

Background: G protein-coupled receptors (GPCRs) exist in dimeric/oligomeric arrays but the structural basis underling this phenomenon is not well understood.
Results: The M3 muscarinic receptor, a prototypic GPCR, can form multiple, structurally distinct dimeric species.
Conclusion: GPCRs may exist in a dynamic equilibrium of multiple dimers or oligomers.
Significance: Our findings may facilitate efforts to target GPCR dimers for therapeutic purposes.
Keywords: Cholinergic Receptor, G Protein-coupled Receptors (GPCR), Neurotransmitter Receptors, Receptor Structure-function, Site-directed Mutagenesis
Abstract
Class A G protein-coupled receptors (GPCRs) are able to form homodimers and/or oligomeric arrays. We recently proposed, based on bioluminescence resonance energy transfer studies with the M3 muscarinic receptor (M3R), a prototypic class A GPCR, that the M3R is able to form multiple, structurally distinct dimers that are probably transient in nature (McMillin, S. M., Heusel, M., Liu, T., Costanzi, S., and Wess, J. (2011) J. Biol. Chem. 286, 28584–28598). To provide more direct experimental support for this concept, we employed a disulfide cross-linking strategy to trap various M3R dimeric species present in a native lipid environment (transfected COS-7 cells). Disulfide cross-linking studies were carried out with many mutant M3Rs containing single cysteine (Cys) substitutions within two distinct cytoplasmic M3R regions, the C-terminal portion of the second intracellular loop (i2) and helix H8 (H8). The pattern of cross-links that we obtained, in combination with molecular modeling studies, was consistent with the existence of two structurally distinct M3R dimer interfaces, one involving i2/i2 contacts (TM4-TM5-i2 interface) and the other one characterized by H8-H8 interactions (TM1-TM2-H8 interface). Specific H8-H8 disulfide cross-links led to significant impairments in M3R-mediated G protein activation, suggesting that changes in the structural orientation or mobility of H8 are critical for efficient receptor-G protein coupling. Our findings provide novel structural and functional insights into the mechanisms involved in M3R dimerization (oligomerization). Because the M3R shows a high degree of sequence similarity with many other class A GPCRs, our findings should be of considerable general interest.
Introduction
Members of the superfamily of G protein-coupled receptors (GPCRs)2 are expressed by virtually every cell type and regulate the activity of all major physiological processes (1). Importantly, GPCRs are the target of a huge number of clinically useful drugs (2). Among the various subclasses of GPCRs, the class A subfamily represents by far the largest subgroup of GPCRs, consisting of ∼670 members in humans (3).
Several studies have demonstrated that class A GPCRs are able to interact with heterotrimeric G proteins in their monomeric forms (4–6). However, a large body of evidence obtained from different experimental approaches indicates that class A GPCRs are able to form dimeric or oligomeric complexes (7–11). Many studies have shown that the formation of such complexes can affect various aspects of GPCR function, including (but not limited to) receptor-G protein coupling efficiency and selectivity (7–11). Interestingly, a pentameric complex between a rhodopsin dimer and transducin has recently been visualized by transmission electron microscopy single particle reconstructions (12).
To better understand how class A GPCRs function at the molecular level, it is essential to identify the structural elements governing the dimerization/oligomerization of this class of receptors. Despite numerous molecular, biochemical, and biophysical studies, many questions remain regarding the molecular mechanisms underlying class A GPCR complex formation. Accumulating evidence suggests that residues located on the “outer” (lipid-facing) surface of the TM helical bundle and various intracellular regions participate in the formation of class A GPCR dimers/oligomers (7–11). However, depending on the specific receptor under investigation, different receptor regions have been implicated in receptor-receptor interactions. As a result, no clear consensus has emerged regarding the relative roles of different TM and extramembranous receptor regions in class A GPCR dimerization. However, such knowledge may prove critical for the development of novel strategies aimed at modulating GPCR dimerization (oligomerization) for therapeutic purposes.
For many years, we have used the M3 muscarinic acetylcholine receptor (M3R) as a model system to investigate different aspects of GPCR function, including the molecular basis of class A GPCR complex formation (for the sake of simplicity, we will refer to receptor dimers and oligomers simply as “receptor dimers” throughout the text). The M3R is a member of the muscarinic receptor family (M1–M5) that preferentially activates G proteins of the Gq family (13). The existence of M3R dimers has been confirmed by different experimental approaches, including mutant M3R complementation (14), disulfide cross-linking (15, 16), and co-immunoprecipitation (15, 16) studies, as well as bioluminescence resonance energy transfer (BRET) techniques (17, 18).
By using a BRET strategy, we recently identified many mutant M3Rs that were impaired in their ability to dimerize (18). The pattern of mutant receptors exhibiting impaired dimerization, together with molecular modeling studies, suggested the existence of multiple energetically favorable M3R dimers endowed with different geometries. Moreover, our data were in good agreement with a model in which these structurally distinct M3R dimers exist in a dynamic equilibrium (18).
In the present study, we used a disulfide cross-linking strategy to provide additional experimental support for the existence of multiple M3R-M3R interfaces and to further define the receptor regions involved in M3R dimerization. Previous studies examining the molecular basis of class A GPCR dimerization have focused primarily on the role of different TM helices in receptor-receptor interactions. In contrast, in the present study, we tested the potential proximity of two intracellular receptor regions in different M3R dimeric species. These regions were the C-terminal portion of the second intracellular loop (i2 loop) where it links to TM4, and helix 8 (H8), the intracellular helix that follows TM7 (Fig. 1). The primary reason for targeting these two intracellular domains was the outcome of the BRET study by McMillin et al. (18). In this study, we predicted the existence of an M3R dimer, which contains an H8-H8 interface and another dimeric species where the two i2 loops of two M3R protomers are located within close proximity of each other.
FIGURE 1.

Scheme highlighting the M3R sequences that were subjected to Cys substitution mutagenesis. Single Cys substitutions were introduced into a modified version of the rat M3R that lacked most endogenous Cys residues (M3′(3C)-Xa; see ”Experimental Procedures“ for details). Endogenous Cys residues were replaced with either serine or alanine (open squares), except for Cys140, Cys220, and Cys532 (19). Solid circles indicate amino acids that were subjected to Cys substitution mutagenesis (Arg176–Thr180 and Lys548–Thr556).
We systematically subjected residues located with the C-terminal portion of the i2 loop (residues Arg176-Thr180) and H8 (residues Lys548–Thr556) of the M3R to cysteine (Cys) substitution mutagenesis, resulting in 14 mutant M3Rs containing singly Cys residues on the intracellular M3R surface (Fig. 1). To explore the possibility that the targeted sites face each other in an M3R complex, we monitored the ability of the different receptor constructs to undergo cross-link formation (under oxidizing conditions). Importantly, these studies were carried out with receptors being present in a native membrane environment (membranes prepared from transfected COS-7 cells). The cross-linking data that we obtained, combined with molecular modeling studies, provide strong experimental support for the existence of multiple M3R dimers or M3R-M3R interfaces. We also noted that agonist treatment of receptor-expressing cells promoted the proximity of residues located within H8 in an M3R complex that was characterized by an H8-H8 interface. Functional data suggested that this conformational change is required for productive receptor-G protein coupling.
The present study provides novel information about the structure and function of M3R dimers. Class A GPCRs share a high degree of structural and functional homology. For this reason, the outcome of the present study should be of broad general relevance.
EXPERIMENTAL PROCEDURES
Materials
Carbamylcholine chloride (carbachol), atropine sulfate, cupric sulfate (CuSO4), 1,10-phenanthroline, and N-ethylmaleimide were obtained from Sigma. [3H]N-Methylscopolamine ([3H]NMS; 85.4 Ci/mmol) and myo-[3H]inositol (20 Ci/mmol) were purchased from PerkinElmer Life Sciences. [35S]GTPγS (1,000–1,200 Ci/mmol) was from PerkinElmer. CuSO4 was mixed with 1,10-phenanthroline at a molar ratio of 1:3. Throughout the text, concentrations given for the Cu(II)-(1,10-phenanthroline)3 complex (CuPhen) refer to molar copper concentrations. Protein A-Sepharose CL-4B was obtained from GE Healthcare. The rabbit Gαq/11 antiserum was generated against the C-terminal Gαq/11 sequence (C)LQLNLKEYNLV (16).
Generation of Cys-substituted Mutant M3R Constructs
Cys substitutions were introduced into a mammalian expression plasmid (pCD) coding for a modified version of the rat M3R (M3′(3C)-Xa; Ref. 19; see Fig. 1). The M3′(3C)-Xa construct lacks most endogenous Cys residues (except for Cys140, Cys220, and Cys532), all five N-glycosylation sites contained within the extracellular N-terminal domain, and the central portion of the third intracellular loop (Ala274–Lys469; this sequence was replaced with two adjacent factor Xa cleavage sites). Single Cys residues were introduced at specific sites of the M3′(3C)-Xa receptor by using the QuikChangeTM site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The entire coding sequences of all mutant M3Rs were sequenced to exclude the presence of any unwanted mutations.
Expression of Mutant M3Rs in COS-7 Cells
All receptor constructs were transiently expressed in COS-7 cells, as described in detail previously (20). Transfected cells were incubated with 1 μm atropine for the last 24 h of culture, to obtain higher receptor densities. About 48 h after transfections, COS-7 cells were harvested, and membranes were prepared for radioligand binding and cross-linking studies as described (20).
Radioligand Binding Studies
Radioligand binding studies were performed with whole cell membrane preparations obtained from transfected COS cells as described previously (20). In brief, binding reactions containing ∼10 μg of membrane protein per tube were carried out for 2 h (22 °C) in 1 ml of binding buffer containing 25 mm sodium phosphate and 5 mm MgCl2 (pH 7.4). In saturation binding assays, we employed six different [3H]NMS concentrations (200 to 7,000 pm). [3H]NMS is a hydrophilic, plasma membrane-impermeable muscarinic antagonist. When we used intact cells in the past, we detected similar numbers of [3H]NMS binding sites as compared with whole cell membrane preparations, indicating that [3H]NMS primarily labels cell surface receptors.3 In competition binding assays, we used a fixed concentration of [3H]NMS (500 pm) in the presence of 10 different concentrations of carbachol, the cold competitor. Nonspecific binding was defined as binding observed in the presence of 10 μm atropine. Binding reactions were terminated by rapid filtration over GF/C Brandel filters, followed by three washes (∼4 ml/wash) with ice-cold distilled water. The amount of radioactivity that remained bound to the filters was determined by liquid scintillation spectrometry. Binding data were analyzed by using the nonlinear curve-fitting program Prism 4.0 (GraphPad Software, San Diego, CA).
Phosphoinositide Assay
To determine whether the different mutant M3Rs retained the ability to couple to G proteins, we studied their ability to mediate agonist-dependent stimulation of phosphoinositide hydrolysis. Assays were carried out as described in detail previously (20). In brief, transfected COS-7 cells were split into 12-well plates 24 h after transfection and labeled with myo-[3H]inositol. Assays were carried out by incubating receptor-expressing cells with increasing concentrations of the muscarinic agonist carbachol. Data were analyzed using the nonlinear curve-fitting program Prism 4.0 (GraphPad Software, San Diego, CA).
Disulfide Cross-linking Experiments
Disulfide cross-linking studies were carried out as described in detail previously (16, 20). In brief, receptor-containing COS-7 cell membranes were incubated with the oxidizing agent, CuPhen (100 μm). Reactions were carried out for 10 min either at room temperature (23 °C) or at 37 °C and then terminated by the addition of EDTA and N-ethylmaleimide (10 mm each), followed by a 10-min incubation on ice. Membrane proteins were then solubilized by incubating samples with 1.2% digitonin (Roche Applied Science) and used either directly for SDS-PAGE or stored at −70 °C until use.
Western Blotting Studies
Immunoblotting experiments were carried out using published protocols (16, 20). In brief, samples containing 20 μg of solubilized membrane protein were incubated with Laemmli loading buffer (30 min at 37 °C) under non-reducing conditions and then loaded onto 10% Tris glycine polyacrylamide gels (Invitrogen) and run at 135 V in the presence of 0.1% SDS. Immunoblots were probed with a rabbit anti-M3R polyclonal antibody directed against the C-terminal 18 amino acids of the rat M3R (16) (Fig. 1). M3R monomers or dimers were visualized by using SuperSignal West Pico Chemiluminescent Substrate (Pierce) and autoradiography.
Co-immunoprecipitation Studies
Co-immunoprecipitation studies were carried out as described by Hu et al. (16). In brief, we first generated modified versions of selected Cys-substituted M3R constructs in which the C-terminal recognition sequence for the anti-M3R antibody was replaced with a FLAG epitope (DYKDDDDK). COS-7 cells were then co-transfected with the non-tagged and FLAG-tagged version of the same mutant M3R. After treatment of COS-7 cell membranes for 10 min with CuPhen (100 μm), solubilized membrane proteins (∼600 μg) were incubated for 1 h at 4 °C with either the anti-M3R antibody or an anti-FLAG monoclonal antibody (GenScript, Piscataway, NJ). After this step, 30 μl of protein A-agarose (anti-M3R antibody) or protein A/G-agarose (anti-FLAG antibody) were added, followed by an additional incubation period of 1 h at 4 °C. Bound immunoreactive proteins were eluted and immunoblotting studies were performed with the monoclonal anti-FLAG antibody or the polyclonal anti-M3R antibody, as described in detail previously (16).
[35S]GTPγS Binding/Immunoprecipitation Assay
To determine the effect of M3R cross-linking on the efficiency of receptor-G protein coupling, we followed the protocol described by Akam et al. (21). In brief, membranes from COS-7 cells expressing different Cys-substituted mutant M3R constructs were first treated with CuPhen (100 μm; 10 min at 37 °C). Control samples were incubated under the same experimental conditions in the absence of CuPhen. Subsequently, receptor-containing membranes (75 μg of protein) were incubated with the muscarinic agonist, carbachol (1 mm) for 2 min at 30 °C in the presence of 1 nm [35S]GTPγS. Following membrane solubilization and pre-clearing, Gαq/11 proteins were immunoprecipitated with a selective anti-Gαq/11-specific antiserum as described (21). Nonspecific binding was determined in the presence of 10 μm GTPγS.
Generation of M3R Dimer Models
On the basis of the crystal structure of the rat M3R (22) we generated models of all possible dimerization topologies through a systematic computational procedure based on a Python program operating within the MAESTRO environment of the Schrodinger package (16, 18, 23). Briefly, the dimer models were built through a systematic revolution of one monomer (the “mobile” monomer) around the other monomer, combined with rotations of the mobile monomer around its axis. Revolution and rotations were performed with increments of 10°. At each revolution step, before the mobile monomer advanced to the next step, a complete rotation of the mobile monomer around its axis was performed. For each rotational step, the two monomers were arranged at a distance that ensured minimum van der Waals energy, calculated according to the OPLS2005 force field. The process was continued until the mobile monomer completed an entire revolution around the other, leading to a total of 1,296 systematically built dimer models.
RESULTS
Introduction of Single Cys Residues into Membrane-Proximal Cytoplasmic Regions of the M3Rs
A modified version of the rat M3R lacking most native Cys residues (M3′(3C)-Xa) served as a template for Cys substitution mutagenesis (Fig. 1). The M3′(3C)-Xa construct displays similar ligand binding and G protein coupling properties as the wild-type (WT) M3R, as reported previously (19). In the present study, we tested the hypothesis that residues residing within the C-terminal portion of the i2 loop (adjacent to TM4) and H8 helix (adjacent to TM7) are located at possible M3R dimer interfaces. Specifically, we generated five mutant M3Rs containing single Cys substitutions within the i2 loop (residues Arg176-Thr180) and nine mutant M3Rs containing single Cys substitutions within H8 (residues Lys548–Thr556) (Fig. 1). After an initial pharmacological characterization of the various receptor constructs, we carried out disulfide cross-linking studies to explore the potential proximity of the targeted residues in M3R dimers.
Ligand Binding and Functional Properties of Cys-substituted Mutant M3Rs
We first examined whether the Cys-substituted mutant M3Rs retained the ability to bind muscarinic ligands with high affinity. [3H]NMS saturation binding studies using membranes prepared from transfected COS-7 cells demonstrated that all mutant M3Rs were able to bind [3H]NMS, a muscarinic antagonist, with high affinity, similar to that observed with the M3′(3C)-Xa construct from which all mutant receptors were derived (range of [3H]NMS KD values: ∼400–750 pm; Tables 1 and 2). Moreover, mutant M3R and M3′(3C)-Xa expression levels ([3H]NMS Bmax values) were in a similar range (∼11–16 pmol/mg; Tables 1 and 2). [3H]NMS/carbachol competition binding studies demonstrated that all mutant M3Rs were able to bind carbachol, a muscarinic agonist, with high affinity, in a fashion similar to the M3′(3C)-Xa construct (range of carbachol Ki values: ∼7–41 μm; Tables 1 and 2).
TABLE 1.
Ligand binding and functional properties of mutant M3Rs containing single Cys substitutions within the C-terminal segment of the i2 loop
COS-7 cells were transiently transfected with the indicated M3′(3C)-Xa-derived Cys-substituted mutant M3R constructs. Radioligand binding and phosphoinositide assays were performed and analyzed as described under ”Experimental Procedures.“ In all cases, carbachol inhibition binding curves were characterized by Hill coefficients ranging from 0.7 to 0.8. Basal inositol monophosphate (IP) levels for the M3′(3C)-Xa construct were 1905 ± 279 dpm (=100%). Data are shown as mean ± S.E. of 2–4 independent experiments, each performed in duplicate.
| Receptor | [3H]NMS binding |
Carbachol binding, |
Carbachol-induced IP production |
|||
|---|---|---|---|---|---|---|
| KD | Bmax | Ki | EC50 | Emax | Basal activity | |
| pm | pmol/mg of protein | μm | nm | Fold above basal | % | |
| M3′(3C)-Xa | 403 ± 128 | 16.2 ± 3.9 | 24.6 ± 7.6 | 11.3 ± 1.1 | 5.5 ± 0.7 | 100 |
| R176C | 484 ± 29 | 11.4 ± 0.4 | 22.0 ± 5.9 | 264 ± 11 | 7.9 ± 1.3 | 76 ± 3 |
| A177C | 521 ± 18 | 12.8 ± 1.9 | 41.3 ± 16.2 | 21.4 ± 5.7 | 5.2 ± 2.7 | 84 ± 9 |
| K178C | 499 ± 46 | 12.1 ± 1.1 | 29.5 ± 8.8 | 40.1 ± 9.6 | 4. 9 ± 2.3 | 109 ± 12 |
| R179C | 493 ± 33 | 11.5 ± 1.1 | 8.2 ± 1.9 | 2.6 ± 0.8 | 2.9 ± 1.4 | 106 ± 1 |
| T180C | 751 ± 27 | 13.0 ± 2.0 | 20.4 ± 3.7 | 22.9 ± 2.3 | 7.7 ± 0.4 | 90 ± 3 |
TABLE 2.
Ligand binding and functional properties of mutant M3Rs containing single Cys substitutions within the H8 helix
Radioligand binding and phosphoinositide assays were performed with COS-7 cells transiently transfected with the indicated M3′(3C)-Xa-derived Cys-substituted mutant M3Rs. Data were analyzed as described under ”Experimental Procedures.“ In all cases, carbachol inhibition binding curves were characterized by Hill coefficients ranging from 0.7 to 0.8. Basal inositol monophosphate (IP) levels for the M3′(3C)-Xa construct were 2683 ± 118 dpm ( = 100%). Data are given as mean ± S.E. of 2–5 independent experiments, each performed in duplicate.
| Receptor | [3H]NMS binding |
Carbachol binding |
Carbachol-induced IP production |
|||
|---|---|---|---|---|---|---|
| KD | Bmax | Ki | EC50 | Emax | Basal activity | |
| pm | pmol/mg of protein | μm | nm | Fold above basal | % | |
| M3′(3C)-Xa | 555 ± 15 | 14.9 ± 3.0 | 23.8 ± 1.2 | 28.0 ± 2.7 | 8.4 ± 0.3 | 100 |
| K548C | 555 ± 92 | 12.4 ± 2.9 | 17.0 ± 1.6 | 23.6 ± 3.27 | 6.9 ± 0.3 | 111 ± 10 |
| T549C | 564 ± 75 | 13.7 ± 0.9 | 12.6 ± 2.2 | 108 ± 5 | 8.5 ± 0.3 | 107 ± 15 |
| F550C | 556 ± 38 | 9.3 ± 1.2 | 6.7 ± 0.1 | 101 ± 22 | 11.9 ± 1.2 | 81 ± 7 |
| R551C | 524 ± 42 | 12.8 ± 0.9 | 19.5 ± 4.7 | 116 ± 24 | 10.9 ± 1.7 | 81 ± 7 |
| T552C | 438 ± 77 | 11.0 ± 0.6 | 14.6 ± 2.3 | 31.3 ± 2.7 | 9.8 ± 2.5 | 73 ± 6 |
| T553C | 548 ± 18 | 11.2 ± 0.7 | 13.7 ± 2.1 | 21.5 ± 2.4 | 5.8 ± 0.4 | 133 ± 17 |
| F554C | 583 ± 53 | 12.0 ± 0.7 | 8.2 ± 2.5 | 46.8 ± 11.2 | 7.7 ± 2.4 | 149 ± 10 |
| K555C | 553 ± 17 | 10.8 ± 1.1 | 18.5 ± 0.8 | 7.2 ± 0.3 | 11.8 ± 2.3 | 76 ± 1 |
| T556C | 462 ± 30 | 13.4 ± 3.0 | 18.2 ± 3.8 | 23.6 ± 2.5 | 9.4 ± 0.2 | 97 ± 3 |
To determine whether the various receptor constructs retained the ability to couple to G proteins of the Gq family, we measured carbachol-induced increases in inositol phosphate (IP) production using transfected COS-7 cells. As shown in Tables 1 and 2, all Cys-substituted mutant receptors, similar to the M3′(3C)-Xa construct, were capable of stimulating IP production in a carbachol-dependent fashion. However, several of the analyzed mutant receptors, including R176C in the i2 loop and T549C, F550C, and R551C in H8 showed significant decreases in carbachol potency (increases in carbachol EC50 values), as compared with M3′(3C)-Xa (∼4–23-fold; Tables 1 and 2). Two of the analyzed mutant receptors, R179C (i2) and T553C (H8), also showed a clear reduction (by ∼30–50%) in maximum IP production, as compared with the M3′(3C)-Xa construct (Tables 1 and 2). These findings are consistent with previous reports that the i2 loop and H8 contain structural elements that are critical for productive M3R/G protein coupling (13, 24).
Disulfide Cross-linking Studies with Cys-substituted Mutant M3Rs
Our initial goal was to detect potential intermolecular M3R-M3R contact sites involving residues contained within the C-terminal portion of the i2 loop preceding TM4 (residues Arg176–Thr180). To address this issue, we exposed membranes prepared from COS-7 cells expressing the R176C, A177C, K178C, R179C, or T180C receptors to the oxidizing agent CuPhen (100 μm) to promote the formation of disulfide bonds between vicinal Cys residues. Cross-linked receptor dimers were detected via Western blotting carried out under non-reducing conditions using a polyclonal antibody directed against the last 18 amino acids of the rat M3R (Fig. 1). As a positive control, we included the L264C mutant receptor in all cross-linking experiments (Leu264 is located at the N terminus of the i3 loop; Fig. 1). In a previous study, we demonstrated that this construct forms cross-linked M3R dimers with very high efficiency (16). When samples were treated with CuPhen at room temperature (23 °C), the R176C-T180C receptors could be detected as both monomeric and putative dimeric species in Western blotting studies (Fig. 2, left panel). Three of these receptors, R176C, R179C, and T180C, showed more pronounced dimer bands, as compared with the corresponding monomer bands. When receptor-containing membranes were treated with CuPhen at 37 °C, all five mutant receptors showed complete (R176C and T180C) or nearly complete (A177C, K178C, and R179C) cross-linking (Fig. 2, right panel). It should be noted that the size of the cross-linked immunoreactive bands observed with the R176C-T180C constructs was somewhat smaller than that of the cross-linked L264C receptor, which served as a positive control (∼70 versus ∼80 kDa, respectively, Fig. 2). The precise reason for this phenomenon remains unclear at present. However, previous studies have shown that branched polypeptides that were cross-linked at different sites showed marked differences in their mobility patterns on SDS gels (25), providing a possible explanation for the observed size differences among the cross-linked mutant M3Rs detected via Western blotting.
FIGURE 2.

Formation of disulfide cross-linked homodimers between mutant M3Rs containing Cys substitutions within the C-terminal portion of the i2 loop. Membranes prepared from COS-7 cells expressing the indicated Cys-substituted, M3′(3C)-Xa-derived receptor constructs were incubated with 100 μm CuPhen at room temperature or at 37 °C for 10 min. Subsequently, membrane lysates were subjected to SDS-PAGE under non-reducing conditions, followed by Western blotting analysis using the polyclonal anti-M3R antibody. As a positive control, we included the L264C-M3′(3C)-Xa construct (16). Representative Western blots are shown. Similar results were obtained in two or three independent experiments. Asterisks, putative receptor monomers; arrows, putative cross-linked receptor dimers.
To explore the possibility that H8 is part of an M3R dimer interface, we tested the ability of the K548C to T556C mutant M3Rs to undergo disulfide cross-linking after CuPhen treatment of receptor-expressing COS-7 cells membranes. When CuPhen treatment was performed at room temperature, three of the nine analyzed mutant receptors, T553C, K555C, and T556C, predominantly migrated as cross-linked dimers on SDS gels run under non-reducing conditions (Fig. 3, left panel). When samples were treated with CuPhen at 37 °C, all nine mutant receptors could be cross-linked with very high efficiency (Fig. 3, right panel). As observed with the R176C-T180C mutant receptors, the electrophoretic mobility of the cross-linked K548C-T556C constructs was significantly greater than that of the L264C receptor (∼60 versus ∼80 kDa, respectively). As already mentioned in the previous paragraph, this unusual running behavior may be due to the fact that the electrophoretic mobility of branched polypeptides critically depends on the actual site of cross-linking (25).
FIGURE 3.

Formation of disulfide cross-linked homodimers between mutant M3Rs containing Cys substitutions within the H8 helix. Cross-linking and Western blotting experiments were carried out as described in the legend to Fig. 2. As a positive control, we included the L264C-M3′(3C)-Xa construct (16). Representative Western blots are shown. Similar results were obtained in two or three independent experiments. Asterisks, putative receptor monomers; arrows, putative cross-linked receptor dimers.
Co-immunoprecipitation Experiments
To confirm that the ∼60–70-kDa immunoreactive species shown in Figs. 2 and 3 represent actual M3R dimers, we carried out a series of co-immunoprecipitation experiments. For these experiments, we chose two representative mutant receptors, R176C (i2) and T556C (H8), which showed efficient disulfide cross-linking even at room temperature. Initially, we generated modified versions of these two constructs in which the C-terminal recognition sequence for the anti-M3R antibody was replaced with a FLAG tag, resulting in R176C-FLAG and T556C-FLAG, respectively. COS-7 cells were transfected with the various constructs alone or co-transfected with R176C and R176C-FLAG (Fig. 4A) or T556C and T556C-FLAG (Fig. 4B). Receptor-expressing membrane samples were then treated with CuPhen (100 μm) to induce receptor cross-linking.
FIGURE 4.

Co-immunoprecipitation studies with representative mutant M3Rs confirming M3R homodimer formation. This figure shows co-immunoprecipitation data for two representative Cys-substituted M3R constructs, R176C (i2 loop) and T556C (H8). A, co-immunoprecipitation of cross-linked R176C and R176C-FLAG mutant M3Rs. B, co-immunoprecipitation of cross-linked T556C and T556C-FLAG mutant M3Rs. For these studies, we generated modified versions of the R176C and T556C constructs in which the C-terminal recognition sequence for the anti-M3R antibody (Fig. 1) was replaced with a FLAG tag, resulting in the R176C-FLAG and T556C-FLAG mutant M3Rs, respectively. Membrane samples prepared from COS-7 cells co-expressing R176C and R176C-FLAG (A) or T556C and T556C-FLAG (B) were treated for 10 min with 100 μm CuPhen as described under “Experimental Procedures.” Solubilized membrane proteins were then incubated with the anti-M3R polyclonal antibody (IP: anti-M3) or a monoclonal anti-FLAG antibody (IP: anti-FLAG). After this step, protein A-agarose was added, and the bound immunoreactive proteins were eluted and blotted with a monoclonal anti-FLAG antibody (immunoblot (IB): anti-FLAG) or the anti-M3R polyclonal antibody (IB: anti-M3) under non-reducing conditions. For comparison, Western blots of membrane proteins prior to the IP step are shown in the left panels (“before IP”). The blots shown are representative of two or three independent experiments. Arrows indicate putative cross-linked receptor dimers.
Following membrane lysis, M3R-containing proteins were immunoprecipitated with the polyclonal anti-M3 antibody, followed by immunoblotting studies using a monoclonal anti-FLAG antibody (Fig. 4, left sides of panels A and B). In a reciprocal fashion, M3R-containing proteins were immunoprecipitated with the monoclonal anti-FLAG antibody, followed by Western blotting analysis using the polyclonal anti-M3 antibody (Fig. 4, right sides of panels A and B). In these co-immunoprecipitation experiments, the ∼70-kDa (R176C) and ∼60-kDa (T556C) bands, respectively, were the only immunoreactive species that could be detected by both the anti-M3 and anti-FLAG antibodies (Fig. 4). These bands were not observed in samples derived from cells transfected with the individual receptor constructs alone. These data strongly support the concept that the ∼60–70-kDa bands observed on Western blots after CuPhen treatment of mutant M3R-containing membranes (Figs. 2 and 3) represent actual M3R dimers.
Cross-linking Studies at Low Mutant M3R Expression Levels
For the studies described above (Figs. 2 and 3), the various M3R constructs were all expressed at relatively high levels (∼9–16 pmol/mg; Tables 1 and 2). To explore whether receptor cross-linking patterns similar to those shown in Figs. 2 and 3 could also be observed at much lower (more physiological) receptor levels, we expressed the R176C-T180C constructs and two representative H8 Cys-substituted mutant receptors, T553C and T556C, at ∼0.2 pmol/mg by reducing the amount of transfected receptor DNA. After treatment of receptor-expressing COS-7 cell membranes with CuPhen (100 μm) for 10 min at room temperature, all analyzed constructs were able to form disulfide cross-linked dimers with very high efficiency, as demonstrated in immunoblotting studies carried out under non-reducing conditions (Fig. 5).
FIGURE 5.

Formation of disulfide cross-linked mutant M3R dimers after drastic reduction of receptor expression levels. To achieve relatively low receptor densities (∼0.2 pmol/mg of membrane protein), COS-7 cells were transfected with reduced amounts of plasmid DNA coding for the indicated receptor constructs (see ”Experimental Procedures“ for details). As a positive control, we included the L264C-M3′(3C)-Xa construct (16). Subsequently, membranes prepared from these cells were incubated with 100 μm CuPhen for 10 min at room temperature. Membrane extracts were then subjected to SDS-PAGE under non-reducing conditions, and receptor proteins were visualized via Western blotting using the polyclonal anti-M3R antibody. The blot shown is representative of three independent experiments. Asterisk, putative receptor monomers; arrows, putative cross-linked receptor dimers.
Effect of Agonist Treatment on Disulfide Cross-linking Patterns
To explore whether receptor activation affected the observed disulfide cross-linking patterns (Figs. 2 and 3), we performed cross-linking studies using membranes prepared from receptor-expressing COS-7 cells, either in the absence or presence of the muscarinic agonist, carbachol (1 mm). To quantitate the effect of carbachol on M3R-M3R cross-linking, we used scanning densitometry to compare the intensities of immunoreactive bands corresponding to putative receptor dimers obtained with carbachol-exposed membranes with those determined with membranes containing the same receptors but not treated with carbachol. The results of representative cross-linking experiments are shown in Figs. 6 and 7. In the case of the R176C-T180C mutant M3Rs, agonist treatment had little or no effect on disulfide cross-linking patterns (Fig. 6). In contrast, carbachol treatment of membranes containing the T553C (H8) and K555C (H8) constructs led to pronounced increases in M3R cross-link formation (Fig. 7). Agonist exposure of T553C- or K555C-containing cell membranes increased dimer formation by 97 ± 17 and 50 ± 10% (mean ± S.E.; n = 3), respectively, as compared with membranes containing the same receptors but not treated with carbachol. Similar results were obtained in co-immunoprecipitation studies (Fig. 8). Agonist treatment of membranes containing several other H8 Cys mutant M3Rs also triggered significant increases in dimer formation, although to a lesser extent (increase in dimer band intensity in %): R551C, 19 ± 9%; T552C, 43 ± 13%; F554C, 12 ± 5% (mean ± S.E.; n = 3; Fig. 7).
FIGURE 6.

Formation of disulfide cross-links between mutant M3Rs containing Cys substitutions within the i2 loop is agonist-independent. Membranes prepared from COS-7 cells expressing the indicated mutant M3Rs containing Cys substitutions within the C-terminal segment of the i2 loop were incubated for 10 min with 100 μm CuPhen (at room temperature) in the absence (−) or presence (+) of 1 mm carbachol (CCh). As a positive control, we included the L264C-M3′(3C)-Xa construct (16). Subsequently, membrane extracts were subjected to SDS-PAGE under non-reducing conditions, and receptor proteins were visualized via Western blotting using the polyclonal anti-M3R antibody. The blot shown is representative of three independent experiments. Asterisk, putative receptor monomers; arrow, putative cross-linked receptor dimers.
FIGURE 7.

Formation of disulfide cross-links is agonist-dependent for several mutant M3Rs containing Cys substitutions within H8. Experiments were carried out in the same fashion as described in the legend to Fig. 6. The blot shown is representative of three independent experiments. Note that carbachol treatment (CCh; 1 mm) facilitated cross-link formation in several cases. Asterisk, putative receptor monomers; arrow, putative cross-linked receptor dimers.
FIGURE 8.

Co-immunoprecipitation studies confirm increased disulfide cross-linking after agonist treatment of T553C- and K555C-containing membranes. This figure shows co-immunoprecipitation data for T553C (A) and K555C (B) mutant M3Rs. Initially, we generated modified versions of T553C and K555C receptors in which the C-terminal recognition sequence for the anti-M3R antibody (Fig. 1) was replaced with a FLAG tag, resulting in the T553C-FLAG and K555C-FLAG constructs, respectively. Membrane samples prepared from COS-7 cells co-expressing T553C and T553C-FLAG (A) or K555C and K555C-FLAG (B) were incubated with (+) or without (−) carbachol (CCh; 1 mm), followed by the addition of 100 μm CuPhen (incubation for 10 min at room temperature). After incubation of solubilized membrane proteins with a monoclonal anti-FLAG antibody (IP: anti-FLAG), protein A/G-agarose was added, and the bound immunoreactive proteins were eluted and blotted with the anti-M3R polyclonal antibody (immunoblot (IB): anti-M3) under non-reducing conditions. In both A and B, Western blots of membrane proteins prior to the IP step are also shown (before IP; left panels). The blots shown are representative of two independent experiments. Putative cross-linked receptor dimers are marked by arrows.
CuPhen Is Required for the Formation of Cross-linked M3R Dimers
To investigate whether cross-linked M3R dimers were able to form even in the absence of the oxidizing agent, CuPhen, we carried out a series of additional cross-linking studies. As shown in Figs. 9 and 10, we observed little or no M3R cross-linking in the absence of CuPhen. This observation indicates that the presence of CuPhen is essential for the formation of disulfide cross-linked dimers observed with several of the investigated Cys-substituted mutant M3Rs.
FIGURE 9.

Effect of CuPhen and atropine on the formation of disulfide cross-linked M3R dimers (i2 loop Cys mutants). Membranes prepared from COS-7 cells expressing the indicated mutant M3Rs containing Cys substitutions within the C-terminal segment of the i2 loop were incubated with (+) or without (−) 100 μm CuPhen for 10 min (at room temperature). CuPhen-treated samples were left either untreated or incubated with atropine (Atr; 1 μm), a muscarinic antagonist/inverse agonist. The L264C-M3′(3C)-Xa construct (16) served as a positive control. Membrane extracts were then subjected to SDS-PAGE under non-reducing conditions, and M3Rs were detected via Western blotting using the polyclonal anti-M3R antibody. The observed pattern of bands indicates that CuPhen is required for efficient M3R-M3R disulfide cross-linking displayed by some of the studied mutant receptors and that atropine has no significant effect on the efficiency of M3R dimer formation. The blots shown are representative of two independent experiments. Asterisk, putative receptor monomers; arrow, putative cross-linked receptor dimers.
FIGURE 10.

Effect of CuPhen and atropine on the formation of disulfide cross-linked M3R dimers (H8 Cys mutants). Experiments were carried out in the same fashion as described in the legend to Fig. 9. In the absence of CuPhen, multiple immunoreactive bands can be detected, including a ∼60-kDa band that is similar in size to that of cross-linked M3R-M3R dimers (H8 Cys mutant M3Rs). However, it is unlikely that this band represents a cross-linked M3R dimer, because it is also observed with the M3′(3C)-Xa construct that lacks Cys residues available for disulfide cross-link formation. The Western blotting data indicate that CuPhen is required for efficient M3R-M3R disulfide cross-linking displayed by some of the H8 Cys mutant receptors. Moreover, with the exception of the 553C construct, atropine treatment (Atr; 1 μm) has no significant effect on the efficiency of M3R dimer formation. Interestingly, atropine selectively facilitates the formation of T553C mutant M3R dimers. The blots shown are representative of two independent experiments. Asterisk, putative receptor monomers; arrow, putative cross-linked receptor dimers.
Effect of Antagonist Treatment on Disulfide Cross-linking Patterns
To examine whether the observed disulfide cross-linking patterns (Figs. 2 and 3) were affected by treatment with a muscarinic antagonist, we carried out cross-linking studies using membranes prepared from receptor-expressing COS-7 cells, either in the absence or presence of the muscarinic antagonist (inverse agonist), atropine (1 μm). The results of representative cross-linking experiments are shown in Figs. 9 and 10. Except for the T553C mutant M3R (Fig. 10, center panel), atropine treatment had essentially no effect on the observed disulfide cross-linking patterns. However, atropine exposure of membranes expressing the T553C construct caused a significant increase (by ∼50%) in M3R cross-link formation (Fig. 10, center panel).
Functional Activity of Cross-linked M3R Dimers
As described above, CuPhen treatment of several Cys-substituted mutant M3Rs resulted in the efficient formation of cross-linked M3R dimers, trapping structurally distinct receptor dimers containing either an i2-i2 or an H8-H8 interface. To explore whether these cross-linked M3R dimers retained the ability to couple to G proteins, we determined receptor-mediated G protein activation by treating non-oxidized or oxidized (CuPhen-treated) membranes with carbachol (1 mm) and then monitored G protein activation using a [35S]GTPγS binding/Gαq/11 immunoprecipitation assay (21). For these studies, we selected five representative mutant M3Rs (R176C, T180C, F550C, T553C, and T556C) that showed highly efficient cross-link formation (Figs. 2 and 3). For these experiments, CuPhen treatment of receptor-expressing membranes was performed at 37 °C (100 μm CuPhen for 10 min), because cross-link formation was usually not complete when CuPhen treatment was carried out at room temperature (Figs. 2 and 3).
As shown in Table 3, the cross-linking procedure had little or no effect on [3H]NMS and carbachol binding affinities. However, we noted that [3H]NMS Bmax values were significantly reduced after treatment of samples with CuPhen at 37 °C (Table 3). Under these experimental conditions, several of the mutant receptors (T180C, F550C, and T553C) were expressed at considerably lower levels than the M3′(3C)-Xa construct from which they were derived. For this reason, we reduced the amount of transfected M3′(3C)-Xa DNA (from 4 to 1 and 0.2 μg of plasmid DNA) to obtain Bmax levels similar to those observed with these mutant receptors (see Table 3).
TABLE 3.
Ligand binding properties of selected Cys-substituted mutant M3Rs cross-linked at 37 °C
COS-7 cells were transiently transfected with the indicated M3′(3C)-Xa-derived Cys-substituted mutant M3R constructs (4 μg of plasmid DNA each). Radioligand binding assays were performed using membranes prepared from these cells after incubation with CuPhen (100 μm) or CuPhen-free buffer for 10 min at 37 °C. To express the M3′(3C)-Xa construct at different densities, we transfected COS-7 cells with three different amounts of plasmid DNA, as indicated. In all cases, carbachol inhibition binding curves were characterized by Hill coefficients ranging from 0.7 to 0.8. Data are shown as mean ± S.E. of 3–7 independent experiments, each performed in duplicate.
| Receptor | [3H]NMS, Bmax |
CuPhen buffer |
||
|---|---|---|---|---|
| CuPhen-free buffer | CuPhen buffer | [3H]NMS, KD | Carbachol, Ki | |
| pmol/mg of protein | nm | μm | ||
| Vector | 0.01 ± 0.03 | 0.01 ± 0.00 | ||
| M3′(3C)-Xa (0.2 μg) | 0.45 ± 0.14 | 0.28 ± 0.08 | ||
| M3′(3C)-Xa (1 μg) | 1.15 ± 0.21 | 0.99 ± 0.25 | ||
| M3′(3C)-Xa (4 μg) | 3.21 ± 0.40 | 1.97 ± 0.32 | 0.44 ± 0.08 | 33.2 ± 7.0 |
| R176C (i2) | 2.44 ± 0.81 | 0.79 ± 0.33 | 0.47 ± 0.06 | 9.0 ± 5.0 |
| T180C (i2) | 1.60 ± 0.86 | 0.31 ± 0.13 | 1.11 ± 0.29 | 27.9 ± 3.3 |
| F550C (H8) | 1.03 ± 0.27 | 0.25 ± 0.04 | 1.15 ± 0.37 | 19.3 ± 5.3 |
| T553C (H8) | 1.66 ± 0.47 | 0.38 ± 0.07 | 1.02 ± 0.27 | 19.7 ± 7.8 |
| T556C (H8) | 2.21 ± 0.84 | 1.26 ± 0.40 | 0.64 ± 0.12 | 41.3 ± 12.6 |
The functional data that we obtained are summarized in Fig. 11. [35S]GTPγS binding/Gαq/11 immunoprecipitation assays demonstrated that carbachol (1 mm) treatment of M3′(3C)-Xa-expressing cells led to a significant increase in [35S]GTPγS binding, indicative of productive receptor-G protein coupling. The magnitude of this response increased with higher M3′(3C)-Xa expression levels (Fig. 11; Table 3). Importantly, CuPhen treatment of M3′(3C)-Xa-expressing membranes had no significant effect on the magnitude of the observed functional responses, indicating the CuPhen itself had no detrimental effects on the efficiency of receptor-G protein coupling under the chosen experimental conditions. Cross-linking of the T180C and T556C constructs did not have a significant effect on the efficacy of agonist-induced G protein activation. In contrast, cross-linking of the R176C, F550C, and T553C receptors led to significant reductions in the magnitude of agonist-stimulated [35S]GTPγS binding, as compared with the corresponding samples that had not been treated with CuPhen (Fig. 11).
FIGURE 11.
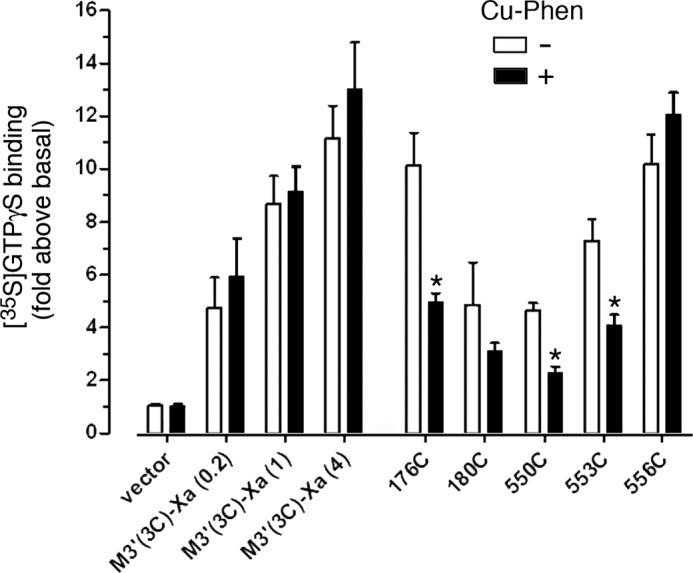
Several cross-linked mutant M3Rs show reduced G protein coupling efficacy. COS-7 cells were transfected with vector DNA, the M3′(3C)-Xa receptor, or the indicated M3′(3C)-Xa-derived Cys-substituted mutant constructs. Subsequently, cell membranes were incubated in the absence or presence of CuPhen (100 μm, 10 min at 37 °C). Cell membranes were then treated with the agonist carbachol (CCh; 1 mm) in the presence of [35S]GTPγS for 2 min at 30 °C. CCh-stimulated [35S]GTPγS binding was assessed by selective immunoprecipitation of [35S]GTPγS-labeled Gαq/11, using an anti-Gαq/11 antiserum. Because several mutant receptors were expressed at lower levels (Bmax) than the M3′(3C)-Xa construct (4 μg of DNA) from which they were derived, we also transfected cells with decreased amounts of M3′(3C)-Xa DNA (0.2 and 1 μg) to achieve a range of M3′(3C)-Xa Bmax values, similar to those observed with some of the mutant constructs (Table 3). Data are expressed as fold-increase in [35S]GTPγS binding above basal levels determined in the absence of agonist. Basal [35S]GTPγS binding activities were similar for the various mutant receptors. Data are given as mean ± S.E. of 3–5 independent experiments, each carried out in duplicate. *, p < 0.05, as compared with the corresponding non-CuPhen-treated samples.
Molecular Modeling Studies
The receptor cross-linking patterns that we observed in the present study strongly support the concept that M3R dimers exist in at least two distinct topologies. The first topology involves a dimerization interface with i2-i2 contacts on the cytosolic side, as well as TM4-TM5 contacts within the lipid bilayer (Fig. 12A). Models with this dimerization interface are most compatible with the cross-linking data obtained with the R176C-T180C constructs.
FIGURE 12.

Molecular models of two structurally distinct M3R-M3R interfaces. A and B, two M3R dimeric species with different M3R-M3R interfaces (view from the cytoplasm). The model in panel A shows i2-i2 contacts on the cytosolic side and TM4-TM5 contacts within the receptor core. The model in panel B shows H8-H8 and H8-TM1 contacts on the cytosolic side and TM1-TM1 interactions within the phospholipid bilayer. The M3R backbone is schematically portrayed as a ribbon with a continuous spectrum of colors ranging from red at the N terminus to purple at the C terminus (TM1 in red/orange, TM2 in orange, TM3 in orange/yellow, TM4 in yellow, TM5 in yellow/green, TM6 in blue, and TM7 in purple). The Cα carbons of the residues that were subjected to Cys substitution mutagenesis are shown as spheres. Green spheres indicate residues where Cys substitutions led to efficient cross-linking at room temperature. Yellow spheres represent residues where Cys substitutions did not promote efficient cross-linking at room temperature.
The second topology involves a dimerization interface with H8-H8 and H8-TM1 contacts on the cytosolic side, as well as additional TM1-TM1 interactions (Fig. 12B). Models with this dimerization interface are most compatible with the cross-linking data obtained with the K548C-T556C constructs. Among all amino acids located within this segment, the two Cα carbons of residue 556 show the shortest distance (6.8 Å) in the model depicted in Fig. 12B. Consistent with this observation, the T556C construct most readily formed cross-linked dimers at room temperature (Fig. 3).
DISCUSSION
Based on the outcome of BRET and molecular modeling studies, we recently predicted the existence of multiple, structurally distinct M3R dimers (18). To provide more direct experimental evidence for this concept, we carried out a series of disulfide cross-linking studies. In the present study, we tested the hypothesis that two cytoplasmic M3R regions, the C-terminal portion of the i2 loop (Ci2 region) and H8, are located at different M3R-M3R interfaces, as predicted by molecular modeling studies (18). Specifically, we subjected M3R residues Arg176-Thr180 (Ci2) and residues Lys548-Thr556 (H8) to Cys substitution mutagenesis and examined whether the resulting mutant M3Rs containing single cytoplasmic Cys residues were able to form disulfide cross-linked dimers under oxidizing conditions. Importantly, these studies were carried out with receptors in a native membrane environment (the membranes used were from receptor-expressing COS-7 cells).
At room temperature, several receptor constructs containing Cys substitutions within the Ci2 region formed cross-linked M3R adducts with high efficiency (R176C, R179C, and T180C) (Fig. 2). When cross-linking studies were carried out at 37 °C, all analyzed Ci2 mutant receptors showed complete (R176C, T180C), or nearly complete (A177C, K178C, and R179C), cross-linking (Fig. 2). The increase in cross-linking efficiency observed at 37 °C is most likely caused by an increase in the conformational mobility of the i2 loop and other receptor regions. Taken together, these data strongly support the concept that the Ci2 region is part of an M3R-M3R interface. In agreement with this notion, the TM helix that lies adjacent to the Ci2, TM4, has been implicated in the dimerization of several class A GPCRs (7–11). TM4 also plays a central role in mediating dimerization within class B GPCRs, including the secretin (26) and calcitonin (27) receptors.
To provide direct experimental evidence that H8 is part of an M3R dimer interface, we carried out cross-linking studies using mutant M3Rs containing single Cys substitutions within the Lys548-Thr556 sequence. At room temperature, three of the nine analyzed H8 mutant receptors, T553C, K555C, and T556C, formed cross-linked M3R dimers with very high efficiency (Fig. 3), indicative of the existence of an M3R dimer characterized by an H8-H8 interface. When cross-linking studies were carried out at 37 °C, all nine Cys-substituted H8 mutant M3Rs showed highly efficient disulfide cross-linking, suggesting that H8 is conformationally highly flexible at this temperature (Fig. 3). Our observation that H8 is located at an M3R dimer interface is consistent with several previous studies analyzing the dimerization pattern of other class A GPCRs (see, for example, Refs. 28–30).
The observed disulfide cross-linking patterns remained unaffected by a drastic reduction of receptor expression levels (to achieve M3R densities similar to those found in native tissues; ∼0.2 pmol/mg of protein; Fig. 5). This observation excludes the possibility that the M3R-M3R cross-links observed in the present study represent artifacts caused by supraphysiologically high receptor expression levels.
To examine whether receptor activation affected the observed disulfide cross-linking patterns, we also performed cross-linking studies in the presence of the muscarinic agonist, carbachol. Agonist treatment had little or no effect on the disulfide cross-linking pattern displayed by the mutant M3Rs containing Cys substitutions within the Ci2 region (Fig. 6), suggesting that this region does not undergo major structural changes following agonist activation. This observation is consistent with x-ray crystallographic data indicating that the position of the adjacent TM4 helix remains virtually unchanged in activated class A GPCRs (31). On the other hand, agonist treatment of several receptors containing Cys substitutions within H8 led to pronounced increases in the efficiency of M3R cross-link formation (Fig. 7).
In a previous disulfide cross-linking study involving mutant M3Rs containing pairs of Cys residues, we provided evidence that the TM7-proximal segment of H8 changes its position relative to the cytoplasmic end of TM1 in an agonist-dependent fashion (20). A similar activity-dependent movement of H8 has also been proposed for bovine rhodopsin on the basis of site-directed spin labeling studies (32). We therefore speculate that such agonist-induced changes in the orientation of H8 may explain the altered cross-linking patterns observed after agonist treatment of the analyzed H8 Cys mutant M3Rs. Many studies have shown that H8 contains residues that play important roles in the efficiency of receptor-G protein interactions (24, 33–35). It is therefore likely that the agonist-induced structural changes at the H8-H8 interface are also of functional relevance.
Interestingly, one of the investigated H8 Cys mutant M3Rs (T553C) showed enhanced disulfide cross-linking after either agonist (carbachol; Fig. 7) or antagonist/inverse agonist exposure (atropine; Fig. 10, center panel). The structural basis underling this phenomenon remains unclear at present. However, it is possible that the two ligands stabilize distinct conformational states of M3R dimers, which favor the formation of T553C-T553C cross-links.
Molecular modeling studies indicated that our cross-linking data are consistent with the existence of two structurally distinct M3R dimer species (Fig. 12, A and B). One of the structures is characterized by a dimerization interface that involves i2/i2 contacts on the cytosolic side as well as TM4-TM5 contacts within the TM receptor core (Fig. 12A). Interestingly, this dimerization interface is analogous to the one described previously for rhodopsin based on atomic force microscopic and molecular modeling studies (36). Moreover, it closely resembles the TM4-TM5-i2 dimer interface recently observed crystallographically for the β1-adrenergic receptor (37). The cross-linking data that we obtained with the H8 Cys mutant receptors are compatible with the existence of a second M3R dimer that involves H8-H8, H8-TM1, as well as TM1-TM1 contacts. The putative structure of this dimeric species is shown in Fig. 12B, as suggested by molecular modeling studies. Notably, this dimerization model is analogous to the one seen crystallographically for the β2-adrenergic receptor (38). The dimer interface shown in Fig. 12B is also very similar, although not identical, to one of the dimer interfaces found crystallographically for the β1-adrenergic receptor (37). In the M3R dimer model shown in Fig. 12B, residues Thr553, Lys555, and Thr556 are predicted to be located directly at the H8-H8 interface, as predicted by our cross-linking data. Fig. 12 clearly demonstrates that the cross-linking patterns that we observed cannot be explained by the existence of a single M3R dimer. However, our data do not exclude the possibility that the different M3R dimeric species are arranged in an oligomeric array. In a related disulfide cross-linking study, we recently identified several mutant M3Rs containing Cys substitutions at the bottom of TM5 and within the N-terminal portion of the i3 loop that could be easily cross-linked under oxidizing conditions (16). Molecular modeling studies demonstrated that the observed cross-linking data were in excellent agreement with the existence of a dimeric M3R species characterized by a TM5-TM5 interface (16). The structural features of this putative M3R dimer are clearly distinct from the two dimeric M3R structures that emerged from the present study. Taken together, our results provide strong experimental support for the existence of multiple M3R dimeric species that may be transient in nature.
Our proposal that the various M3R dimers may coexist in a dynamic equilibrium are consistent with several recent reports suggesting that interactions among certain class A GPCRs may not be as stable as previously assumed (39). For example, receptor immobilization experiments showed that interactions among β1-adrenergic (40) or D2 dopamine receptors (41) are transient in nature, in agreement with molecular dynamics simulations of the δ-opioid receptor (42). Similar results have been obtained in recent single molecule microscopic studies of the M1 muscarinic receptor (43), the N-formyl peptide receptor (44), and the β1- and β2-adrenergic receptors (45). Using single molecule microscopy, Nenasheva et al. (46) also demonstrated the existence of reversible M2 receptor dimers on the surface of mouse cardiomyocytes. Taken together, these data strongly support our proposal that the structurally distinct M3R dimers identified by cross-linking analysis are transient in nature and may coexist in a dynamic equilibrium (18).
To explore whether M3R dimers cross-linked at either the i2/i2 or the H8-H8 interface retained the ability to couple to G proteins, we determined receptor-mediated G protein activation by treating non-oxidized or oxidized (CuPhen-treated) membranes with a maximally effective concentration of carbachol and then monitored G protein activation using a [35S]GTPγS binding/Gαq/11 immunoprecipitation assay (16, 21). For these experiments, we selected five representative Cys mutant M3Rs that showed very high cross-link efficiency (R176C, T180C, F550C, T553C, and T556C). We found that cross-linking of the R176C (i2), F550C (H8), and T553C (H8) mutant receptors led to a pronounced reduction in the magnitude of agonist-stimulated [35S]GTPγS binding, as compared with the corresponding control samples that had not been exposed to the oxidizing agent (Fig. 11).
As already discussed above, the i2 loop is critically involved in receptor-mediated G protein activation (24, 47, 48). It is therefore likely that the reduced G protein coupling efficiency of the cross-linked R176C receptor (note that Arg176 is located in the center of the i2 loop) interferes with productive M3R-G protein interactions via steric constraints caused by the presence of the disulfide bond. Also, as mentioned earlier, considerable evidence suggests that the TM7-proximal portion of H8 undergoes activity-dependent conformational changes in class A GPCRs (20, 32). We therefore speculate that the reduced functional activity observed with the cross-linked F550C and T553C constructs are caused by the fact that the H8-H8 cross-links interfere with the agonist-induced conformational reorientation of H8 that is required for productive receptor-G protein interactions. However, this concept needs to be tested more rigorously in future experiments.
Recently, several class A GPCR dimers (or oligomers) have been observed in x-ray crystallographic studies. High resolution structures of the CXCR4 (49) and the μ-opioid (50) receptors featured dimers comprised of an interface involving TM5 and TM6. Interestingly, the x-ray structure of the μ-opioid receptor revealed a second dimer interface involving interactions between residues located on TM1, TM2, and H8 (50), allowing for the formation of higher order receptor oligomers. On the other hand, the κ-opioid receptor was crystallized as a dimer that only featured the TM1-TM2-H8 interface (51). Recently, the ligand-free β1-adrenergic receptor was crystallized in a lipid membrane-like environment (37). X-ray crystallographic data revealed the presence of two dimeric interfaces, one involving TM1-TM2-H8 and the other consisting of interactions between TM4, TM5, and the i2 loop (37). These observations suggest that the TM1-TM2-H8 interface is well conserved among class A GPCRs, whereas the dimer-oligomer interfaces involving other TM regions appear structurally more diverse (TM4-TM5, TM5-TM6, etc.). As already mentioned above, the two M3R interfaces identified in the present cross-linking study closely resemble those described for the β1-adrenergic receptor in x-ray crystallographic studies (37). This similarity in quaternary structure between these two receptor subtypes may reflect the fact that both receptors belong to the biogenic amine subfamily of GPCRs, which show a high degree of structural homology. The observation that two independent experimental approaches arrived at similar conclusions regarding the structure of biogenic amine GPCR dimer interfaces strongly supports the concept that such dimeric/oligomeric receptor species are also likely to exist under physiological conditions.
Consistent with the results of the present study showing extensive cross-linking among M3R constructs containing single Cys residues in the i2 loop, many residues of the i2 loop of the ligand-free β1-adrenergic receptor contributed to the formation of the TM4-TM5-i2 dimer interface (37). Because the i2 loop plays a central role in receptor mediated-G protein activation (24, 47, 48), the close proximity of two adjacent i2 loops in GPCR dimers (or oligomers) may have important functional implications.
There are several key questions that emerge from the outcome of the present study. For example, do the various M3R dimer species differ in their pharmacological and functional properties and do similar receptor adducts also form in native tissues? What is the relative abundance of the different dimeric species and are there any cellular factors that regulate their formation? Answers to these questions may pave the way to the development of new strategies aimed at interfering with GPCR dimer formation for therapeutic purposes.
In conclusion, this study provides novel information about various structural and functional aspects involved in M3R dimerization. In general, family A GPCRs share a high degree of structural homology. It is therefore likely that the data presented in this study are of rather broad general relevance.
Acknowledgment
We thank Helen Slusher of American University for assistance in the preparation of the molecular modeling figures.
This work was supported by the Intramural Research Program of the National Institutes of Health, NIDDK, and the American University.
J. Wess, unpublished data.
- GPCR
- G protein-coupled receptor
- BRET
- bioluminescence resonance energy transfer
- Ci2
- C-terminal portion of the second intracellular loop
- CuPhen
- Cu(II)-(1,10-phenanthroline)3
- i2 and i3 loop
- second and third intracellular loop
- H8
- helix 8
- [3H]NMS
- N-[3H]methylscopolamine
- IP
- inositol monophosphate
- M3R
- M3 muscarinic receptor
- PI
- (poly)phosphoinositide
- TM1–7
- transmembrane helices 1–7.
REFERENCES
- 1. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 2. Drews J. (2000) Drug discovery. A historical perspective. Science 287, 1960–1964 [DOI] [PubMed] [Google Scholar]
- 3. Lagerström M. C., Schiöth H. B. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357 [DOI] [PubMed] [Google Scholar]
- 4. Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., Sunahara R. K. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Whorton M. R., Jastrzebska B., Park P. S., Fotiadis D., Engel A., Palczewski K., Sunahara R. K. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 283, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. White J. F., Grodnitzky J., Louis J. M., Trinh L. B., Shiloach J., Gutierrez J., Northup J. K., Grisshammer R. (2007) Dimerization of the class A G protein-coupled neurotensin receptor NTS1 alters G protein interaction. Proc. Natl. Acad. Sci. U.S.A. 104, 12199–12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angers S., Salahpour A., Bouvier M. (2002) Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu. Rev. Pharmacol. Toxicol. 42, 409–435 [DOI] [PubMed] [Google Scholar]
- 8. Javitch J. A. (2004) The ants go marching two by two. Oligomeric structure of G-protein-coupled receptors. Mol. Pharmacol. 66, 1077–1082 [DOI] [PubMed] [Google Scholar]
- 9. Milligan G. (2007) G protein-coupled receptor dimerisation. Molecular basis and relevance to function. Biochim. Biophys. Acta 1768, 825–835 [DOI] [PubMed] [Google Scholar]
- 10. Palczewski K. (2010) Oligomeric forms of G protein-coupled receptors (GPCRs). Trends Biochem. Sci. 35, 595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Milligan G. (2013) The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol. Pharmacol. 84, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jastrzebska B., Ringler P., Palczewski K., Engel A. (2013) The rhodopsin-transducin complex houses two distinct rhodopsin molecules. J. Struct. Biol. 182, 164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wess J. (1996) Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 10, 69–99 [DOI] [PubMed] [Google Scholar]
- 14. Maggio R., Vogel Z., Wess J. (1993) Coexpression studies with mutant muscarinic/adrenergic receptors provide evidence for intermolecular crosstalk between G protein-linked receptors. Proc. Natl. Acad. Sci. U.S.A. 90, 3103–3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zeng F. Y., Wess J. (1999) Identification and molecular characterization of M3 muscarinic receptor dimers. J. Biol. Chem. 274, 19487–19497 [DOI] [PubMed] [Google Scholar]
- 16. Hu J., Thor D., Zhou Y., Liu T., Wang Y., McMillin S. M., Mistry R., Challiss R. A., Costanzi S., Wess J. (2012) Structural aspects of M3 muscarinic acetylcholine receptor dimer formation and activation. FASEB J. 26, 604–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goin J. C., Nathanson N. M. (2006) Quantitative analysis of muscarinic acetylcholine receptor homo- and heterodimerization in live cells. Regulation of receptor down-regulation by heterodimerization. J. Biol. Chem. 281, 5416–5425 [DOI] [PubMed] [Google Scholar]
- 18. McMillin S. M., Heusel M., Liu T., Costanzi S., Wess J. (2011) Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 286, 28584–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zeng F. Y., Hopp A., Soldner A., Wess J. (1999) Use of a disulfide cross-linking strategy to study muscarinic receptor structure and mechanisms of activation. J. Biol. Chem. 274, 16629–16640 [DOI] [PubMed] [Google Scholar]
- 20. Li J. H., Han S. J., Hamdan F. F., Kim S. K., Jacobson K. A., Bloodworth L. M., Zhang X., Wess J. (2007) Distinct structural changes in a G protein-coupled receptor caused by different classes of agonist ligands. J. Biol. Chem. 282, 26284–26293 [DOI] [PubMed] [Google Scholar]
- 21. Akam E. C., Challiss R. A., Nahorski S. R. (2001) Gq/11 and Gi/o activation profiles in CHO cells expressing human muscarinic acetylcholine receptors. Dependence on agonist as well as receptor-subtype. Br. J. Pharmacol. 132, 950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kruse A. C., Hu J., Pan A. C., Arlow D. H., Rosenbaum D. M., Rosemond E., Green H. F., Liu T., Chae P. S., Dror R. O., Shaw D. E., Weis W. I., Wess J., Kobilka B. K. (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chemical Computing Group (2010) The molecular operating environment (MOE), version 2010.10, Chemical Computing Group, Inc., Montreal, QC Canada [Google Scholar]
- 24. Wess J. (1998) Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol. Ther. 80, 231–264 [DOI] [PubMed] [Google Scholar]
- 25. Griffith I. P. (1972) The effect of cross-links on the mobility of proteins in dodecyl sulphate-polyacrylamide gels. Biochem. J. 126, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harikumar K. G., Pinon D. I., Miller L. J. (2007) Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J. Biol. Chem. 282, 30363–30372 [DOI] [PubMed] [Google Scholar]
- 27. Harikumar K. G., Ball A. M., Sexton P. M., Miller L. J. (2010) Importance of lipid-exposed residues in transmembrane segment four for family B calcitonin receptor homo-dimerization. Regul. Pept. 164, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salom D., Lodowski D. T., Stenkamp R. E., Le Trong I., Golczak M., Jastrzebska B., Harris T., Ballesteros J. A., Palczewski K. (2006) Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 16123–16128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo W., Urizar E., Kralikova M., Mobarec J. C., Shi L., Filizola M., Javitch J. A. (2008) Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 27, 2293–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Knepp A. M., Periole X., Marrink S. J., Sakmar T. P., Huber T. (2012) Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes. Biochemistry 51, 1819–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Katritch V., Cherezov V., Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang K., Farrens D. L., Altenbach C., Farahbakhsh Z. T., Hubbell W. L., Khorana H. G. (1996) Structure and function in rhodopsin. Cysteines 65 and 316 are in proximity in a rhodopsin mutant as indicated by disulfide formation and interactions between attached spin labels. Biochemistry 35, 14040–14046 [DOI] [PubMed] [Google Scholar]
- 33. Cai K., Klein-Seetharaman J., Farrens D., Zhang C., Altenbach C., Hubbell W. L., Khorana H. G. (1999) Single-cysteine substitution mutants at amino acid positions 306–321 in rhodopsin, the sequence between the cytoplasmic end of helix VII and the palmitoylation sites. Sulfhydryl reactivity and transducin activation reveal a tertiary structure. Biochemistry 38, 7925–7930 [DOI] [PubMed] [Google Scholar]
- 34. Ernst O. P., Meyer C. K., Marin E. P., Henklein P., Fu W. Y., Sakmar T. P., Hofmann K. P. (2000) Mutation of the fourth cytoplasmic loop of rhodopsin affects binding of transducin and peptides derived from the carboxyl-terminal sequences of transducin α and γ subunits. J. Biol. Chem. 275, 1937–1943 [DOI] [PubMed] [Google Scholar]
- 35. Swift S., Leger A. J., Talavera J., Zhang L., Bohm A., Kuliopulos A. (2006) Role of the PAR1 receptor 8th helix in signaling the 7-8-1 receptor activation mechanism. J. Biol. Chem. 281, 4109–4116 [DOI] [PubMed] [Google Scholar]
- 36. Liang Y., Fotiadis D., Filipek S., Saperstein D. A., Palczewski K., Engel A. (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 278, 21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang J., Chen S., Zhang J. J., Huang X. Y. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. (2007) High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lambert N. A. (2010) GPCR dimers fall apart. Sci. Signal. 3, pe12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dorsch S., Klotz K. N., Engelhardt S., Lohse M. J., Bünemann M. (2009) Analysis of receptor oligomerization by FRAP microscopy. Nat. Methods 6, 225–230 [DOI] [PubMed] [Google Scholar]
- 41. Fonseca J. M., Lambert N. A. (2009) Instability of a class a G protein-coupled receptor oligomer interface. Mol. Pharmacol. 75, 1296–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Provasi D., Johnston J. M., Filizola M. (2010) Lessons from free energy simulations of δ-opioid receptor homodimers involving the fourth transmembrane helix. Biochemistry 49, 6771–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hern J. A., Baig A. H., Mashanov G. I., Birdsall B., Corrie J. E., Lazareno S., Molloy J. E., Birdsall N. J. (2010) Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. U.S.A. 107, 2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kasai R. S., Suzuki K. G., Prossnitz E. R., Koyama-Honda I., Nakada C., Fujiwara T. K., Kusumi A. (2011) Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 192, 463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Calebiro D., Rieken F., Wagner J., Sungkaworn T., Zabel U., Borzi A., Cocucci E., Zürn A., Lohse M. J. (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc. Natl. Acad. Sci. U.S.A. 110, 743–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nenasheva T. A., Neary M., Mashanov G. I., Birdsall N. J., Breckenridge R. A., Molloy J. E. (2013) Abundance, distribution, mobility and oligomeric state of M2 muscarinic acetylcholine receptors in live cardiac muscle. J. Mol. Cell. Cardiol. 57, 129–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu J., Wang Y., Zhang X., Lloyd J. R., Li J. H., Karpiak J., Costanzi S., Wess J. (2010) Structural basis of G protein-coupled receptor-G protein interactions. Nat. Chem. Biol. 6, 541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., Granier S. (2012) Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu H., Wacker D., Mileni M., Katritch V., Han G.W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S.W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]