathway. Mol. Biol. Cell 15, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Honnappa S., Gouveia S. M., Weisbrich A., Damberger F. F., Bhavesh N. S., Jawhari H., Grigoriev I., van Rijssel F. J., Buey R. M., Lawera A., Jelesarov I., Winkler F. K., Wüthrich K., Akhmanova A., Steinmetz M. O. (2009) An EB1-binding motif acts as a microtubule tip localization signal. Cell 138, 366–376 [DOI] [PubMed] [Google Scholar]
- 26. Koval A. P., Karas M., Zick Y., LeRoith D. (1998) Interplay of the proto-oncogene proteins CrkL and CrkII in insulin-like growth factor-I receptor-mediated signal transduction. J. Biol. Chem. 273, 14780–14787 [DOI] [PubMed] [Google Scholar]
- 27. de Jong R., ten Hoeve J., Heisterkamp N., Groffen J. (1997) Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene 14, 507–513 [DOI] [PubMed] [Google Scholar]
- 28. Raymond B., Young J. C., Pallett M., Endres R. G., Clements A., Frankel G. (2013) Subversion of trafficking, apoptosis, and innate immunity by type III secretion system effectors. Trends Microbiol. 21, 430–441 [DOI] [PubMed] [Google Scholar]
- 29. ten Hoeve J., Morris C., Heisterkamp N., Groffen J. (1993) Isolation and chromosomal localization of CRKL, a human crk-like gene. Oncogene 8, 2469–2474 [PubMed] [Google Scholar]
- 30. Barber D. L., Mason J. M., Fukazawa T., Reedquist K. A., Druker B. J., Band H., D'Andrea A. D. (1997) Erythropoietin and interleukin-3 activate tyrosine phosphorylation of CBL and association with CRK adaptor proteins. Blood 89, 3166–3174 [PubMed] [Google Scholar]
- 31. Sattler M., Salgia R., Shrikhande G., Verma S., Pisick E., Prasad K. V., Griffin J. D. (1997) Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing c-Kit, phosphatidylinositol 3-kinase, and p120CBL. J. Biol. Chem. 272, 10248–10253 [DOI] [PubMed] [Google Scholar]
- 32. Manié S. N., Beck A. R., Astier A., Law S. F., Canty T., Hirai H., Druker B. J., Avraham H., Haghayeghi N., Sattler M., Salgia R., Griffin J. D., Golemis E. A., Freedman A. S. (1997) Involvement of p130Cas) and p105HEF1, a novel Cas-like docking protein, in a cytoskeleton-dependent signaling pathway initiated by ligation of integrin or antigen receptor on human B cells. J. Biol. Chem. 272, 4230–4236 [DOI] [PubMed] [Google Scholar]
- 33. Sattler M., Salgia R. (1997) Activation of hematopoietic growth factor signal transduction pathways by the human oncogene BCR/ABL. Cytokine Growth Factor Rev. 8, 63–79 [DOI] [PubMed] [Google Scholar]
- 34. Sattler M., Salgia R., Shrikhande G., Verma S., Uemura N., Law S. F., Golemis E. A., Griffin J. D. (1997) Differential signaling after β1 integrin ligation is mediated through binding of CRKL to p120CBL and p110HEF1. J. Biol. Chem. 272, 14320–14326 [DOI] [PubMed] [Google Scholar]
- 35. Burton E. A., Plattner R., Pendergast A. M. (2003) Abl tyrosine kinases are required for infection by Shigella flexneri. EMBO J. 22, 5471–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hrincius E. R., Wixler V., Wolff T., Wagner R., Ludwig S., Ehrhardt C. (2010) CRK adaptor protein expression is required for efficient replication of avian influenza A viruses and controls JNK-mediated apoptotic responses. Cell. Microbiol. 12, 831–843 [DOI] [PubMed] [Google Scholar]