

Background: An RNAi screen identified UBE3C as a key player in degradation of a model unfolded protein.
Results: UBE3C knockdown results in incomplete degradation of relatively stable substrates.
Conclusion: UBE3C enhances proteasome processivity to prevent the accumulation of potentially harmful protein fragments.
Significance: This advances our understanding of proteasome processivity and the consequences of defects therein.
Keywords: E3 Ubiquitin Ligase, Proteasome, Protein Degradation, Protein Processing, Protein Stability
Abstract
To maintain protein homeostasis, cells must balance protein synthesis with protein degradation. Accumulation of misfolded or partially degraded proteins can lead to the formation of pathological protein aggregates. Here we report the use of destabilizing domains, proteins whose folding state can be reversibly tuned using a high affinity ligand, as model substrates to interrogate cellular protein quality control mechanisms in mammalian cells using a forward genetic screen. Upon knockdown of UBE3C, an E3 ubiquitin ligase, a reporter protein consisting of a destabilizing domain fused to GFP is degraded more slowly and incompletely by the proteasome. Partial proteolysis is also observed when UBE3C is present but cannot ubiquitinate substrates because its active site has been mutated, it is unable to bind to the proteasome, or the substrate lacks lysine residues. UBE3C knockdown also results in less substrate polyubiquitination. Finally, knockdown renders cells more susceptible to the Hsp90 inhibitor 17-AAG, suggesting that UBE3C protects against the harmful accumulation of protein fragments arising from incompletely degraded proteasome substrates.
Introduction
Modern understanding of the mechanisms that govern the transcription and translation of the genetic code have led to well developed models for protein synthesis. However, maintenance of protein homeostasis achieved through degradation of these proteins remains less thoroughly understood. Although we may understand how proteins are turned over in some individual cases, the general principles governing recognition and degradation of misfolded or unfolded proteins are not well characterized. Proteins can misfold for a number of reasons, including chemical or heat stress, transcription or translation errors, viral infection, and mutations in the encoding DNA. Such events commonly expose hydrophobic regions that are prone to aggregation (1). Regardless of the source of this insult, cells must contend with the resulting misfolded protein via degradation, sequestration, or secretion as aggregation of these proteins can impair cellular function. Protein aggregates have been implicated in many human pathologies including Huntington disease, Parkinson disease, and amyotrophic lateral sclerosis (2). Understanding the fundamental mechanisms of cellular protein quality control and degradation is central to basic biology and may provide insights into human disease.
The ubiquitin-proteasome system is one of the most well characterized pathways for protein degradation (3, 4). Protein substrates are typically acylated on lysine residues by the C terminus of the 8-kDa protein, ubiquitin (Ub).2 Successive Ub moieties can be linked to existing mono- or polyUb chains through one of the ubiquitin seven lysine residues or on its N terminus. Depending on the lysine residue used for linkages, the polyUb chain encodes different signals (5–8), with Lys-48-linked chains commonly associated with proteasomal targeting and degradation (9, 10). Substrates are tagged with Ub by E3 Ub ligases after a series of Ub transfer events from E1-activating to E2-conjugating enzymes. E3 ligases participate in the transfer of Ub to substrates either by facilitating ubiquitination by an associated E2 enzyme (RING finger family) or by accepting Ub from the E2 and transferring it to substrates directly (HECT family) (11, 12). Although this general mechanism has been well documented in the literature, the specific roles of the roughly 600 E3 ligases in the human proteome remain poorly annotated.
The proteasome is a multisubunit protease responsible for the degradation of proteins within the cytoplasm and nucleus (13, 14). Although it can independently degrade misfolded or oxidized proteins, most of the proteasome substrates are delivered through polyUb targeting. It is composed of a catalytic core (the 20 S proteasome) and one or two regulatory particles (the 19 S) that facilitate substrate unfolding, Ub recycling, and substrate trafficking. Although the combination of the 20 S and two 19 S particles, called the 26 S proteasome, is considered a “complete” proteasome unit, other accessory proteins are known to associate at sub-stoichiometric levels and assist in proteasome function (15–17). Proteasomes normally degrade proteins beginning from an unstructured initiation region in a processive manner in that substrates targeted for degradation are completely degraded. However, this is not always the case, as it has been reported that the stability of a substrate can influence whether it is completely degraded. Furthermore, the domain architecture of substrates can influence the nature of processivity, as a substrate with a particularly stable domain downstream of a degradation initiation region can “protect” less stable downstream domains that would be degraded were their positions to be swapped (18–20).
We have developed a class of chemical biological tools called destabilizing domains (DDs) that confer small molecule ligand-dependent stability to any fused protein of interest (21). In the absence of stabilizing ligand, the DDs become unfolded, ubiquitinated, and then rapidly processed by the proteasome in a processive manner that results in degradation of the entire fusion (22) (Fig. 1A). The addition (and/or subsequent removal) of different amounts of ligand allows DD levels to be reversibly tuned. Three such domains, based on human FK506-binding protein (FKBP), Escherichia coli dihydrofolate reductase (DHFR), and human estrogen receptor ligand binding domain have been developed to date (21, 23, 24).
FIGURE 1.

Identification and validation of UBE3C as important in DD degradation. A, DDs fused to GFP are stable in the presence of ligand but become unstable and ultimately degraded upon ligand withdrawal. B, an siRNA screen was conducted in cells expressing mCherry and a DD-GFP construct. Ligand withdrawal results in loss of GFP signal without loss of mCherry signal. Compromised DD degradation due to RNAi will result in slower loss of GFP fluorescence after washout without a corresponding loss of mCherry signal. C, all scores from the ligand washout screen are plotted in ascending order. D, raw flow cytometry data from the screen show GFP signal versus mCherry signal. Populations were treated with the indicated siRNA and incubated with stabilizing ligand unless otherwise indicated (-S1). Ligand was removed 0 or 3 h before analysis. E, cells were treated with siUBE3C or NTsi and analyzed by immunoblotting against UBE3C. F, wild-type or catalytically inactive (C1051A) UBE3C was expressed in DD-GFP HeLa cells. Cells were treated with siUBE3C or NTsi in the presence of stabilizing ligand and analyzed by flow cytometry at the indicated times after ligand removal. DD-GFP cells expressing no exogenous UBE3C were included as control. Signal is expressed as a percentage of starting GFP/mCherry fluorescence. G, codon-wobbled wild-type or catalytically inactive (C1051A) UBE3C was expressed in DD-GFP HeLa cells. Cells were treated with siUBE3C or NTsi and analyzed by immunoblotting against UBE3C.
Although these domains have been used in a broad range of contexts for the study of the function of specific proteins (25–31), we saw an opportunity to use the DDs themselves as model substrates for protein degradation. The use of small molecules gives us precise control over how and when our substrate unfolds without the need for other nonspecific perturbations such as heat shock, chemical stressors, or translation inhibition. In an effort to better understand the process of DD degradation from recognition of the unfolded state to ubiquitination to degradation, we conducted a focused siRNA screen including all putative members of the chaperone family, the ubiquitin conjugation pathway, and motor proteins. We found that depletion of the protein UBE3C significantly reduces the rate of DD degradation.
UBE3C is a 126-kDa member of the HECT family of E3 ubiquitin ligases. These ligases form a covalent thioester bond with Ub using a conserved cysteine residue in the HECT domain before transferring Ub to substrates. Prior studies reported that UBE3C associates with the proteasome and assembles Lys-29- and Lys-48-linked polyUb chains (32–34). The yeast ortholog, Hul5p, ubiquitinates substrates at the proteasome to mediate protein turnover (15, 16). The physiological relevance of this activity was demonstrated when deletion of Hul5 was found to compromise the recovery rate of yeast after heat shock (35). In both yeast and mammalian cells, previous work has shown that in the absence of Hul5p/UBE3C, the proteasome is less able to completely degrade particularly stable proteins, leaving behind a truncated product (36–38). Although it has been reported that UBE3C acts on client substrate proteins TIP120B, IRF3, and IRF7 (34, 39), we find that it also plays a more general role in protein homeostasis. Furthermore, a mutation in the HECT domain of UBE3C was recently identified in a study of autism spectrum disorder (ASD) mutations (40). We report that UBE3C contributes to proteasome processivity by ubiquitinating substrates that are difficult for the proteasome to process, facilitating their complete degradation. This activity is important for cell fitness under conditions of protein folding stress, and an attenuation of this activity may be pathologically relevant in ASD.
EXPERIMENTAL PROCEDURES
Cell Culture and Viral Transduction
HeLa S3 (ATCC CCL-2.2) and ΦNX-eco cells (Nolan lab, Stanford University) were cultured in growth media (DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mm glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin) (Invitrogen) at 37 °C and 5% CO2. Retrovirus was produced by transfecting ΦNX-eco cells with pBMN and pCL-ampho vectors at a 2:1 mass ratio using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Viral supernatants were harvested 48 h after transfection and concentrated 10-fold using Amicon Ultra centrifugal filter units (100 kDa molecular weight cutoff). HeLa S3 cells were infected at 37 °C overnight with concentrated viral supernatants containing 4 μg/ml Polybrene (Sigma). Infected cells were either sorted by FACS or selected using appropriate drug 48–72 h post-infection.
siRNA Reverse Transfection
siRNA was purchased as On-Target Plus SMARTpools from Dharmacon with the exception of siUBE3C, where use of the pool was replaced with one specific siRNA reagent after pool deconvolution (J-007183-07-0002). Dharmafect IV (Dharmacon) was added to Opti-MEM (Invitrogen) and incubated at room temperature for 5 min. An equal volume of Opti-MEM containing siRNA was mixed with the Dharmafect solution to give a siRNA concentration of 50 nm and incubated for 30 min at room temperature. This mixture was then mixed with an equal volume of growth media containing 80,000 cells/ml to achieve a final siRNA concentration of 25 nm, then plated. 96-, 24-, or 12-well plates contained 0.25, 1, or 2 μl of Dharmafect in a final well volume of 100, 500, or 1000 μl, respectively. Medium was changed 12–24 h after transfection.
Flow Cytometry
Cells were trypsinized and resuspended in culture media, then analyzed at the Stanford Shared FACS Facility using LSRII with 10,000 events represented. For cells in 96-well format, samples were brought to final volumes of 50 μl and transferred to 96-well U-bottom plates, then analyzed using the BD Biosciences High-Throughput Sampler. Typically a 10-μl sample volume was collected (representing 1000–5000 events). For cells in 24-well or larger format, samples were brought to final volumes of 200–300 μl and analyzed in FACS tubes.
siRNA Screen Scoring
The GFP/mCherry ratio for each siRNA sample was divided by the GFP/mCherry ratio for non-targeting siRNA (NTsi). This -fold increase over control was used as the score.
Ligand Washout
Cells were washed once with conditioned media (growth media supplemented with purified recombinant protein) then incubated with conditioned media for the remainder of the washout period. For DD-GFP cells, conditioned media contained 5 μm FKBP protein with the F36V mutation (22). For DD*-GFP cells, conditioned media contained 5 μm E. coli DHFR protein. For HA-DD-GFP-DD*-FLAG cells, conditioned media contained either 5 μm FKBP(F36V) protein and 10 μm trimethoprim (TMP; for S1 washout) or 5 μm E. coli DHFR protein and 1 μm S1 (for TMP washout). For DD-DHFR cells, conditioned media contained 5 μm FKBP(F36V) protein and 5 μm methotrexate (MTX) or 0.1% DMSO.
Immunoblotting
Cells were harvested, and lysates were resolved by SDS-PAGE as described previously (22). Antibodies were anti-FLAG (mouse, F1804, Sigma), anti-GAPDH (mouse, 6C5, Abcam), anti-GFP (mouse, JL-8, Clontech), anti-HA (rat, 3F10, Rochel), anti-tubulin (mouse, DM1A, Sigma), and anti-UBE3C (rabbit, N1N3, GeneTex).
Coimmunoprecipitation of UBE3C and Proteasomes
Confluent cells in 10-cm dishes were washed with PBS then cross-linked using 2 ml of 0.25% formaldehyde in PBS for 7 min at room temperature. Cross-linking was quenched by removing the formaldehyde solution and adding 2 ml of ice-cold 1.25 m glycine in PBS and incubating for 5 min. Cells were harvested and lysed in 1 ml of radioimmune precipitation assay buffer on ice for 45 min. Insoluble debris was removed by centrifugation at 15,000 rpm for 10 min. Lysates were precleared by incubating with 10 μl of washed Pierce Protein A/G-agarose beads for 2 h at 4 °C. The supernatant was then incubated with 10 μg of rabbit polyclonal anti-PSMD2 antibody (Bethyl A303–853A) for 4 h at 4 °C. The antibody complex was then added to 10 μl of washed Pierce Protein A/G-agarose beads and incubated overnight at 4 °C. The unbound protein fraction was reserved for analysis, then beads were washed 3 times with PBS. Protein was eluted, and cross-linking was reversed by the addition of 2× SDS sample buffer and incubation at 95 °C for 30 min. Samples were then analyzed by immunoblotting.
Ubiquitin-associated (UBA) Domain Pulldown
Cells expressing DD-GFP were harvested, lysed (with or without Usp2 treatment), incubated with agarose beads conjugated to hPLIC2 UBA domain, washed, and eluted as previously described (22).
Growth Competition Assay
Cells expressing either GFP or mCherry were transfected in 24-well format with siUBE3C or NTsi, respectively, and left overnight in transfection media. Cells were then trypsinized and mixed in 12-well plates. DMSO or 100 nm 17-AAG was added to wells at various times so that all cells could be analyzed by flow cytometry together 72 h after transfection. The ratios of the number of GFP-positive cells to the number of mCherry-positive cells was normalized to the starting ratio.
Generation of Diced siRNA Pools
Total RNA was prepared from ∼5 × 106 HeLa cells using RNeasy (Qiagen). SuperScript III (Invitrogen) was then used to generate a cDNA library. DyNAzyme EXT (Thermo) was then used to amplify a region of each gene of interest by PCR. A second PCR amplification with DyNAzyme was then carried out to add T7 polymerase sites to each gene of interest. T7 mixed with pyrophosphatase (Meyer laboratory, Stanford University) was then used to transcribe dsRNA, which was then treated with DNase I (Invitrogen) and diced with Giardia Dicer (Teruel lab, Stanford University). Diced RNA products were purified using Purelink RNA Mini columns (Ambion) and eluted in water.
Reverse Transfection of Diced siRNA Pools
In a 96-well U-bottom plate, diced siRNA was mixed with 0.2 μl of Lipofectamine 2000 (Invitrogen) in 50 μl of Opti-MEM (Invitrogen) at a final RNA concentration of 5 nm and incubated at room temperature for 20 min. 50 μl of growth media containing 5000 HeLa cells was then mixed with the lipid complex in a 96-well culture plate and incubated for 15 min at room temperature. Cells were then cultured normally. 24 h post-transfection, media was replaced with growth media. 72 h post-transfection, cells were analyzed.
1-Anilinonaphthalene-8-sulfonic acid (ANS) Staining and Microscopy
1-Anilinonaphthalene-8-sulfonic acid (Sigma) was dissolved in DMSO at 75 mm, then diluted in PBS to 10 mm. This working stock was added to culture media to reach a final concentration of 250 μm. HeLa cells were stained with ANS for 1 h, then imaged on a Zeiss Axioskop 2 microscope.
Sample Preparation for Mass Spectrometry Analysis
Frozen cell pellets (containing 107 HeLa cells each) were resuspended in 500 μl of 8 m urea, then sonicated (Bioruptor UCD-200) for 15 min (high power, 15 s on, 15 s off). The samples were then agitated at room temperature for 15 min and then sonicated again as before. A 100-μl aliquot of the sample was removed for processing, and the rest was stored at −80 °C. The cell lysate was then diluted to 2 m urea with 50 mm ammonium bicarbonate, and the total protein was quantified with a BCA kit (Thermo Fischer Scientific). 1 m tris(2-carboxyethyl)phosphine (TCEP) (Soltec Ventures) was then added to a final concentration of 10 mm, and samples were agitated for 30 min at 37 °C, then cooled to room temperature. Alkylation was performed by adding 1 m iodoacetamide (Sigma) to a final concentration of 15 mm, then agitating samples for 30 min in the dark at room temperature. Next, the lysate was diluted to 1 m urea with 50 mm ammonium bicarbonate, upon which heavy standard peptides and sequencing grade trypsin (Promega, catalog #V5113) were added (1 μl of synthetic peptide mixture/10 μg of protein, and 1 μg of trypsin/100 μg of protein). The protein solution was agitated overnight at 37 °C. Peptides were purified using Sep-Pak C18 cartridges (Waters) and lyophilized before being solubilized in 2% acetonitrile and 0.1% formic acid at a concentration of 1 μg/μl.
Selected Reaction Monitoring Mass Spectrometry-based Quantification
Peptides (either two or three unique per protein, JPT Peptide Technologies) were separated on an EASY-nLC Nano-HPLC system (Proxeon, Odense, Denmark) and were introduced to the TSQ Vantage triple quadrupole MS system (Thermo Fisher Scientific, Bremen, Germany) via electrospray ionization and analyzed in selected reaction monitoring mode. The peptide separation was carried out using a 25 × 0.1-mm C18 trapping column (MICHROM C18, 5 μm, 120 Å) and a 200 × 0.075-mm diameter reverse-phase C18 capillary column (Maisch C18, 3 μm, 120 Å). Peptides (2 μg of total protein digest) were separated with a linear gradient from 0 to 45% acetonitrile in 70 min at a flow rate of 300 nl/min. Precursor and fragment ions were targeted for their corresponding heavy and light peptides. Endogenous peptides were quantified by the ratio of the areas between the light (endogenous) and heavy (synthetic standard) peptides peaks, with five biological replicates.
RESULTS
Identification and Validation of UBE3C as Important for DD Degradation
Given that DDs are known to be substrates of the ubiquitin-proteasome system (21, 22), we designed a focused siRNA screen to identify proteins involved in the degradation of DDs. We designed a custom siRNA library of Dharmacon SMARTpools containing all putative motor proteins, chaperones, and members of the ubiquitin conjugation system (supplemental Table S1). We used MMLV retrovirus to create a reporter HeLa S3 cell line stably expressing an FKBP-DD fused to Superfolder GFP (21, 41) with mCherry expression driven by an internal ribosome entry site (DD-GFP HeLa cells). In the event of compromised DD degradation, we reasoned that we would be able to use flow cytometry to detect increases in GFP signal. Cells incubated with stabilizing ligand would have high levels of DD-GFP fluorescence. Upon ligand removal, DD-GFP levels would be expected to diminish as the unfolded DD is recognized, ubiquitinated, and degraded by the proteasome. Compromised degradation should result in slower decay of this signal (Fig. 1B). In the nonpermissive (no stabilizing ligand) steady state, compromised degradation should be detectable as an increase in the basal fluorescence levels of the DD-GFP construct. Additionally, mCherry expression could be used ratiometrically to filter out false positives that have nonspecific effects on protein levels as well as to account for cell-to-cell variation in overall expression levels of the transgenic construct.
We conducted the siRNA screen in two formats mirroring the ligand washout and nonpermissive steady-state conditions described above. In the ligand washout format, cells were reverse-transfected with library siRNAs and incubated with stabilizing ligand for the duration of the 72 h knockdown. Ligand was washed out 3 h before analysis by flow cytometry (Fig. 1B). In the ligand-absent format, cells were reverse-transfected with library siRNAs then analyzed 72 h later by flow cytometry. Both screens were performed in duplicate. The ratio of GFP signal to mCherry signal for each sample was compared with the same ratio in cells transfected with NTsi. Each siRNA was scored as the -fold change in this ratio compared with NTsi (Fig. 1B).
Twenty-one SMARTpools that scored highly in both the ligand-absent and ligand-present screens were further validated by repeating the two screening experiments using deconvoluted pools as well as homemade diced siRNA pools. Because the DD used in the screening and subsequent deconvolution experiments was derived from FKBP (21), we further validated the top 21 hits using a DD derived from E. coli DHFR (DD*) (23). Obtaining similar results with both DD and DD* constructs would provide evidence that these hits are not interacting specifically with FKBP and that they are relevant to general protein quality control or degradation. We established a new cell line stably expressing DD* fused to Superfolder GFP (DD*-GFP HeLa cells) that was transfected with the deconvoluted pools and homemade diced siRNA pools and analyzed as before. The DD and DD* constructs yielded comparable results in these experiments, demonstrating that these siRNA-induced effects on the rates of DD degradation are not FKBP-specific (data not shown).
In both screening experiments, siRNA against UBE3C (siUBE3C) scored highest (Fig. 1C and supplemental Table S1), with a clearly visible shift in the siUBE3C-treated population toward higher GFP signal when compared with control (Fig. 1D). This difference is maintained even 48 h after knockdown, consistent with the increase in fluorescence observed in siUBE3C-treated cells cultured in the absence of ligand (data not shown). To validate knockdown specificity, we first performed an immunoblot to confirm a difference in UBE3C protein levels with and without siRNA treatment (Fig. 1E). We then complemented knockdown of UBE3C by stably expressing either wild-type or a catalytically inactive mutant (C1051A) UBE3C protein in our screening cell line. These constructs were codon-wobbled at the siRNA targeting site to escape RNAi-mediated knockdown. Transfecting these cells with NTsi and siUBE3C and examining them by flow cytometry 1.5 and 3 h after ligand washout revealed that the wild-type, but not catalytically inactive, form of UBE3C is able to complement the knockdown phenotype of endogenous UBE3C (Fig. 1F). Immunoblotting confirms that the exogenous UBE3C constructs express at higher levels than wild-type endogenous UBE3C (Fig. 1G).
Proteasome Processivity Is Compromised in the Absence of UBE3C
In yeast, deletion of Hul5, the ortholog of UBE3C, results in incomplete processing of some proteasome substrates, specifically those featuring an unstable domain fused to a very stable one (36). Mammalian cells behave in a similar manner upon UBE3C knockdown (37). To determine whether UBE3C knockdown in HeLa cells would result in incomplete degradation of DD-GFP, cells were transfected with NTsi or siUBE3C and analyzed by immunoblot against GFP 0, 2, and 4 h after withdrawal of the stabilizing ligand (Fig. 2A). Levels of the full-length fusion protein correlated well with expected results from flow cytometry (Fig. 1F). In addition, we observed a strong lower molecular weight band in samples with UBE3C knockdown and ligand washout (Fig. 2A).
FIGURE 2.

Proteasome processivity is compromised in the absence of UBE3C. A and E, DD-GFP cells were treated with siUBE3C or NTsi in the presence of stabilizing ligand, then harvested for immunoblotting (IB) at the indicated times following ligand removal. -S indicates cells that were incubated without stabilizing ligand. B and C, DD-GFP or DD*-GFP cells were treated and examined as in A. Cells receiving bortezomib treatment were incubated with the drug for the duration of ligand washout. D, wild-type or catalytically inactive (C1051A) UBE3C was expressed in DD-GFP HeLa cells. Cells were treated and examined as in A. DD-GFP cells expressing no exogenous UBE3C were included as control.
To assess whether this lower molecular weight band was the result of incomplete degradation by the proteasome, we repeated the experiment and treated a population of cells with the proteasome inhibitor bortezomib for the duration of the ligand washout. The truncation product was absent when the proteasome was blocked (Fig. 2B). To eliminate the possibility that incomplete degradation was an effect specific to the FKBP-derived DD, we repeated the experiment with DD*-GFP HeLa cells and consistently observed incomplete degradation of the DD*-GFP substrate with UBE3C knockdown (Fig. 2C).
Next we asked whether the catalytic function of UBE3C is relevant to the formation of this fragment. DD-GFP HeLa cells expressing codon-wobbled wild-type or catalytically inactive UBE3C were transfected with NTsi or siUBE3C and examined by immunoblot after ligand washout. Complete processing requires the presence of catalytically active UBE3C, as expression of wild-type enzyme led to complete degradation of the substrate, but expression of inactive enzyme did not (Fig. 2D). Furthermore, this truncation product is not an acute phenomenon brought about by sudden high levels of proteasome substrate that would appear due to ligand washout. The lower molecular weight truncation product was observed long after ligand washout and even during steady-state, nonpermissive conditions (Fig. 2E). Taken together, the above results suggest a role for UBE3C in the ubiquitination of proteasome substrates, leading to their complete degradation.
Based on the above results, we envisioned a model for UBE3C-assisted degradation of substrates in which substrates that are entering the proteasome via an unstructured region, such as a DD (18, 19), may become partially processed in the absence of UBE3C if the proteasome encounters a domain that is particularly stable, as GFP is known to be (41, 42). We reasoned that we should be able to influence the type of partial processing product by controlling how a substrate enters the proteasome (43). To this end we created a HeLa S3 cell line expressing a GFP fusion protein with DD fused to its N terminus and DD* fused to its C terminus (Fig. 3A). To facilitate detection, an HA tag was added to the N terminus of DD, and a FLAG tag was added to the C terminus of DD*. Removal of the stabilizing ligand for DD, Shield-1, should cause the protein to enter the proteasome via its N terminus and, in the absence of UBE3C, result in a truncation product detectable with antibodies against GFP or FLAG but not HA. Similarly, removal of the stabilizing ligand for DD*, TMP, should result in a truncation product detectable with antibodies against GFP or HA but not FLAG.
FIGURE 3.

UBE3C affects proteasomal degradation regardless of substrate orientation. A, schematic of the HA-DD-GFP-DD*-FLAG fusion protein with DD-ligand pairs shown bound together. B, flow cytometry was used to analyze the behavior of the fusion protein. Cells were incubated with the indicated ligand(s) for 24 h. In some samples, ligand(s) were then removed, and cells were analyzed 4 h later along with samples that underwent no washout. Error bars show S.E. C, cells treated with the indicated siRNAs were incubated with both stabilizing ligands for 24 h. The indicated ligand was then washed out, and cells were harvested for immunoblotting (IB) 3 h later.
First we analyzed behavior of the construct using flow cytometry to confirm that removal of either ligand is sufficient to destabilize the entire protein. Incubation with both ligands results in high GFP levels, but the absence of either suffices to induce degradation (Fig. 3B). Then, to examine the types of truncation products formed under various conditions, we knocked down UBE3C, performed ligand washout, and immunoblotted cell lysates using antibodies against GFP, HA, and FLAG. In the absence of UBE3C, two different truncation products appear depending on the ligand removed. We observe the expected pattern of detection, with FLAG antibody unable to detect the product of TMP removal and HA antibody unable to detect the product of Shield-1 washout (Fig. 3C). Unexpectedly, we observed partial processing even without UBE3C knockdown when Shield-1 is removed but not when TMP is removed, perhaps indicating some bias in the directionality of proteasome processing or asymmetric GFP processing difficulty. However, the amount of substrate fragment is greater in cells treated with siUBE3C. These results support our hypothesis that the substrate will enter the proteasome via its unstructured region and also show that UBE3C is active regardless of the direction of degradation, although there may be some bias.
Substrate Fragments Do Not Inhibit the Proteasome
We hypothesized based on the above results that we observe a substrate fragment because of the highly stable, well folded nature of GFP proteins (41, 42) and that perhaps a less stable protein might be fully degraded even in the absence of UBE3C, and similarly, a more stable protein might not be fully degraded even in its presence. To address this question, we created a cell line expressing DD fused to wild-type E. coli DHFR, a protein that becomes more stably folded upon binding to the small molecule methotrexate (MTX) (18, 44). Knockdown of UBE3C in this cell line had no effect on fragment formation in this cell line as we observed fragment formation upon ligand removal was dependent solely upon the presence or absence of MTX (Fig. 4A). Furthermore, there appears to be no difference in intensity in the fragment band in cells treated with MTX but with different siRNA. This suggests that there is likely some range of substrate stability for which UBE3C is useful in addressing partial degradation; easily degraded substrates do not require UBE3C involvement, and UBE3C activity is insufficient for very stable substrates.
FIGURE 4.
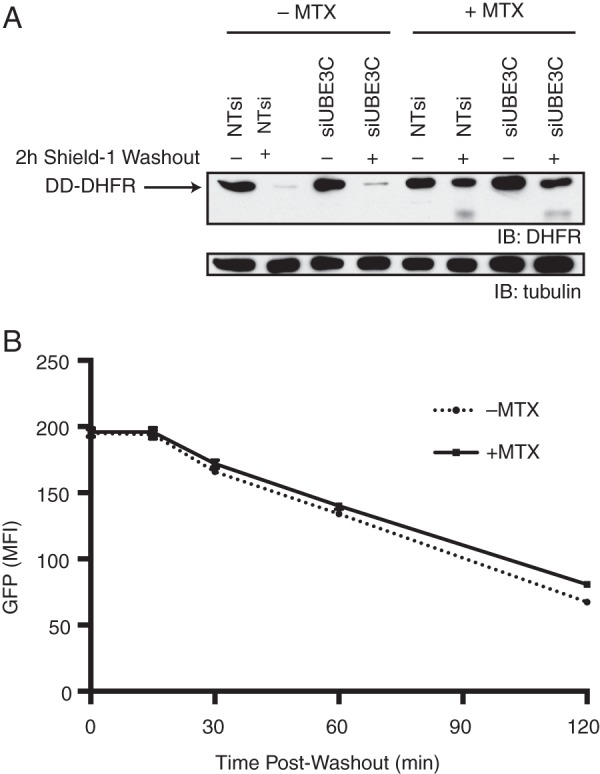
Substrate fragments do not inhibit the proteasome. A, HeLa S3 cells expressing DD-DHFR were treated with or without siUBE3C and MTX, then harvested and analyzed by immunoblot (IB) at 0 or 2 h after ligand withdrawal. B, HeLa S3 cells expressing DD-DHFR and DD-sfGFP were cultured in the presence of stabilizing ligand and treated with or without MTX, then analyzed by flow cytometry at the indicated times after ligand washout. MFI, mean fluorescence intensity. Error bars show S.E.
We observe that UBE3C knockdown results not only in incomplete degradation of DD-sfGFP but also in its slower degradation (Figs. 1 and 2A). We asked whether these two phenotypes might be related; could the fragments be inhibiting the proteasome and, therefore, cause the slower degradation of the full-length protein? We hypothesized that if we could cause fragments to appear using one DD construct, we should be able to slow the degradation of another DD construct. To test this, we transduced our DD-DHFR cell line with the DD-sfGFP construct. We wanted to use the DD-DHFR construct because we are able to cause fragments to appear even in the presence of UBE3C (Fig. 4A) so that we could get a clean answer as to whether it is the fragments that cause slower degradation of the full-length protein or some other effect of UBE3C knockdown. Treatment of these cells with MTX and subsequent ligand removal should cause fragments to form that could impair degradation of full-length DD-sfGFP. However, this is not what we observe, as MTX treatment has no effect upon the rate of degradation of DD-sfGFP (Fig. 4B). This result suggests that fragment formation alone does not impair the proteasome.
Binding of UBE3C to the Proteasome Is Not Sufficient For Its Activity
It has been previously reported that UBE3C binds to the proteasome, specifically the PSMD2 subunit, and that its binding is attenuated by the deletion of its N-terminal 132 amino acids (34). However, this shortened UBE3C remains able to extend Ub chains in vitro (33). We wanted to build upon this work and asked whether the N-terminal 132 residues are necessary for UBE3C function in the more physiologically relevant context of mammalian cells. To address this question, we deleted these residues from UBE3C, leaving only the C-terminal portion of the protein (ΔN132-UBE3C), and expressed it in DD-GFP HeLa cells (Fig. 5B). DD-GFP degradation kinetics were examined as before by flow cytometry. We found that expression of ΔN132-UBE3C is not sufficient to restore wild-type degradation kinetics when UBE3C is knocked down (Fig. 5C).
FIGURE 5.

Binding of UBE3C to the proteasome is not sufficient for its activity. A, HeLa cell lines expressing the indicated exogenous UBE3C construct were immunoprecipitated (IP) with antibody against proteasome subunit PSMD2 using protein A/G-agarose beads. Input and bead-bound fractions were then analyzed by immunoblot (IB) against UBE3C. B, schematics of UBE3C constructs. UBE3C was expressed in cells with UBE3CNterm intact, removed, or replaced with the UBL domain of hHR23A. C, DD-GFP HeLa cells were stably transduced with no exogenous UBE3C or with the constructs shown in A. Cells were treated with siUBE3C or NTsi in the presence of stabilizing ligand and analyzed by flow cytometry at 3 h after ligand removal. DD-GFP cells expressing no exogenous UBE3C were included as control. Error bars show S.E.
We next asked whether the sole purpose of the N-terminal region is proteasome-binding. If so, then replacement of this region with another proteasome binding domain should be sufficient to complement knockdown of UBE3C. For this purpose we chose the UBL domain of hHR23A (UBL), which has also been shown to bind to PSMD2 (45–47). The UBL-ΔN132-UBE3C chimeric protein was expressed in DD-GFP HeLa cells and analyzed as above. It was also unable to complement UBE3C knockdown (Fig. 5C), suggesting that the N-terminal 132 residues play a role in UBE3C function beyond simple PSMD2 binding or that the N-terminal region facilitates binding in a very specific location and/or manner. We confirmed that UBL-ΔN132-UBE3C and wild-type UBE3C coimmunoprecipitate with the proteasome and that ΔN132-UBE3C does not (Fig. 5A).
UBE3C Aids Proteasome Processivity By Ubiquitinating Substrates
Based on the observation that active UBE3C enzyme is required for complete processing of proteasome substrate, we hypothesized that UBE3C ubiquitinates partially processed substrates to aid in their complete degradation. We reasoned that if ubiquitination was crucial to UBE3C activity, then we might be able to detect partially processed substrates even in the presence of UBE3C if they were sufficiently difficult to ubiquitinate. To test this hypothesis, we designed GFP variants with all (GFPK0) or all but one (GFPK1) of the 20 lysine residues mutated to arginine, as the majority of ubiquitination happens at lysine residues (4). Examination of the crystal structure of Superfolder GFP (PDB code 2B3P) revealed that all 20 of the lysine residues are either on the surface of the protein or poorly resolved and, therefore, probably solvent-exposed. This observation, coupled with previous evidence that GFP is highly tolerant of mutations to its surface residues (48), made us optimistic that a lysine-free mutant of sfGFP would fold properly and retain some of its fluorescent properties. For GFPK1, we chose the most C-terminal lysine (position 213) to leave intact, as this would establish the greatest distance between the unstructured DD and the site for UBE3C to act if the protein were unfolded at the proteasome.
The size of the truncation products we observe is always slightly larger than the expected size of GFP alone, suggesting that a C-terminal fragment of the DD remains attached to the GFP domain. If we were to fuse a DD to the N terminus of these GFP variants, the presence of any lysine residues in the DD-derived portion of this fragment would compromise our goal of testing a substrate lacking lysine. For this reason, we used DD*, where the last lysine residue occurs at position 109 of 159, giving us a better chance of generating a lysine-free fragment. For the FKBP-derived DD, a lysine is present at position 105 of 107 (21, 23), rendering it less suitable for this experiment.
HeLa S3 cells expressing DD* fused to wild-type GFPK20, GFPK0, or GFPK1 with C-terminal HA tags were transfected with NTsi or siUBE3C and examined by immunoblot 0, 2, 4, and 6 h after withdrawal of the stabilizing ligand. As expected, incomplete degradation was observed in all three cell lines when UBE3C is knocked down. However, in cells transfected with NTsi, incomplete degradation was observed in the GFPK0 cell line but not in the GFPK20 or GFPK1 lines (Fig. 6A), suggesting that the presence of even a single lysine residue on a proteasome substrate is sufficient for UBE3C to function. These results support the idea that UBE3C ubiquitinates proteasome substrates to ensure complete degradation.
FIGURE 6.
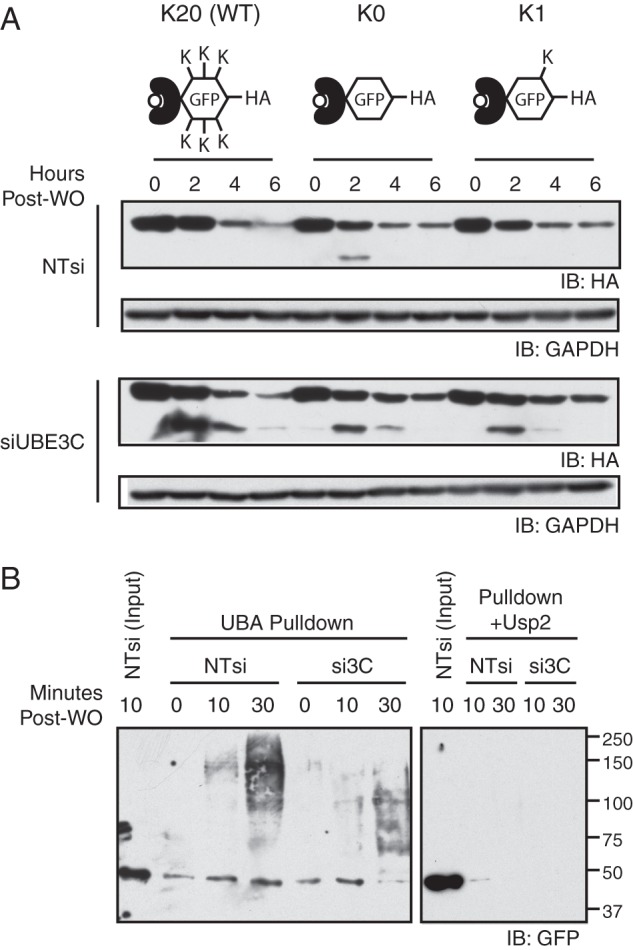
UBE3C aids proteasome processivity by ubiquitinating substrates. A, DD* was fused to GFP containing 20 (WT), 0 (K0), or 1 (K1) lysine residues and stably expressed in HeLa cells. Cells were treated with siUBE3C or NTsi in the presence of stabilizing ligand and harvested for immunoblotting (IB) at the indicated times after ligand removal. B, DD-GFP cells were treated with the indicated siRNA in the presence of stabilizing ligand, then harvested and lysed at the indicated time points after ligand withdrawal. Ubiquitinated species were enriched from cell lysates using UBA domain affinity chromatography, and the bound fractions were analyzed by immunoblot (left panel). As a control, some samples were treated with recombinant Usp2 enzyme before incubation with UBA domain beads (right panel).
If UBE3C ubiquitinates substrates, then we would expect that the ubiquitination state of destabilized DD-GFP constructs over time should reveal a difference in cells expressing versus lacking UBE3C. We harvested and lysed cells treated with NTsi or siUBE3C shortly after ligand withdrawal and enriched for ubiquitinated species using UBA domain affinity chromatography, then examined these species by immunoblot. UBE3C knockdown results in the molecular weight of ubiquitinated DD-GFP species to be significantly smaller than those observed in cells expressing UBE3C (Fig. 6B, left panel). DD-GFP is still ubiquitinated in the absence of UBE3C, but UBE3C appears to rapidly ubiquitinate these Ub chains on the substrate. A similar experiment involving pretreatment of cell lysates with the deubiquitinating enzyme Usp2 confirms that the signal in the left panel comes from ubiquitinated species (Fig. 6B, right panel).
UBE3C Contributes to Cellular Fitness during Protein Folding Stress
Up to this point we observed incomplete degradation of DD-GFP substrates in the absence of UBE3C but have only monitored the degradation of DD-GFP. In the absence of UBE3C, we would expect that many other cellular proteasome substrates would be incompletely degraded. We hypothesized that UBE3C Ub ligase activity might be important to cellular fitness if the proteasome is faced with a large load of proteins to degrade as buildup of these fragments may result in cytotoxicity. To address this hypothesis we developed an assay to interrogate cellular fitness as a function of UBE3C expression.
We created HeLa cell lines stably expressing either GFP or mCherry. We reasoned that if one of these lines was treated with siUBE3C and the other was treated with NTsi and the two populations were mixed and cultured together, that they would grow at the same rate in the absence of any additional stress. However, under certain stress conditions such as unfolded protein stress, incompletely degraded protein fragments may accumulate in cells lacking UBE3C, and this effect may manifest itself in a co-culture experiment as an effect on the rate of proliferation. Culturing these cells together with an appropriate stressor would result in a change over time in the ratio of GFP-positive to mCherry-positive cells observed by flow cytometry.
HeLa cells expressing GFP or mCherry were cultured separately and transfected with either siUBE3C (GFP HeLa) or NTsi (mCherry HeLa). Cells were then mixed and co-cultured in the presence or absence of a panel of chemical stressors including translational inhibitors, Hsp90 inhibitors, proteasome inhibitors, a microtubule poison, a DNA damaging agent, and a reducing agent. A 30-min heat shock was also tested. Cells were analyzed by flow cytometry 48 h after stress treatment, and the ratio of GFP-positive to mCherry-positive cells was compared with cells treated with vehicle (DMSO). Of the stressors tested, only Hsp90 inhibitors caused siUBE3C-treated cells to proliferate more slowly than NTsi-treated cells (data not shown).
We chose to characterize the behavior of 17-AAG, an Hsp90 inhibitor (49, 50), more in depth by treating cells for varying lengths of time before analysis by flow cytometry. Cells treated with vehicle showed little change in GFP-positive/mCherry-positive cell ratio, whereas we observed a steady decline in this ratio in cells treated with 17-AAG from 24 to 48 h. The magnitude of this decrease from 48 to 72 h of treatment did not increase appreciably (Fig. 7A). These results support our hypothesis that UBE3C E3 ligase function is important for proteasome function and cellular fitness in response to unfolded protein stress.
FIGURE 7.

UBE3C contributes to cellular fitness during protein folding stress. A, HeLa cells expressing GFP or mCherry were transfected with siUBE3C or NTsi, respectively. 24 h after transfection, GFP and mCherry cells were mixed and cultured for an additional 48 h, then analyzed by flow cytometry. Drug treatment was staggered such that all cells were analyzed 72 h post-transfection. Error bars show S.E. B and C, HeLa cells were treated with the indicated siRNA, then stained with ANS 72 h after siRNA transfection. Cells were then either imaged (B) or analyzed by flow cytometry (C).
Based on the above results, we hypothesized that the observed fitness defect might be the result of the accumulation of partially degraded proteasome substrates that are not well folded. We examined HeLa cells by microscopy after treatment with siUBE3C or NTsi by staining them with the dye ANS, a fluorescent molecular probe for hydrophobicity and, by extension, protein folding (51, 52). UBE3C knockdown resulted in the appearance of strongly fluorescent ANS-stained structures not visible in control cells (Fig. 7B). This increase in brightness is quantifiable by flow cytometry (Fig. 7C) with median ANS signal increasing nearly 2-fold in UBE3C knockdown cells versus control. These results taken together with the fitness defect observed with 17-AAG treatment suggest that UBE3C knockdown results in the accumulation of harmful protein products that can contribute to stress to the degradation machinery.
A UBE3C Mutation Associated With Autism Spectrum Disorders Attenuates Activity
A point mutation in UBE3C was recently implicated in ASD, specifically the mutation of serine 845 to phenylalanine (40). Lacking a crystal structure for UBE3C, we turned instead to the crystal structure of UBE3A, a related HECT E3 ligase. Residue 845 of UBE3C maps onto arginine 616 of UBE3A, which was shown by Huang et al. (53) to be highly solvent-inaccessible. Both of these residues lie in the HECT domains of their respective proteins. The serine at position 845 in UBE3C should be similarly buried in the core of the protein, so mutation of this residue to a much bulkier phenylalanine residue could compromise the ability of the protein to fold the HECT domain into an active conformation.
We next asked whether expression of the S845F mutant is able to complement knockdown of endogenous UBE3C to the same extent as wild-type enzyme. DD-GFP cells expressing codon-wobbled wild-type or S845F UBE3C were treated with NTsi or siUBE3C and examined by immunoblot after ligand washout. The wild-type enzyme is able to fully complement knockdown (no fragment is visible); however, the mutant enzyme seems to have attenuated activity, as a less intense fragment band is visible after longer exposure times (Fig. 8). This result suggests that the S845F mutation is somewhat deleterious to UBE3C function.
FIGURE 8.

A UBE3C mutation associated with autism spectrum disorders attenuates activity. DD-GFP cells expressing the indicated wobbled UBE3C construct were treated with siUBE3C or NTsi in the presence of stabilizing ligand, then harvested for immunoblotting (IB) either 1 h after ligand removal or without ligand removal.
UBE3C Is Not a Stoichiometric Proteasome Subunit
Although we and others found that UBE3C binds to the proteasome (34), it is not clear whether it is a stoichiometric proteasome subunit. To address this question, we employed selected reaction monitoring to determine the number of copies of UBE3C present in HeLa cells and compared the result with the number of copies of proteasome core subunits and proteasome regulatory particle subunits present in the same cells. We found that UBE3C is only present at 3–4% that of the level of core proteasome subunits (Table 1); therefore, UBE3C cannot be a stoichiometric subunit.
TABLE 1.
Quantitation of UBE3C and proteasome subunit abundance
Selected reaction monitoring was used to determine the mean ± S.D. of the number of copies of UBE3C and various representative proteasome subunits present in HeLa cells.
| Gene symbol | Copies/cell mean | Copies/cell S.D. | Stoichiometry |
|---|---|---|---|
| UBE3C | 41,000 | 5,700 | 1 |
| PSMA6a | 1,300,000 | 120,000 | 33 |
| PSMB6a | 1,100,000 | 170,000 | 27 |
| PSMD11b | 520,000 | 60,000 | 13 |
| PSMD2b | 530,000 | 34,000 | 13 |
a Proteasome core subunit.
b Proteasome regulatory particle subunit.
DISCUSSION
The DD technology was originally envisioned as a tool that would provide biologists small molecule control over protein function without the often prohibitively difficult or resource-consuming process of identifying such a molecule de novo for each protein of interest. As we learned more about how DDs are degraded, we began to appreciate their potential value as model substrates for determining the fate of unfolded proteins in cells. Upon withdrawal of the stabilizing ligand, DDs are rapidly unfolded and ubiquitinated, leading to Ub- and proteasome-mediated decreases in protein levels that occur within minutes (22). We saw an opportunity to learn more about protein quality control and the ubiquitin-proteasome system using DDs.
We set out to use RNAi screening to identify proteins involved in protein quality control or degradation. When we designed the screen, we hoped to find among a panel of putative E3 ligases or chaperones a “first responder” that recognizes the newly unfolded DD. We imagined that loss of such a protein would dramatically diminish cell ability to process these model substrates. To our surprise, proteins that are reportedly involved in such roles, such as CHIP (54–57) and Hsp70 (1, 58, 59) family proteins, did not emerge as strong hits from our screen (supplemental Table S1), suggesting some degree of functional redundancy among first responders. We would have expected an effect of much larger magnitude than any that we observed had a single protein been responsible for this type of quality control. This conclusion regarding the functional redundancy of putative first responders comes with a handful of caveats. First, we did not rigorously test knockdown efficiency of every gene in the screen. Furthermore, we cannot be certain that we covered every E3 ligase and chaperone; a first responder may fall outside of the focus of our screen, and knockdown of only one member of a family of closely related genes would not suffice if all members must be removed before a defect is revealed. However, the decision to follow up on UBE3C was straightforward, as it consistently produced a robust signal of significantly larger magnitude than any other gene included in our experiments (Fig. 1, C and D).
Upon knockdown of UBE3C, we consistently observed that the appearance of a truncation product resulting from partial proteasomal degradation of a DD fusion protein always coincides with a slower rate of full-length protein degradation (Figs. 1D, 2, and 4A). The incomplete degradation is likely caused by the proteasome failing to properly unfold relatively stable domains such as sfGFP. UBE3C can aid this process in some cases but is unnecessary or incapable in others (Fig. 4A). We have determined that slower disappearance of the full-length protein is unlikely to be an effect of fragment formation, as fragments of one protein do not inhibit degradation of another (Fig. 4B). However, it has been reported that the proteasome is able to concurrently degrade multiple polypeptide chains, so the presence of a stalled fragment may not inhibit degradation of other proteins (60). Because overexpression of UBE3C does not accelerate degradation (Fig. 1F), suggesting that UBE3C must not be rate-limiting, it is likely that ubiquitination of substrates by UBE3C is necessary for the rapid degradation of only some fraction of proteins that is relatively difficult to degrade by virtue of high folding energy, sheer bulk, or other biophysical property that challenges proteasomes. Loss of UBE3C causes these substrates to require longer periods of time in the proteasome to degrade. As substrates dwell longer on proteasomes, they may become more predisposed to dissociation (with observable fragment formation), just as stalling of unclamped DNA polymerases often leads to their dissociation from DNA (61). The same holds true even if UBE3C is present, but the substrate remains difficult to degrade, as was the case with DD-DHFR in the presence of MTX (Fig. 4A).
Building on the work of You and Pickart (33), we also demonstrated the importance of proteasome binding for UBE3C function. We were unable to complement knockdown of UBE3C via overexpression of ΔN132-UBE3C, which lacks the ability to bind to the proteasome but remains an active ligase (Fig. 5). Furthermore, overexpression of UBL-ΔN132-UBE3C, a chimeric protein that binds to the same proteasome subunit as UBE3C (34, 46), cannot complement UBE3C knockdown (Fig. 5). These results suggest that simple binding of UBE3C to the proteasome subunit PSMD2 is not sufficient for UBE3C to exert its activity. It appears that the N-terminal 132 residues may play a somewhat more complex role than simply targeting UBE3C to proteasomes. Binding of UBE3C to the proteasome may be governed by more strict requirements regarding orientation, conformation, or kinetics. Although the UBL domain of the chimeric UBL-ΔN132-UBE3C protein may successfully target the UBE3C domain to the proper proteasome subunit, the ligase may bind in the wrong region or in the wrong orientation. Beyond simple structural considerations, it is also possible that the UBL domain associates with different biophysical parameters (i.e. too tightly or too loosely) relative to the UBE3C N-terminal 132 residues.
Our work also led to insights regarding the nature of UBE3C E3 ligase activity. We found that active enzyme (Figs. 1D and 2D) as well as accessible substrate lysine residues (Fig. 6A) are necessary for efficient substrate processing. This led us to investigate the effect of UBE3C knockdown on substrate ubiquitination in a time-dependent fashion, where we found that knockdown resulted in smaller ubiquitin conjugates being formed over time compared with control samples (Fig. 6B). These two pieces of evidence lead us to believe that similar to the yeast ortholog Hul5 (15, 36), UBE3C rapidly ubiquitinates substrates that have already been ubiquitinated by another E3 ligase as polyubiquitination and degradation of our substrate is slowed but not abolished by UBE3C knockdown.
Because UBE3C activity is not necessary for complete degradation of some substrates (Fig. 4A), we propose that UBE3C polices primarily recalcitrant proteasome-bound substrates by ubiquitinating them. To ensure processive degradation for all substrates, because UBE3C is substoichiometric relative to proteasomes (Table 1), it is likely that either UBE3C interacts with proteasomes in a dynamic fashion or substrates that are difficult to degrade are somehow retargeted to proteasomes with UBE3C bound.
We sought to determine the relevance of UBE3C in the context of protein homeostasis. Although we observe the formation of DD-GFP substrate fragments in the absence of UBE3C, it is safe to assume that GFP is not the only cellular proteasome substrate capable of forming such fragments, as we observe increased ANS staining upon UBE3C knockdown in wild-type HeLa cells (Fig. 7, B and C). Furthermore, it would not be surprising if some fraction of these fragments might be deleterious to cell health. Although we noticed no obvious defects in cell health upon UBE3C knockdown, we showed by growth competition that cells lacking UBE3C are at a fitness disadvantage compared with wild-type cells in the presence of the Hsp90 inhibitor 17-AAG (Fig. 7A). Hsp90 inhibitors are known to cause release and subsequent proteasomal degradation of Hsp90 client proteins (49), resulting in their cytotoxic accumulation. Cells expressing UBE3C would be expected to be better equipped to deal with higher proteasome load. Surprisingly, similar treatment with proteasome inhibitor did not yield results similar to treatment with 17-AAG. Treatment with either drug is known to cause a heat shock response in cells (20, 60). Induction of this response may be sufficient to compensate for the loss of UBE3C activity in the case of proteasome inhibition. The inability of cells to compensate during Hsp90 inhibition suggests that either the drug dose used was not high enough to induce a heat shock response or that Hsp90 activity is crucial to the cell ability to compensate for the loss of UBE3C activity.
Although we have demonstrated the importance of UBE3C ubiquitin ligase activity, it is not entirely clear how ubiquitination facilitates substrate degradation. A conservative explanation might be that polyubiquitin simply constitutively retargets these substrates to the proteasome to ensure complete degradation. Alternatively, this ubiquitination may more directly aid the degradation process by actively promoting protein unfolding, as the addition of a polyubiquitin chain can serve not only signaling roles but also can alter the biophysical properties of a protein, causing it to become destabilized (62, 63). The targeting and unfolding models presented above are not mutually exclusive but, rather, are two points along a spectrum of possibilities. It is likely that polyubiquitination by UBE3C fills both of these roles to some extent, although more experiments are required to determine where along this spectrum it falls.
The appearance of a UBE3C mutation in studies of ASD (40) hints at one facet of the developmental defects that occurs in ASD patients. Because cells deficient in UBE3C activity have a fitness defect with respect to protein folding stress (Fig. 7A) and the S845F mutation attenuates UBE3C activity (Fig. 8), we can imagine that patients suffering from a UBE3C mutation might be rendered susceptible to stresses to protein degradation machinery encountered during development. Our work provides the first clue toward a better understanding of the contribution of this mutation to ASD.
In this study we showed that DDs could be used as model substrates to interrogate protein quality control and protein degradation. We identified UBE3C as a protein that plays an important role in proteasome processivity, which is consistent with what is known about its ortholog in yeast, Hul5p. We present evidence that UBE3C ubiquitinates substrates that are difficult to process and that lack of this activity can be deleterious to cell fitness. We believe the primary contribution of UBE3C to protein homeostasis and cellular fitness arises from the ligase ability to prevent the formation of pathological protein fragments that arise from incomplete proteolysis of substrates by the proteasome. Because protein fragmentation is known to lead to aggregation (64) and aggregation is correlated with if not causative of many pathological states (2, 65–67), one can conclude that the ability of cells to prevent the formation of protein fragments is important for fitness. Thus UBE3C may play a central protective role in maintaining protein homeostasis by ensuring complete degradation of proteasome substrates that might otherwise aggregate.
Acknowledgments
We thank the Stanford Shared FACS Facility and Michael Rape for helpful advice, the Meyer laboratory for reagents and advice, and Dan Jarosz for valuable comments on this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM073046.

This article contains supplemental Table S1.
- Ub
- ubiquitin
- DD
- destabilizing domain
- FKBP
- FK506-binding protein
- DHFR
- dihydrofolate reductase
- ASD
- autism spectrum disorder
- NTsi
- non-targeting siRNA
- ANS
- 1-anilinonaphthalene-8-sulfonic acid
- TMP
- trimethoprim
- MTX
- methotrexate
- UBA
- ubiquitin-associated
- 17-AAG
- 17-N-allylamino-17-demethoxygeldanamycin
- sfGFP
- superfolder GFP.
REFERENCES
- 1. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 2. Ross C. A., Poirier M. A. (2005) Opinion. What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 6, 891–898 [DOI] [PubMed] [Google Scholar]
- 3. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 4. Varshavsky A. (2012) The ubiquitin system, an immense realm. Annu. Rev. Biochem. 81, 167–176 [DOI] [PubMed] [Google Scholar]
- 5. Hayden M. S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 6. Ikeda F., Dikic I. (2008) Atypical ubiquitin chains. New molecular signals. “Protein modifications. Beyond the usual suspects” review series. EMBO Rep. 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mocciaro A., Rape M. (2012) Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. J. Cell Sci. 125, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wickliffe K. E., Williamson A., Meyer H.-J., Kelly A., Rape M. (2011) K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol. 21, 656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pickart C. M. (1997) Targeting of substrates to the 26 S proteasome. FASEB J. 11, 1055–1066 [DOI] [PubMed] [Google Scholar]
- 10. Thrower J. S., Hoffman L., Rechsteiner M., Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 12. Kee Y., Huibregtse J. M. (2007) Regulation of catalytic activities of HECT ubiquitin ligases. Biochem. Biophys. Res. Commun. 354, 329–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomko R. J., Jr., Hochstrasser M. (2011) Order of the proteasomal ATPases and eukaryotic proteasome assembly. Cell Biochem. Biophys. 60, 13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crosas B., Hanna J., Kirkpatrick D. S., Zhang D. P., Tone Y., Hathaway N. A., Buecker C., Leggett D. S., Schmidt M., King R. W., Gygi S. P., Finley D. (2006) Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127, 1401–1413 [DOI] [PubMed] [Google Scholar]
- 16. Leggett D. S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R. T., Walz T., Ploegh H., Finley D. (2002) Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10, 495–507 [DOI] [PubMed] [Google Scholar]
- 17. Wang X., Huang L. (2008) Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell. Proteomics 7, 46–57 [DOI] [PubMed] [Google Scholar]
- 18. Lee C., Schwartz M. P., Prakash S., Iwakura M., Matouschek A. (2001) ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell 7, 627–637 [DOI] [PubMed] [Google Scholar]
- 19. Prakash S., Tian L., Ratliff K. S., Lehotzky R. E., Matouschek A. (2004) An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol. 11, 830–837 [DOI] [PubMed] [Google Scholar]
- 20. Prakash S., Inobe T., Hatch A. J., Matouschek A. (2009) Substrate selection by the proteasome during degradation of protein complexes. Nat. Chem. Biol. 5, 29–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Banaszynski L. A., Chen L.-C., Maynard-Smith L. A., Ooi A. G., Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Egeler E. L., Urner L. M., Rakhit R., Liu C. W., Wandless T. J. (2011) Ligand-switchable substrates for a ubiquitin-proteasome system. J. Biol. Chem. 286, 31328–31336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwamoto M., Björklund T., Lundberg C., Kirik D., Wandless T. J. (2010) A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 17, 981–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyazaki Y., Imoto H., Chen L.-C., Wandless T. J. (2012) Destabilizing domains derived from the human estrogen receptor. J. Am. Chem. Soc. 134, 3942–3945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dolan B. P., Sharma A. A., Gibbs J. S., Cunningham T. J., Bennink J. R., Yewdell J. W. (2012) MHC class I antigen processing distinguishes endogenous antigens based on their translation from cellular vs. viral mRNA. Proc. Natl. Acad. Sci. U.S.A. 109, 7025–7030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dvorin J. D., Martyn D. C., Patel S. D., Grimley J. S., Collins C. R., Hopp C. S., Bright A. T., Westenberger S., Winzeler E., Blackman M. J., Baker D. A., Wandless T. J., Duraisingh M. T. (2010) A plant-like kinase in Plasmodium falciparum regulates parasite egress from erythrocytes. Science 328, 910–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glass M., Busche A., Wagner K., Messerle M., Borst E. M. (2009) Conditional and reversible disruption of essential herpesvirus proteins. Nat. Methods 6, 577–579 [DOI] [PubMed] [Google Scholar]
- 28. Gong Y., de Lange T. (2010) A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell 40, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Madeira da Silva L., Owens K. L., Murta S. M., Beverley S. M. (2009) Regulated expression of the Leishmania major surface virulence factor lipophosphoglycan using conditionally destabilized fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 106, 7583–7588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pruett-Miller S. M., Reading D. W., Porter S. N., Porteus M. H. (2009) Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet. 5, e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Russo I., Oksman A., Vaupel B., Goldberg D. E. (2009) A calpain unique to alveolates is essential in Plasmodium falciparum and its knockdown reveals an involvement in pre-S-phase development. Proc. Natl. Acad. Sci. U.S.A. 106, 1554–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mastrandrea L. D., You J., Niles E. G., Pickart C. M. (1999) E2/E3-mediated assembly of lysine 29-linked polyubiquitin chains. J. Biol. Chem. 274, 27299–27306 [DOI] [PubMed] [Google Scholar]
- 33. You J., Pickart C. M. (2001) A HECT domain E3 enzyme assembles novel polyubiquitin chains. J. Biol. Chem. 276, 19871–19878 [DOI] [PubMed] [Google Scholar]
- 34. You J., Wang M., Aoki T., Tamura T.-A., Pickart C. M. (2003) Proteolytic targeting of transcriptional regulator TIP120B by a HECT domain E3 ligase. J. Biol. Chem. 278, 23369–23375 [DOI] [PubMed] [Google Scholar]
- 35. Fang N. N., Ng A. H., Measday V., Mayor T. (2011) Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol. 13, 1344–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aviram S., Kornitzer D. (2010) The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol. Cell. Biol. 30, 985–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martínez-Noël G., Galligan J. T., Sowa M. E., Arndt V., Overton T. M., Harper J. W., Howley P. M. (2012) Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell. Biol. 32, 3095–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kohlmann S., Schäfer A., Wolf D. H. (2008) Ubiquitin ligase Hul5 is required for fragment-specific substrate degradation in endoplasmic reticulum-associated degradation. J. Biol. Chem. 283, 16374–16383 [DOI] [PubMed] [Google Scholar]
- 39. Yu Y., Hayward G. S. (2010) The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. O'Roak B. J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B. P., Levy R., Ko A., Lee C., Smith J. D., Turner E. H., Stanaway I. B., Vernot B., Malig M., Baker C., Reilly B., Akey J. M., Borenstein E., Rieder M. J., Nickerson D. A., Bernier R., Shendure J., Eichler E. E. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T. C., Waldo G. S. (2006) Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88 [DOI] [PubMed] [Google Scholar]
- 42. Martin A., Baker T. A., Sauer R. T. (2008) Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat. Struct. Mol. Biol. 15, 139–145 [DOI] [PubMed] [Google Scholar]
- 43. Berko D., Tabachnick-Cherny S., Shental-Bechor D., Cascio P., Mioletti S., Levy Y., Admon A., Ziv T., Tirosh B., Goldberg A. L., Navon A. (2012) The direction of protein entry into the proteasome determines the variety of products and depends on the force needed to unfold its two termini. Mol. Cell 48, 601–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kraut D. A., Israeli E., Schrader E. K., Patil A., Nakai K., Nanavati D., Inobe T., Matouschek A. (2012) Sequence and species dependence of proteasomal processivity. ACS Chem. Biol. 7, 1444–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schauber C., Chen L., Tongaonkar P., Vega I., Lambertson D., Potts W., Madura K. (1998) Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391, 715–718 [DOI] [PubMed] [Google Scholar]
- 46. Wang Q., Goh A. M., Howley P. M., Walters K. J. (2003) Ubiquitin recognition by the DNA repair protein hHR23a. Biochemistry 42, 13529–13535 [DOI] [PubMed] [Google Scholar]
- 47. Watkins J. F., Sung P., Prakash L., Prakash S. (1993) The Saccharomyces cerevisiae DNA repair gene RAD23 encodes a nuclear protein containing a ubiquitin-like domain required for biological function. Mol. Cell. Biol. 13, 7757–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lawrence M. S., Phillips K. J., Liu D. R. (2007) Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 129, 10110–10112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whitesell L., Lindquist S. L. (2005) HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 [DOI] [PubMed] [Google Scholar]
- 50. Whitesell L., Mimnaugh E. G., De Costa B., Myers C. E., Neckers L. M. (1994) Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins. Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. U.S.A. 91, 8324–8328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Semisotnov G. V., Rodionova N. A., Razgulyaev O. I., Uversky V. N., Gripas' A. F., Gilmanshin R. I. (1991) Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers 31, 119–128 [DOI] [PubMed] [Google Scholar]
- 52. Slavik J., Vondrejs V. (1981) 1-Anilinonaphthalene-8-sulphonate as a fluorescent probe for temperature-induced changes in Escherichia coli membranes. Folia Microbiol. (Praha) 26, 176–178 [DOI] [PubMed] [Google Scholar]
- 53. Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P. M., Huibregtse J. M., Pavletich N. P. (1999) Structure of an E6AP-UbcH7 complex. Insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 [DOI] [PubMed] [Google Scholar]
- 54. Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Höhfeld J., Patterson C. (2001) The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 3, 93–96 [DOI] [PubMed] [Google Scholar]
- 55. Dickey C. A., Patterson C., Dickson D., Petrucelli L. (2007) Brain CHIP. Removing the culprits in neurodegenerative disease. Trends Mol. Med. 13, 32–38 [DOI] [PubMed] [Google Scholar]
- 56. Jana N. R., Dikshit P., Goswami A., Kotliarova S., Murata S., Tanaka K., Nukina N. (2005) Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640 [DOI] [PubMed] [Google Scholar]
- 57. Scaglione K. M., Zavodszky E., Todi S. V., Patury S., Xu P., Rodríguez-Lebrón E., Fischer S., Konen J., Djarmati A., Peng J., Gestwicki J. E., Paulson H. L. (2011) Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol. Cell 43, 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Buchberger A., Bukau B., Sommer T. (2010) Protein quality control in the cytosol and the endoplasmic reticulum. Brothers in arms. Mol. Cell 40, 238–252 [DOI] [PubMed] [Google Scholar]
- 59. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 60. Lee C., Prakash S., Matouschek A. (2002) Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J. Biol. Chem. 277, 34760–34765 [DOI] [PubMed] [Google Scholar]
- 61. Bennett E. J., Harper J. W. (2008) DNA damage. Ubiquitin marks the spot. Nat. Struct. Mol. Biol. 15, 20–22 [DOI] [PubMed] [Google Scholar]
- 62. Goloubinoff P., De Los Rios P. (2007) The mechanism of Hsp70 chaperones. (Entropic) pulling the models together. Trends Biochem. Sci. 32, 372–380 [DOI] [PubMed] [Google Scholar]
- 63. Hagai T., Levy Y. (2010) Ubiquitin not only serves as a tag but also assists degradation by inducing protein unfolding. Proc. Natl. Acad. Sci. U.S.A. 107, 2001–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 65. Kopito R. R. (1999) Biosynthesis and degradation of CFTR. Physiol. Rev. 79, S167–S173 [DOI] [PubMed] [Google Scholar]
- 66. Landles C., Bates G. P. (2004) Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 5, 958–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. David D. C., Ollikainen N., Trinidad J. C., Cary M. P., Burlingame A. L., Kenyon C. (2010) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 8, e1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]