

Background: Intestinal production of TGF-β is decreased in neonatal necrotizing enterocolitis (NEC).
Results: Oral administration of TGF-β (TGF-β1) inhibited inflammation in intestinal epithelium and systemic production of IL-6 and IFN-γ as well as NEC incidence in an animal model.
Conclusion: Oral administration of TGF-β1 can compensate for TGF-β1 deficiency in gut diseases.
Significance: TGF-β1 can potentially be used to prevent and treat gut diseases.
Keywords: Chromatin Immunoprecipitation (ChIP), Inflammation, Innate Immunity, Intestinal Epithelium, NF-kappa B (NF-KB), SMAD Transcription Factor, Transforming Growth Factor Beta (TGFbeta)
Abstract
Inflammatory immune responses play an important role in mucosal homeostasis and gut diseases. Nuclear factor κB (NF-κB), central to the proinflammatory cascade, is activated in necrotizing enterocolitis (NEC), a devastating condition of intestinal injury with extensive inflammation in premature infants. TGF-β is a strong immune suppressor and a factor in breast milk, which has been shown to be protective against NEC. In an NEC animal model, oral administration of the isoform TGF-β1 activated the downstream effector Smad2 in intestine and significantly reduced NEC incidence. In addition, TGF-β1 suppressed NF-κB activation, maintained levels of the NF-κB inhibitor IκBα in the intestinal epithelium, and systemically decreased serum levels of IL-6 and IFN-γ. The immature human fetal intestinal epithelial cell line H4 was used as a reductionistic model of the immature enterocyte to investigate mechanism. TGF-β1 pretreatment inhibited the TNF-α-induced IκBα phosphorylation that targets the IκBα protein for degradation and inhibited NF-κB activation. Chromatin immunoprecipitation (ChIP) assays demonstrated decreased NF-κB binding to the promoters of IL-6, IL-8, and IκBα in response to TNF-α with TGF-β1 pretreatment. These TGF-β1 effects appear to be mediated through the canonical Smad pathway as silencing of the TGF-β central mediator Smad4 resulted in loss of the TGF-β1 effects. Thus, TGF-β1 is capable of eliciting anti-inflammatory effects by inhibiting NF-κB specifically in the intestinal epithelium as well as by decreasing systemic IL-6 and IFN-γ levels. Oral administration of TGF-β1 therefore can potentially be used to protect against gastrointestinal diseases.
Introduction
Necrotizing enterocolitis (NEC)2 is a deadly inflammatory bowel disease affecting ∼12% of premature infants with a birth weight <1500 grams (1). It is the most common gastrointestinal surgical emergency in premature neonates. The pathophysiology behind NEC is poorly understood; however, the primary risk factors are prematurity, enteral feeding, and inappropriate bacterial colonization, which collectively lead to mucosal barrier disruption, transmural intestinal injury, and significant inflammatory responses typically affecting the terminal ileum and colon (2).
Several aspects of intestinal immaturity place the preterm gut at risk for NEC. In particular, studies in human tissues and animal models have shown that intestinal NF-κB is strongly activated at birth but is down-regulated as the intestine matures (3, 4). This developmental down-regulation of NF-κB may not be seen in preterm infants at risk for NEC, presumably due to immaturity of the gut. The NF-κB family represents a group of structurally related proteins that promote transcription of a wide variety of proinflammatory cytokines. In their resting state, homo or hetero NF-κB dimers are bound to the inhibitory κB proteins (IκB) and are thus functionally and physically restricted to the cytoplasm. Extracellular stimulation can trigger signal transduction pathways, which in turn phosphorylate IκBα, targeting it for ubiquitination and subsequent proteasome-mediated degradation (5). The NF-κB thus liberated moves to the nucleus, where it activates proinflammatory cytokine transcription. Our laboratory has specifically shown that the underlying mechanism for the increased activation of NF-κB activity in the immature intestine is decreased expression of the NF-κB inhibitor IκBα (3). Means of protecting or increasing IκBα and thus decreasing NF-κB signaling may provide opportunities to prevent NEC in the immature preterm gut.
TGF-β is a multifunctional factor that regulates cell growth, adhesion, and differentiation in a wide variety of cell types (6). It is also a strong immune suppressor (7). TGF-β null mice remain healthy for 2 weeks after birth but develop an excessive inflammatory response leading to a lethal multifocal inflammatory disease (7). TGF-β transduces signaling through a transmembrane type II receptor, which in turn recruits and activates/phosphorylates the intracellular type I receptor. The activated type I receptor then activates/phosphorylates the downstream effectors, Smad2 and Smad3, which subsequently form complexes with the common Smad mediator, Smad4, to translocate to the nucleus. The Smad complexes regulate transcription of TGF-β target genes in conjunction with various transcriptional or co-transcriptional regulators. In addition to the canonical Smad pathway, other signaling pathways, including the extracellular signal-regulated kinases (ERK1/2), mitogen-activated protein kinase (p38), Src, and phosphatidylinositol 3-kinase (PI3K) pathways, have been reported to mediate TGF-β effects in a context-dependent manner (8).
Previous studies have shown that TGF-β can inhibit NF-κB activation by promoting mRNA expression and protein stabilization of IκBα (9–11). We thus hypothesized that oral administration of TGF-β1, one of the TGF-β isoforms, would address a critical aspect of intestinal immaturity by suppressing NF-κB signaling through sustained IκBα, thereby protecting against NEC in vivo. In a rat pup model of NEC, oral administration of TGF-β1 activated Smad2, suppressed NF-κB activation, preserved IκBα expression specifically in intestinal epithelium, and reduced NEC incidence. In addition, TGF-β1 reduced serum proinflammatory cytokines IL-6 and IFN-γ, two proinflammatory cytokines elevated in NEC. Correspondingly, in a human fetal nontransformed immature cell line used as a model of the preterm gut, TGF-β1 suppressed TNF-α-induced NF-κB activation by down-regulating IκBα phosphorylation, a necessary process preceding IκBα degradation. Chromatin immunoprecipitation assays demonstrate that TGF-β1 also suppressed NF-κB binding to IκBα, suggesting TGF-β1 did not enhance NF-κB-mediated IκBα transcription to maintain IκBα expression in response to TNF-α in immature enterocytes. We observed involvement of the canonical Smad4 pathway in mediating the TGF-β1 anti-inflammatory effect in immature intestinal epithelial cells (IEC) by inhibitor and Smad4 silencing studies. This is the first study to demonstrate the anti-inflammatory effect of TGF-β1 by oral administration specifically in intestinal epithelium of an immature gut, resulting in resistance to intestinal injury in an animal model.
MATERIALS AND METHODS
Cell Cultures and Reagents
H4 cells (a generous gift from Dr. W. Allan Walker, Massachusetts General Hospital, Harvard Medical School) are a human fetal nontransformed primary intestinal epithelial cell line used as a model of immature IEC (12). H4 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum, 1% glutamine, 1% sodium pyruvate, 1% amino acids, 1% HEPES, 50 units/ml penicillin, 50 μg/ml streptomycin, and 0.2 units/ml insulin at 37 °C in a 5% CO2 atmosphere. Cell passages 11–21 were used. Recombinant TGF-β1 and TNF-α were purchased from R&D Systems (Minneapolis, MN). Recombinant human IFN-γ was from PeproTech (Rocky Hill, NJ). Inhibitors used to inhibit activation of intracellular pathways (PD98059 for the ERK pathway, and LY294002 for the PI3K/Akt pathway) were from Calbiochem.
Neonatal Rat NEC Model
All animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the University of Chicago Institutional Animal Care and Use Committee (IACUC) under Animal Protocol Number 71557. Cesarean sections were performed under isoflurane anesthesia, and all efforts were made to minimize suffering. If a rat pup showed illness during the course of the study, the animal was humanely euthanized, and death was not used as an end point.
Animal experiments were conducted following a well described rat NEC animal model with modifications as follows (13). Neonatal rat pups were delivered on embryonic day 20 by cesarean section from time-dated pregnant Sprague-Dawley dams. Once stabilized, pups were colonized with 107 colony-forming units each of Serratia marcescens, Klebsiella pneumoniae, and Streptococcus viridans, fed with Esbilac puppy formula every 3 h via an orogastric feeding catheter, and stressed with hypoxia (5% oxygen and 95% nitrogen for 10 min) three times a day to induce NEC. Naturally born and dam-fed pups were included as healthy controls. Animals were sacrificed when ill or at the end of the experiment on day 5. H&E-stained intestinal sections were scored by a pathologist blinded to treatment group using a previously published NEC scoring system to evaluate the degree of intestinal injury on a “0–4” scale. Scores ≥2 are defined as NEC (13).
Immunohistochemistry Staining
Intestinal (ileal) segments were formalin-fixed and paraffin-embedded. Sections were deparaffinized in xylene and hydrated with ethanol. For antigen unmasking, slides were heated in 10 mm sodium citrate buffer (pH 6.0) before treatment with 0.3% hydrogen peroxide. The specimens were blocked with 5% BSA in TBST (Tris-buffered saline with 0.05% v/v Tween 20) followed by 4 °C overnight incubation with p-NF-κB, IκBα, and p-Smad2 antibodies (Cell Signaling) or control IgGs isolated before immunization from the same animal species used to generate the antibodies. After washing, the sections were incubated with polymer-HRP secondary antibodies (Dako, Carpentaria, CA). Positive staining and nuclei were visualized with diaminobenzidine chromogen and hematoxylin counterstaining, respectively. For quantitation of NF-κB activity, nuclear p-NF-κB-positive IEC versus total IEC from three fields of each slide (n = 3) were counted and presented as means of percentages of p-NF-κB-positive IEC over total IEC ± S.E. For quantitation of IκBα and p-Smad2 staining, IHC intensity was scored on a 0–4 scale with 0 being the lowest and 4 being the highest intensity among all samples examined (14). Results are presented as means of scores ± S.E.
Immunoblotting
For tissue lysate preparation, 2-cm distal intestine (ileum) was homogenized with a hand-held homogenizer (Pellet Pestle, Kimble/Kontes, Vineland, NJ) in lysis buffer (50 mm Tris·HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% SDS, 50 mm DTT, 50 μg/ml aprotinin, 50 μg/ml leupeptin, and 5 mm PMSF). For cell culture lysate preparation, cells on plates were rinsed with cold PBS and lysed in the same lysis buffer as for tissues. Immunoblotting was carried out as described previously (15) with antibodies from Cell Signaling against NF-κB (p65), p-NF-κB, IκBα, p-IκBα, poly(ADP-ribose) polymerase, lactate dehydrogenase, Erk1/2, p-Erk1/2, Akt, p-Akt, p38, p-p38, JNK, p-JNK, Smad2, p-Smad2, and Smad4.
Isolation of IEC from Animals
Intestine was collected, cut into small pieces, and incubated in 7 ml of Ca2+-free RPMI medium containing 1% serum, 5 mm EGTA, and 1.5 mm MgCl2 for 45 min at 37 °C with shaking to wash off IEC and leukocytes from intestine. Tissue debris was separated from cell suspension by filtration with mesh and washed again to collect another 7 ml of cell suspension. A total of 14 ml of cell suspension was subjected to centrifugation at 1600 rpm for 10 min, and cell pellets were then resuspended in 44% Percoll in 1× Hanks' balanced salt solution and centrifuged at 2000 rpm for 20 min. The cloudy fractions on top of the Percoll as well as the pellets were collected as IEC and intestinal leukocytes, respectively. The IEC fraction was collected, pelleted, washed with PBS, pelleted again, and lysed in radioimmune precipitation buffer for immunoblotting for Smad2 and phospho-Smad2.
Subcellular Fractionation
Cytoplasmic and nuclear fractions were harvested using a method modified from Inan et al. (16). Briefly, cells were washed with PBS and then lysed in a lysis buffer (10 mm Hepes, 10 mm KCl, 0.1 mm EDTA, 2 mm MgCl2, 0.5 mm sucrose, 0.1% Nonidet P-40, 0.5 mm PMSF, 1 mm DTT, 1× complete protease inhibitor). Lysates were collected by scraping and centrifuged at 2000 × g for 5 min at 4 °C to collect cytoplasmic (supernatant) and nuclei (pellets). The cytoplasmic fraction was spun at 12,000 rpm for 10 min to remove debris and saved in a new microcentrifuge. Nuclei were washed with lysis buffer without Nonidet P-40, centrifuged, lysed in high salt buffer (20 mm Hepes, pH 7.4, 1.5 mm MgCl2, 420 mm NaCl, 0.2 mm EDTA, 5% glycerol, 1 mm DTT, 0.5 mm PMSF, protease inhibitors), and either sonicated briefly or incubated on ice for 40 min to disrupt nuclei. The nuclear fraction was collected following centrifugation and combined with the same volume of low salt buffer (20 mm Hepes, pH 7.4, 50 mm NaCl, 0.2 mm EDTA, 20% glycerol, 1 mm DTT, 0.5 mm PMSF, protease inhibitors). Protein concentrations were determined by Bio-Rad protein assay.
NF-κB DNA Binding Activity Assay
Activation of NF-κB p65 was determined by an ELISA-based DNA binding TransAM assay (Active Motif) using nuclear fractions as per the manufacturer's instructions. Assays were performed in triplicate, and data are presented as means of intensity ± S.E. * depicts p < 0.05 between the groups by Student's t test.
RNA Interference
To perform Smad4 silencing, 70% confluent cells on 6-well plates were transiently transfected with 200 nm of a pool of four scrambled control or Smad4 siRNAs (Dharmacon, Lafayette, CO) using Oligofectamine (Invitrogen) according to the manufacturer's guidelines. Medium containing 30% serum was directly added to wells 4 h following transfection. On the next day, cells were cultured back in regular serum medium, treated with 30 ng/ml TGF-β1 overnight, and then treated with 10 ng/ml TNF-α for 1 h before lysates were collected for Smad4, IκBα, and phospho-IκBα immunoblotting. Successful gene silencing was confirmed by immunoblotting for Smad4.
Multiplex Cytokine Assay
Serum was separated from blood of experimental NEC animals by centrifugation, and 25 μl of serum was diluted in 100 μl of PBS. 60 μl of diluted serum was added in black wall microplates in duplicate, subjected to a flow-assisted and bead-based multiplex cytokine assay (Bio-Rad), and analyzed with a standard curve generated using the five-parameter curve-fitting parameter of the Bioplex Manager 6.0 software (Bio-Rad). The cytokine concentration in serum is expressed as picograms of cytokine per ml of serum on log scale (Log (pg/ml)).
Chromatin Immunoprecipitation (ChIP) Assay
A two-step cross-linking ChIP assay was performed as described previously with modifications (17). Briefly, 2 × 106 H4 cells were collected by trypsinization and first fixed in 2 mm disuccinimidyl glutarate for 45 min at room temperature and then in 1% formaldehyde for 8 min followed by a quenching of formaldehyde using 0.125 m glycine. Cells were washed with PBS and lysed in cell lysis buffer (50 mm Tris-HCl, pH 8.0, 2 mm EDTA, 0.1% IGEPAL® 630, 10% glycerol, 1 mm DTT) supplemented with protease inhibitors on ice for 10 min to release cytoplasm. Nuclei were collected by centrifugation at 2500 × g at 4 °C and lysed in 200 μl of SDS lysis buffer (50 mm Tris, pH 8.1, 10 mm EDTA, 1% SDS plus phosphatase, and protease inhibitors) on ice for 10 min. Nuclei were briefly disrupted by sonication for 10 s, and chromatin/DNA contents were measured using a NanoDrop. The same amount of chromatin from each treatment was sheared under identical conditions using a Bioruptor (Diagenode Inc.) on high power and four cycles of 10 min of 30 s on/30 s off to an average size of 500–1000 bp. Sheared chromatin were diluted 1:10 in immunoprecipitation buffer (20 mm Tris, pH 8.1, 2 mm EDTA, 150 mm NaCl, 1% Triton X-100) and cleared first by centrifugation at 12,000 × g for 10 min at 4 °C and then with 60 μl of protein A/G beads for 1 h at 4 °C. 1% of diluted chromatin lysate for immunoprecipitation was saved as input. Immunoprecipitation was performed with 4 μg of an NF-κB antibody (Santa Cruz Biotechnology sc-372) overnight at 4 °C with rocking and then with 60 μl of protein A/G beads for 2 h. The beads were washed three times with immunoprecipitation wash buffer I (20 mm Tris, pH 8.1, 2 mm EDTA, 50 mm NaCl, 1% Triton X-100, 0.1% SDS), one time with immunoprecipitation wash buffer II (10 mm Tris, pH 8.1, 1 mm EDTA, 0.25 m LiCl, 1% Nonidet P-40, 1% deoxycholic acid), and finally three times with Tris-EDTA. DNA-protein complexes were eluted from agarose in 250 μl of freshly prepared elution buffer (100 mm sodium bicarbonate, 1% SDS, 0.05 μg/μl RNase A) at 65 °C for 20 min twice to collect a total of 500 μl of DNA elute. NaCl and Tris-HCl, pH 6.8, EDTA, and proteinase K solution were added to combined elutes (immunoprecipitated chromatin) and input chromatin to final concentrations of 200 mm NaCl, 50 mm Tris-HCl, pH 6.8, 10 mm EDTA, 200 μg/ml proteinase K and uncross-linked at 65 °C overnight. DNA was extracted by phenol-chloroform-isoamyl alcohol (25:24:1) and ethanol-precipitated in the presence of 20 ng/ml linear acrylamide. Purified immunoprecipitated and input DNA were dissolved in 60 μl, and 3 μl was used in TaqMan quantitative PCR reactions using TaqMan Fast Advanced master mix (Applied Biosystems) and primers and probes (Table 1) designed and prepared from Integrated DNA Technologies, Inc. that target NF-κB elements on the promoters of IL-6, IL-8, and IκBα (18–20). The PCR conditions were 50 °C for 2 min, 95 °C for 20 s, and 40 cycles of 95 °C for 1 s and 60 °C for 1 min on an Applied Biosystems 7900HT fast real-time PCR system. Immunoprecipitated and input DNA concentrations were automatically calculated by SDS software using a standard curve generated with 5, 0.5, and 0.05 ng/μl genomic DNA. Input-normalized immunoprecipitated DNA concentrations represent relative levels of NF-κB binding to promoters and are presented as -fold changes to the untreated groups in the figures.
TABLE 1.
Primers and probes used in NF-κB ChIP quantitative PCR

* Double-quenched probes with two nonfluorescent quenchers, an internal ZEN quencher, and a 3′-Iowa Black FQ (IBFQ) quencher.
Statistical Analysis
Statistical analysis was performed using a χ2 test for NEC incidence, Student's t test for paired data, or one-way analysis of variance with a Bonferroni's correction for multiple comparisons with GraphPad InStat software. All data are presented as means ± S.E., and differences were considered to be significant with p values <0.05.
RESULTS
Oral Administration of TGF-β1 Activated Smad2 in Intestinal Epithelium and Decreased NEC Severity and Incidence
TGF-β is a strong immune suppressor. The effect of TGF-β1, one of the TGF-β isoforms, on the pathophysiology of NEC through oral administration was examined in an experimental rodent model with intestinal injury similar to NEC. Animal studies were conducted as described under “Materials and Methods” with nonstressed and naturally born dam-fed pups as healthy controls. TGF-β1 at 30 ng/ml was used for oral administration as this is within the physiologic concentration found in breast milk (21). Whether TGF-β1 can initiate signal transduction in the intestine through oral administration was first examined. IHC staining of the active/phosphorylated form of the intracellular TGF-β effector Smad2 in intestinal tissues shows significantly enhanced Smad2 activation/phosphorylation in TGF-β1-treated as compared with vehicle-treated experimental NEC animals. Activation of Smad2 was readily detected in healthy dam-fed control pups, indicating a basal activation of TGF-β signaling in pups receiving breast milk (Fig. 1, A and B). The absence of background staining in the negative control for phospho-Smad2 staining indicates that the phospho-Smad2 staining is specific. Immunoblotting of phospo-Smad2 with lysates of purified IEC shows that TGF-β1 elicited signaling in the intestine by 20 h following oral administration (Fig. 1C). The degree of intestinal injury was scored on a 0–4 scale with scores ≥2 defined as NEC. All dam-fed healthy controls had score 0, and both vehicle-treated and TGF-β1-treated experimental NEC pups had scores 0–3. However, the TGF-β1-treated group had fewer pups with score 3 as compared with the vehicle-treated group. A statistical analysis confirmed that TGF-β1 significantly reduced disease severity as well as overall incidence (Fig. 2A). In the animal model, there was no NEC in the dam-fed control group (n = 20) but 52% NEC incidence in the vehicle-treated experimental NEC group (n = 48). Oral administration of TGF-β1 reduced NEC incidence from 52 to 32% (n = 48) (p < 0.05, Fig. 2B).
FIGURE 1.

Oral administration of TGF-β1 activated TGF-β signaling in IEC in experimental NEC. An animal model of NEC was conducted as described under “Materials and Methods.” Intestinal sections from experimental NEC pups treated with vehicle or 30 ng/ml TGF-β1 and from healthy dam-fed controls at the end of experiments on day 5 were IHC-stained for the active/phosphorylated form of the TGF-β intracellular mediator Smad2. A, representative staining shows increased p-Smad2 in IEC in experimental NEC pups gavaged with TGF-β1 as compared with the vehicle-treated control. Healthy dam-fed controls had basal activation of TGF-β signaling reflected by basal p-Smad2 levels. “−”, staining control of IHC was performed without using the primary antibody and shows the absence of background staining of p-Smad2. B, staining from A was quantified by scoring p-Smad2 on a 0–4 scale in three fields of each intestinal section (n = 3) as described under “Materials and Methods.” * depicts p < 0.05 by Student's t test. C, Smad2 was activated in IEC by 20 h following oral TGF-β1 administration. Intestine was collected at 0, 2, and 20 h after the first gavage of TGF-β1. IEC were isolated from intestine as described under “Materials and Methods,” lysed, and subjected to immunoblotting for total and phospho-Smad2.
FIGURE 2.
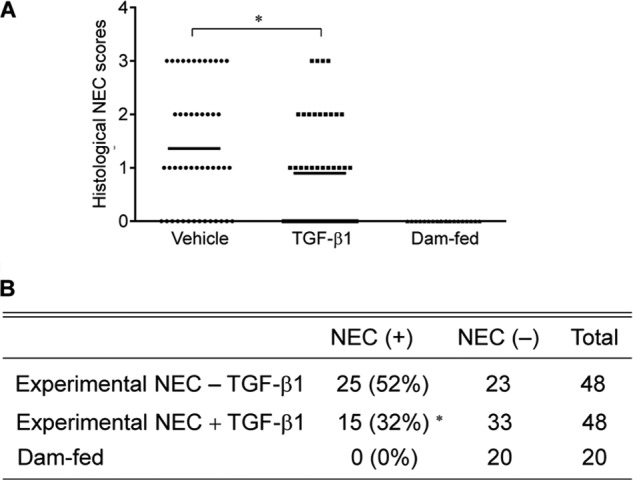
Histological severity of intestinal injury and disease incidence of experimental NEC pups. A, TGF-β1 significantly decreased the severity of NEC injury in an animal model. A neonatal rat NEC model and NEC scoring were conducted as described under “Materials and Methods.” Injury of intestine from healthy dam-fed controls and experimental NEC pups treated with vehicle or 30 ng/ml TGF-β1 was evaluated on a 0–4 scale by a pathologist blinded to experiments. The healthy dam-fed pups show no intestinal injury. * depicts p < 0.05 by one-way analysis of variance with Bonferroni's correction. Bars represent mean values. B, TGF-β1 decreased NEC incidence in an animal model. NEC is defined as scores ≥2, and the NEC incidence is determined from the same experiment as in A. * depicts p < 0.05 between the experimental NEC groups by χ2 test.
TGF-β1 Inhibited NF-κB Activation, Maintained IκBα Expression, and Decreased Proinflammatory Cytokine Production in Experimental NEC
To determine whether TGF-β1 protection against NEC was associated with suppressed NF-κB signaling, activation status of NF-κB (p-NF-κB) was examined by IHC. Fig. 3A shows increased p-NF-κB-positive IEC in experimental NEC as compared with healthy dam-fed pups. Oral administration of TGF-β1 significantly decreased numbers of p-NF-κB-positive IEC in experimental NEC (Fig. 3, A and B). The absence of background staining in the negative control for p-NF-κB staining indicates that the p-NF-κB staining is specific. IκBα expression was also examined by IHC to determine whether TGF-β1-suppressed NF-κB activation was associated with protected expression of this NF-κB inhibitor. IκBα was readily detected in the intestines of healthy dam-fed pups, but was significantly diminished in experimental NEC intestines (Fig. 3C). TGF-β1 administration significantly protected IκBα expression in experimental NEC pups. The absence of background staining in the negative control for IκBα indicates that the IκBα staining is specific. IL-6 and IFN-γ are proinflammatory cytokines and NF-κB targets that have been shown to increase in humans with NEC and animal models of NEC. Oral administration of TGF-β1 significantly decreased their levels in serum (Fig. 4).
FIGURE 3.

TGF-β1 suppressed NF-κB activation and preserved IκBα expression in experimental NEC. A and B, intestinal sections from experimental NEC pups treated with vehicle or 30 ng/ml TGF-β1 and from healthy dam-fed controls were IHC-stained for the active/phosphorylated form of NF-κB (p-NF-κB) (A) and IκBα (B). A, TGF-β1 suppressed NF-κB activation in experimental NEC. Representative staining shows increased intestinal p-NF-κB in vehicle-treated experimental NEC pups as compared with healthy controls and TGF-β1-suppressed NF-κB activation. “−”, staining control of IHC was performed without using the primary antibody and shows the absence of background staining of p-NF-κB. B, staining from A was quantified by counting p-NF-κB-positive IEC in three fields of each intestinal section, and numbers are presented as means of percentages of p-NF-κB-positive IEC ± S.E. C, TGF-β1-protected IκBα protein expression in experimental NEC. Representative staining shows decreased intestinal IκBα expression in vehicle-treated experimental NEC pups as compared with healthy controls and TGF-β1-protected IκBα expression in experimental NEC. “−”, staining control of IHC was performed without using the primary antibody and shows the absence of background staining of IκBα. D, staining from C was scored on a 0–4 scale in three fields of each intestinal section, and numbers are presented as means of intensity ± S.E. * depicts p < 0.05 by Student's t test.
FIGURE 4.
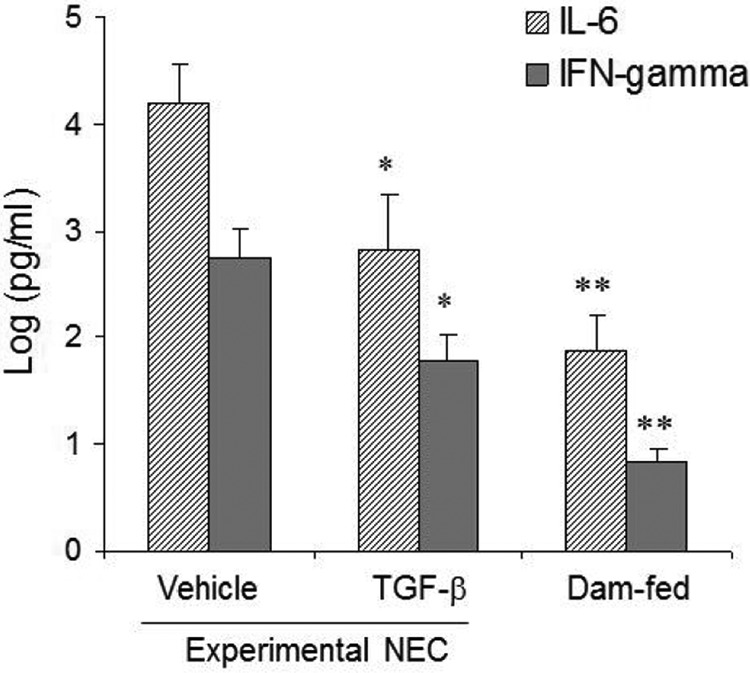
TGF-β1 decreased serum IL-6 and IFN-γ levels in experimental NEC. Blood was collected from vehicle- or TGF-β1-treated experimental pups with the same degree of intestinal injury and from healthy dam-fed controls. Serum was prepared and subjected to a multiplex cytokine assay. Data (n ≥ 5) are presented as means ± S.E. * and ** depict p < 0.05 and 0.01, respectively, as compared with the vehicle-treated group by one-way analysis of variance with Bonferroni's correction.
TGF-β1 Suppressed IκBα Phosphorylation in Human Immature IEC
TGF-β1 reduced NEC incidence, inhibited NF-κB activation, and maintained IκBα expression in vivo (Table 1 and Fig. 3). We next studied the mechanism behind decreased NF-κB activation and protected IκBα expression using the human fetal intestinal epithelial cell line H4 as an in vitro model of immature human IEC (12). TNF-α was used as a proinflammatory cytokine relevant to NEC. Immunoblotting and an ELISA-based DNA binding assay with nuclear and cytoplasmic fractions show that TNF-α induced NF-κB activation/nuclear translocation and DNA binding activity as early as 15 min and sustained this until at least 2 h following treatment (Fig. 5, A and B). TGF-β1 pretreatment suppressed both the TNF-α-induced nuclear translocation and the DNA binding activity of NF-κB (Fig. 5, A and B) with corresponding greater cytoplasmic retention of NF-κB in the cells (Fig. 5A). Consistently, we observed dose-dependent suppression of TNFα-induced IκBα phosphorylation by TGF-β1 pretreatment (Fig. 5C).
FIGURE 5.

TGF-β1 suppression of NF-κB nuclear translocation, DNA binding, and phosphorylation was associated with decreased IκBα phosphorylation and degradation in immature IEC. A, TGF-β1 suppressed NF-κB nuclear translocation induced by TNF-α. Immature human fetal H4 IEC were pretreated with 30 ng/ml TGF-β1 overnight and then 10 ng/ml TNF-α for different intervals as indicated. Nuclear and cytoplasmic fractions were isolated and subjected to immunoblotting for NF-κB, a nuclear marker poly(ADP-ribose) polymerase (PARP), and a cytoplasmic marker lactate dehydrogenase (LDH). B, TGF-β1 suppressed NF-κB DNA binding activity induced by TNF-α. Nuclear fractions from A were subjected to an ELISA-based NF-κB DNA binding assay as described under “Materials and Methods.” Results are presented as means ± S.E., n = 3. * depicts p < 0.05 by Student's t test. OD, optical density. C, TGF-β1 suppressed NF-κB phosphorylation and IκBα phosphorylation and degradation induced by TNF-α. Immature human fetal H4 IEC were pretreated with TGF-β1 at 30 ng/ml overnight and then with increasing doses of TNF-α as indicated for 1 h. Whole cell extracts were isolated and subjected to immunoblotting for total and phospho-IκBα (p-IκBα) and total and phospho-NF-κB (p-NF-κB).
TGF-β1 Suppressed TNF-α-induced NF-κB Binding to IL-8 and IκBα Promoters
TNF-α has been shown to stimulate IL-8 production in H4 cells (22), and oral administration of TGF-β1 decreased IL-6 levels in serum in experimental NEC (Fig. 4). Whether TGF-β1 decreased NF-κB binding to the promoters of IL-8 and IL-6 was determined by ChIP assays. Fig. 6 shows that NF-κB binding to IL-6 and IL-8 promoters was significantly increased by TNF-α treatment. TGF-β1 decreased TNF-α-induced NF-κB binding to these promoters. This effect of TGF-β1 appears to be specific as erythropoietin, also a breast milk factor and a control to TGF-β1 in the assay, did not inhibit the TNF-α-enhanced NF-κB binding to IL-6 and IL-8 promoters. IκBα is an early response gene downstream of NF-κB, and NF-κB induction of IκBα regulates NF-κB activation by auto-feedback inhibition (23). A ChIP assay shows that TNF-α enhanced NF-κB binding to the IκBα promoter but that this binding was also decreased by TGF-β1 (Fig. 6).
FIGURE 6.
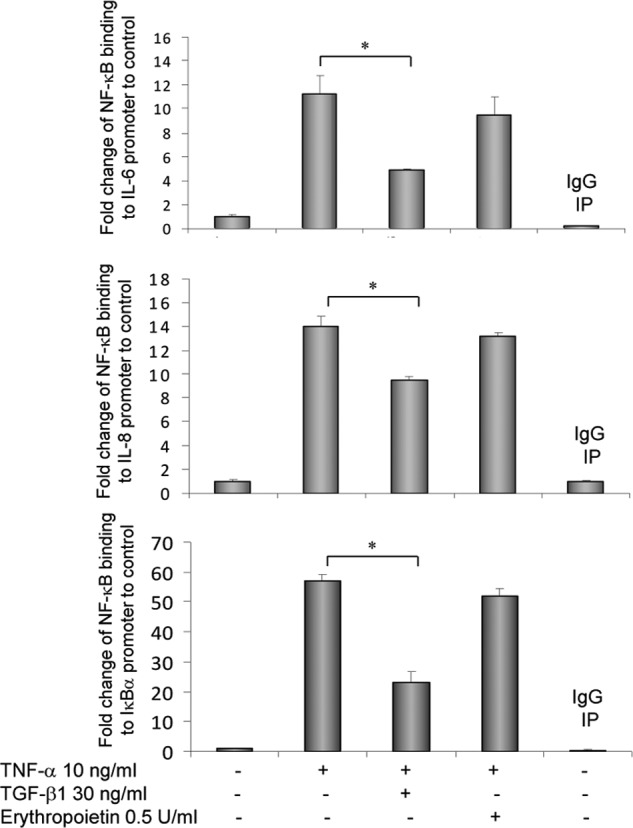
TGF-β1 decreased TNF-α-induced NF-κB binding to IL-6, IL-8, and IκBα promoters in immature IEC. H4 cells were treated overnight with 30 ng/ml TGF-β1 or 0.5 units/ml erythropoietin, as a control for TGF-β, and then with 10 ng/ml TNF-α for 1 h. Cells were collected and subjected to ChIP assay using an NF-κB antibody (Santa Cruz Biotechnology sc-372) and a normal IgG as a negative control antibody to the NF-κB antibody. Data are derived as described under “Materials and Methods” and presented as -fold changes to untreated group as means ± S.E. * depicts p < 0.05 by one-way analysis of variance with Bonferroni's correction. IP, immunoprecipitation.
TGF-β1 Suppressed NF-κB Activation and IκBα Phosphorylation via the Smad Pathway in Immature IEC
TGF-β can initiate signal transduction by activating the canonical Smad pathway as well as non-Smad pathways, including the JNK, p38 mitogen-activated protein kinase, Erk1/2, and PI3K/Akt pathways. The canonical signaling of TGF-β involves phosphorylation of Smad2 and Smad3 by the serine/threonine kinase TGF type I receptor, upon ligand binding. To determine which signaling pathway was involved in our findings, cells were pretreated with TGF-β1 overnight and then stimulated with TNF-α. Immunoblotting for key signaling molecules shows that TGF-β1 pretreatment did not significantly alter TNF-α-induced activation of p38 or JNK (Fig. 7A). Instead, TGF-β1 pretreatment stimulated activation/phosphorylation of the Akt and Smad pathways and prevented TNF-α-induced transient suppression of Erk1/2 activation (Fig. 7A). These data suggest the possible involvement of the Akt, Erk1/2, and Smad pathways in mediating the TGF-β1 immune suppressive effect.
FIGURE 7.

The Smad pathway mediated TGF-β1 suppression of NF-κB activation and IκBα phosphorylation in immature IEC. A, TGF-β1 stimulated Erk1/2, Akt, and Smad pathways in the immature human fetal H4 IEC. Cells were pretreated with 30 ng/ml TGF-β1 overnight and then with 10 ng/ml TNF-α for different intervals as indicated. Cells were then lysed for immunoblotting for activation of effectors downstream of various pathways as indicated. B, inhibition of Erk1/2 and Akt activation did not block TGF-β1 suppression of NF-κB and IκBα phosphorylation. Cells were pretreated with the inhibitors PD98059 (to the Erk1/2 pathway) and LY294002 (to the Akt pathway) for 30 min and then together with TGF-β1 overnight. Cells were then treated with 10 ng/ml TNF-α for 1 h and lysed for immunoblotting for total and phospho-proteins as indicated. C, Smad4 knockdown blocked TGF-β1 suppression of IκBα phosphorylation induced by TNF-α. Cells at 80% confluence were transfected with 200 nm of a pool of four Smad4 or scrambled control siRNAs. 40 h later, cells were pretreated with 30 ng/ml TGF-β1 overnight and then together with 10 ng/ml TNF-α for 1 h before lysis for immunoblotting. All experiments were repeated at least twice with consistent results.
Inhibitor assays were next performed using the MEK inhibitor (PD98059) to inhibit the Erk1/2 pathway and the PI3K inhibitor LY294002 to inhibit the Akt pathway. Fig. 7B shows effective suppression of Erk1/2 and Akt activation by PD98059 and LY294002, respectively, as both basal phosphorylation and TGF-β1-induced phosphorylation of Erk1/2 and Akt were decreased. However, even with effective inhibition, TGF-β1 was still able to suppress phosphorylation of NF-κB and IκBα, demonstrating that these pathways are not critical for TGF-β1 suppression of NF-κB.
Smad4 acts as a common partner in the Smad signaling pathway. Smad4 forms complexes with ligand-activated receptor Smads (Smad2 and Smad3 for TGF-β ligands) and facilitates the translocation of the heteromeric complexes into the nucleus to affect transcriptional regulation. To determine whether the Smad pathway was mediating the TGF-β1 immune suppressive effect, TGF-β1 suppression of IκBα phosphorylation was examined in H4 cells with or without Smad4 knockdown by Smad4 and control siRNA transfection, respectively. Fig. 7C shows undetected Smad4 protein in Smad4 siRNA-transfected as compared with control siRNA-transfected or parental H4 cells. Blockade of the Smad pathway by Smad4 knockdown inhibited TGF-β1 down-regulation of phospho-IκBα, demonstrating that TGF-β1 protection of IκBα from phosphorylation/degradation and subsequent suppression of the inflammatory response are mediated via the Smad pathway in immature IEC.
DISCUSSION
NEC is an inflammatory disorder with significant morbidity and mortality, associated with intestinal immaturity in preterm infants (24). Studies in human tissues and animal models have shown that intestinal NF-κB is strongly activated at birth and is down-regulated as the intestine matures in a healthy gut; however, this developmental inhibition of NF-κB is not yet present in preterm infants at risk for NEC (4, 25). Our laboratory has previously reported that the underlying mechanism for the loss of developmental inhibition of NF-κB activity in immature intestinal epithelial cells is decreased expression of the NF-κB inhibitor IκBα (3). Thus, agents that protect or increase IκBα expression and concomitantly decrease NF-κB activity could have a protective role against injury in immature gut.
TGF-β has been shown to inhibit NF-κB-dependent inflammatory responses in multiple cell types. IκBα is an immediate early gene downstream of NF-κB, and transcription is highly induced in response to NF-κB activation, keeping the NF-κB signaling under control. Previous studies have shown that TGF-β can inhibit NF-κB activity through induction of IκBα mRNA and protein expression as well as through prevention of IκBα degradation in different cell contexts (9, 11, 26). In our study, TGF-β1 did not further enhance NF-κB binding to the IκBα promoter to promote IκBα transcription in response to TNF-α as the ChIP result shows suppressed NF-κB binding to the IκBα promoter in response to TGF-β1. Instead, we observed that TGF-β1 suppressed IκBα phosphorylation, resulting in inhibited degradation and subsequent protection of IκBα protein in response to TNF-α in immature IEC.
Inhibition of NF-κB, an important transcription factor in the regulation of cellular inflammation, has been studied in many diseases. Agents to inhibit NF-κB to suppress inflammation in an NEC animal model have been investigated. A previous study reported that administration of the NEMO-binding domain (NBD) peptide (where NEMO is NF-κB essential modulator) that selectively inhibits the critical upstream IκB kinase (IKK) decreased mortality and bowel injury in an NEC model (25). Their findings therefore indicate that selective NF-κB inhibition represents a promising therapeutic strategy for NEC. However, there is a potential advantage to using TGF-β1 to prevent or ameliorate the development of NEC. Besides the innate immune suppressive effect in IEC in an immature gut from our studies, there are other aspects of TGF-β1, i.e. enhancing epithelial maturation and restitution to maintain the integrity of intestine, that can additionally protect an immature gut from NEC injury. It is noted that although TGF-β1 directly suppressed innate immunity of intestinal epithelium and decreased disease incidence, TGF-β1 was not completely protective as some of the TGF-β1-treated pups still went on to develop NEC regardless of increased IκBα expression and decreased NF-κB activation. NEC is a multifactorial disease, and presumably other risk factors or mechanisms may have contributed to the development of NEC in TGF-β1-treated pups that still developed disease. Indeed, intestinal barrier disruption is an important process during the development of NEC, and we have previously reported that oral administration of TGF-β1 does not protect intestinal barrier integrity (27). It is possible that a combination of anti-inflammatory and barrier-protective agents would have a greater protective effect.
TGF-β family members include isoforms TGF-β1, TGF-β2, and TGF-β3. Although highly homologous in structure and similar in biological activities in many bioassay systems, the TGF-β isoforms are encoded by different genes and have different distributions and biological actions within the body (28). Although TGF-β2 is the most abundant isoform in body fluids, including the aqueous and vitreous fluid of the eye, saliva, amniotic fluid, and breast milk, TGF-β1 is the most abundant isoform in all tissues and is also found in breast milk. TGF-β3 is usually the least abundant isoform in both tissues and body fluids (29). The different physiological functions of TGF-β isoforms have been demonstrated in gene knock-out mice. TGF-β1 null mice can survive in the perinatal period when nursed by their dams but die of excessive inflammatory reactions after weaning (30). In contrast, TGF-β2 null mice display a wide range of developmental heart, lung, craniofacial, limb, spinal column, eye, inner ear, and urogenital defects (31). It has been shown that TGF-β1 is more effective than TGF-β2 in inducing migration of epithelial cells across a wound edge and enhancing epithelial restitution in wounded IEC-6 monolayers (32). Furthermore, TGF-β1 inhibits LPS-induced NF-κB activation in both intestinal epithelial cells and microglial cells (33, 34). Thus, for this study, we specifically investigated the effect of TGF-β1 against the pathophysiology of NEC using an animal model.
Others have studied the impact of TGF-β2 on the immature gut and NEC (35). These studies found that supplementation of TGF-β2 in the feed formula suppresses mucosal injury in two NEC animal models through a mechanism involving suppressed innate immunity in macrophages, whereas our study observed TGF-β1 suppression of innate immunity in epithelium. In addition, two animal models of NEC used in the same previous study (35) are different from the one used in this current study. In one of the NEC models used in the study using TGF-β2 (35), gut mucosal injury was induced in 10–12-day-old mouse pups by intraperitoneal administration of platelet-activating factor and LPS in an acute fashion, and mice were sacrificed 2 h immediately after platelet-activating factor and LPS administration. In the other NEC model used in the same study, 10-day-old mouse pups were separated from the dam, formula-fed, and exposed to hypoxia (5% oxygen for 2 min) twice daily before feedings to induce NEC, and pups were sacrificed 4 days later. In contrast, in our animal model, premature pups were obtained by cesarean section on embryonic day 20, 1 day before the completion of normal gestation, and thus were not exposed to breast milk. Using animals without exposure to breast milk is important to study the effect of oral administration of TGF-β in the gut as TGF-β is a breast milk factor and activation of Smad2 signaling is readily detected in dam-fed pups, presumably by active TGF-β in the breast milk (Fig. 1A). Thus, we used immature pups that had not been exposed to breast milk or factors present in the breast milk to directly examine the effect of oral administration of TGF-β1 in an NEC model.
TGF-β is expressed by intestine and intestinal epithelial cells at detectable levels (35, 36). Therefore the low basal level of phospho-Smad2 staining in intestine of vehicle-treated experimental NEC pups is presumably due to activation of TGF-β signaling by endogenous TGF-β produced by intestine. The absence of background staining in the negative control for phospho-Smad2 staining in Fig. 1A indicates that the phospho-Smad2 staining is specific. The TGF-β1-fed pups and dam-fed pups that were exposed to abundant TGF-β in breast milk had much stronger staining of phospho-Smad2 in the intestine as compared with the vehicle-fed group, indicating specific staining of phospho-Smad2 in tissues. Difference in technique leads to this basal Smad2 phosphorylation not being seen in Fig. 1C, which shows data in intestinal lysate by immunoblotting. However, the robust TGF-β1-stimulated Smad2 phosphorylation is confirmed and evident 20 h after TGF-β1 oral administration.
Mechanistically, the TGF-β1 inhibition of inflammatory NF-κB signaling appears to be mediated through the canonical Smad pathway as TGF-β suppressed IκBα phosphorylation, which results in protected IκBα, and restricted NF-κB activation is lost in cells with Smad4 knockdown in the in vitro study. An in vivo study to confirm the observed in vitro mechanism for the TGF-β1 will be an important next step but was beyond the scope of this present study; thus, immature intestinal epithelial cells were used as an in vitro model with biological and clinical relevance.
In conclusion, oral administration of TGF-β1 reduces the incidence of NEC through an immune suppressive effect directly on intestinal epithelium in vivo and decreased systemic IL-6 and IFN-γ levels. Mechanistic in vitro studies in the human immature fetal H4 IEC show that TGF-β1 suppressed NF-κB activation by protecting IκBα phosphorylation and subsequent degradation in a Smad-dependent manner. Our study provides useful insights into molecular mechanisms and strategies to control innate immune responses of the gut and suggest potential use of oral administration of TGF-β1 in treating or preventing gastrointestinal diseases.
Acknowledgment
We thank Zhe Liu, Ph.D., Department of Statistics, the University of Chicago, for assistance in analyzing multiplex cytokine data.
This work was supported, in whole or in part, by National Institutes of Health National Research Service Award (NRSA) F32 Fellowship Grants HD062144 (to S. S.) and HD059123 (to E. C. C.) and by the Digestive Disease Research Core Center of the University of Chicago (supported by National Institutes of Health Grant DK42086).
- NEC
- necrotizing enterocolitis
- IEC
- intestinal epithelial cell(s)
- IHC
- immunohistochemistry
- p
- phospho.
REFERENCES
- 1. Lemons J. A., Bauer C. R., Oh W., Korones S. B., Papile L. A., Stoll B. J., Verter J., Temprosa M., Wright L. L., Ehrenkranz R. A., Fanaroff A. A., Stark A., Carlo W., Tyson J. E., Donovan E. F., Shankaran S., Stevenson D. K. (2001) Very low birth weight outcomes of the National Institute of Child Health and Human Development Neonatal Research Network, January 1995 through December 1996. NICHD Neonatal Research Network. Pediatrics 107, E1. [DOI] [PubMed] [Google Scholar]
- 2. Claud E. C., Walker W. A. (2001) Hypothesis: inappropriate colonization of the premature intestine can cause neonatal necrotizing enterocolitis. FASEB J. 15, 1398–1403 [DOI] [PubMed] [Google Scholar]
- 3. Claud E. C., Lu L., Anton P. M., Savidge T., Walker W. A., Cherayil B. J. (2004) Developmentally regulated IκB expression in intestinal epithelium and susceptibility to flagellin-induced inflammation. Proc. Natl. Acad. Sci. U.S.A. 101, 7404–7408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frost B. L., Jilling T., Caplan M. S. (2008) The importance of pro-inflammatory signaling in neonatal necrotizing enterocolitis. Semin. Perinatol. 32, 100–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Solt L. A., May M. J. (2008) The IκB kinase complex: master regulator of NF-κB signaling. Immunol. Res. 42, 3–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Massagué J. (2008) TGFβ in cancer. Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kulkarni A. B., Huh C. G., Becker D., Geiser A., Lyght M., Flanders K. C., Roberts A. B., Sporn M. B., Ward J. M., Karlsson S. (1993) Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U.S.A. 90, 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yeganeh B., Mukherjee S., Moir L. M., Kumawat K., Kashani H. H., Bagchi R. A., Baarsma H. A., Gosens R., Ghavami S. (2013) Novel non-canonical TGF-β signaling networks: emerging roles in airway smooth muscle phenotype and function. Pulm. Pharmacol. Ther. 26, 50–63 [DOI] [PubMed] [Google Scholar]
- 9. Rautava S., Lu L., Nanthakumar N. N., Dubert-Ferrandon A., Walker W. A. (2012) TGF-β2 induces maturation of immature human intestinal epithelial cells and inhibits inflammatory cytokine responses induced via the NF-κB pathway. J. Pediatr. Gastroenterol. Nutr. 54, 630–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cavin L. G., Romieu-Mourez R., Panta G. R., Sun J., Factor V. M., Thorgeirsson S. S., Sonenshein G. E., Arsura M. (2003) Inhibition of CK2 activity by TGF-β1 promotes IκB-α protein stabilization and apoptosis of immortalized hepatocytes. Hepatology 38, 1540–1551 [DOI] [PubMed] [Google Scholar]
- 11. Azuma M., Motegi K., Aota K., Yamashita T., Yoshida H., Sato M. (1999) TGF-β1 inhibits NF-κB activity through induction of IκB-α expression in human salivary gland cells: a possible mechanism of growth suppression by TGF-β1. Exp. Cell Res. 250, 213–222 [DOI] [PubMed] [Google Scholar]
- 12. Sanderson I. R., Walker W. A. (1995) Establishment of a human fetal small intestinal epithelial cell line. Int. Arch. Allergy Immunol. 107, 396–397 [DOI] [PubMed] [Google Scholar]
- 13. Caplan M. S., Hedlund E., Adler L., Hsueh W. (1994) Role of asphyxia and feeding in a neonatal rat model of necrotizing enterocolitis. Pediatr. Pathol. 14, 1017–1028 [DOI] [PubMed] [Google Scholar]
- 14. Mackinnon A. C., Tretiakova M., Henderson L., Mehta R. G., Yan B. C., Joseph L., Krausz T., Husain A. N., Reid M. E., Salgia R. (2011) Paxillin expression and amplification in early lung lesions of high-risk patients, lung adenocarcinoma and metastatic disease. J. Clin. Pathol. 64, 16–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shiou S. R., Datta P. K., Dhawan P., Law B. K., Yingling J. M., Dixon D. A., Beauchamp R. D. (2006) Smad4-dependent regulation of urokinase plasminogen activator secretion and RNA stability associated with invasiveness by autocrine and paracrine transforming growth factor-β. J. Biol. Chem. 281, 33971–33981 [DOI] [PubMed] [Google Scholar]
- 16. Inan M. S., Rasoulpour R. J., Yin L., Hubbard A. K., Rosenberg D. W., Giardina C. (2000) The luminal short-chain fatty acid butyrate modulates NF-κB activity in a human colonic epithelial cell line. Gastroenterology 118, 724–734 [DOI] [PubMed] [Google Scholar]
- 17. Nowak D. E., Tian B., Brasier A. R. (2005) Two-step cross-linking method for identification of NF-κB gene network by chromatin immunoprecipitation. BioTechniques 39, 715–725 [DOI] [PubMed] [Google Scholar]
- 18. Li D. H., Kumanogoh A., Cao T. M., Parnes J. R., Cullen J. M. (2004) Woodchuck interleukin-6 gene: structure, characterization, and biologic activity. Gene 342, 157–164 [DOI] [PubMed] [Google Scholar]
- 19. Kim K. S., Rajagopal V., Gonsalves C., Johnson C., Kalra V. K. (2006) A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J. Immunol. 177, 7211–7224 [DOI] [PubMed] [Google Scholar]
- 20. Rupec R. A., Poujol D., Grosgeorge J., Carle G. F., Livolsi A., Peyron J. F., Schmid R. M., Baeuerle P. A., Messer G. (1999) Structural analysis, expression, and chromosomal localization of the mouse ikba gene. Immunogenetics 49, 395–403 [DOI] [PubMed] [Google Scholar]
- 21. Saito S., Yoshida M., Ichijo M., Ishizaka S., Tsujii T. (1993) Transforming growth factor-β (TGF-β) in human milk. Clin. Exp. Immunol. 94, 220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Claud E. C., Savidge T., Walker W. A. (2003) Modulation of human intestinal epithelial cell IL-8 secretion by human milk factors. Pediatr. Res. 53, 419–425 [DOI] [PubMed] [Google Scholar]
- 23. Sun S. C., Ganchi P. A., Ballard D. W., Greene W. C. (1993) NF-κB controls expression of inhibitor IκBα: evidence for an inducible autoregulatory pathway. Science 259, 1912–1915 [DOI] [PubMed] [Google Scholar]
- 24. Kafetzis D. A., Skevaki C., Costalos C. (2003) Neonatal necrotizing enterocolitis: an overview. Curr. Opin. Infect. Dis. 16, 349–355 [DOI] [PubMed] [Google Scholar]
- 25. De Plaen I. G., Liu S. X., Tian R., Neequaye I., May M. J., Han X. B., Hsueh W., Jilling T., Lu J., Caplan M. S. (2007) Inhibition of nuclear factor-κB ameliorates bowel injury and prolongs survival in a neonatal rat model of necrotizing enterocolitis. Pediatr. Res. 61, 716–721 [DOI] [PubMed] [Google Scholar]
- 26. Cho M. L., Min S. Y., Chang S. H., Kim K. W., Heo S. B., Lee S. H., Park S. H., Cho C. S., Kim H. Y. (2006) Transforming growth factor β 1(TGF-β1) down-regulates TNFα-induced RANTES production in rheumatoid synovial fibroblasts through NF-κB-mediated transcriptional repression. Immunol. Lett. 105, 159–166 [DOI] [PubMed] [Google Scholar]
- 27. Shiou S. R., Yu Y., Chen S., Ciancio M. J., Petrof E. O., Sun J., Claud E. C. (2011) Erythropoietin protects intestinal epithelial barrier function and lowers the incidence of experimental neonatal necrotizing enterocolitis. J. Biol. Chem. 286, 12123–12132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blobe G. C., Schiemann W. P., Lodish H. F. (2000) Role of transforming growth factor β in human disease. N. Engl. J. Med. 342, 1350–1358 [DOI] [PubMed] [Google Scholar]
- 29. Massagué J. (1990) The transforming growth factor-β family. Annu. Rev. Cell Biol. 6, 597–641 [DOI] [PubMed] [Google Scholar]
- 30. Shull M. M., Ormsby I., Kier A. B., Pawlowski S., Diebold R. J., Yin M., Allen R., Sidman C., Proetzel G., Calvin D., et al. (1992) Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 359, 693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., Doetschman T. (1997) TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKaig B. C., Hughes K., Tighe P. J., Mahida Y. R. (2002) Differential expression of TGF-β isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am. J. Physiol. Cell Physiol. 282, C172–C182 [DOI] [PubMed] [Google Scholar]
- 33. Haller D., Holt L., Kim S. C., Schwabe R. F., Sartor R. B., Jobin C. (2003) Transforming growth factor-β1 inhibits non-pathogenic Gram negative bacteria-induced NF-κB recruitment to the interleukin-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J. Biol. Chem. 278, 23851–23860 [DOI] [PubMed] [Google Scholar]
- 34. Le Y., Iribarren P., Gong W., Cui Y., Zhang X., Wang J. M. (2004) TGF-β1 disrupts endotoxin signaling in microglial cells through Smad3 and MAPK pathways. J. Immunol. 173, 962–968 [DOI] [PubMed] [Google Scholar]
- 35. Maheshwari A., Kelly D. R., Nicola T., Ambalavanan N., Jain S. K., Murphy-Ullrich J., Athar M., Shimamura M., Bhandari V., Aprahamian C., Dimmitt R. A., Serra R., Ohls R. K. (2011) TGF-β2 suppresses macrophage cytokine production and mucosal inflammatory responses in the developing intestine. Gastroenterology 140, 242–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chung H. L., Hwang J. B., Park J. J., Kim S. G. (2002) Expression of transforming growth factor β1, transforming growth factor type I and II receptors, and TNF-α in the mucosa of the small intestine in infants with food protein-induced enterocolitis syndrome. J. Allergy Clin. Immunol. 109, 150–154 [DOI] [PubMed] [Google Scholar]