

Background: NQO2 protects against γ radiation-induced myeloproliferative disease, but the mechanism remains unknown.
Results: Radiation-induced NQO2, independent of NQO1, competes with the 20 S proteasome for interaction with C/EBPα region Ser-268 to Val-279 to stabilize C/EBPα, leading to protection against myeloproliferative disease.
Conclusion: NQO2 stabilizes C/EBPα against 20 S degradation to protect against myeloproliferative disease.
Significance: Stress-responsive NQO2 functions as an endogenous factor against myeloproliferative diseases.
Keywords: Chemoprevention, Protein Degradation, Protein Stability, Protein-Protein Interactions, Signal Transduction, 20 S Proteasome, C/EBPα, Myeloproliferative Diseases, NQO2
Abstract
NRH:quinone oxidoreductase 2 (NQO2) is a flavoprotein that protects cells against radiation and chemical-induced oxidative stress. Disruption of the NQO2 gene in mice leads to γ radiation-induced myeloproliferative diseases. In this report, we showed that the 20 S proteasome and NQO2 both interact with myeloid differentiation factor CCAAT-enhancer-binding protein α (C/EBPα). The interaction of the 20 S proteasome with C/EBPα led to the degradation of C/EBPα. NQO2, in the presence of its cofactor NRH, protected C/EBPα against 20 S degradation. Deletion and site-directed mutagenesis demonstrated that NQO2 and 20 S competed for the same binding region of S(268)GAGAGKAKKSV(279) in C/EBPα. Exposure of mice and HL-60 cells to γ radiation enhanced the levels of NQO2, which led to an increased NQO2 interaction with C/EBPα and decreased 20 S interaction with C/EBPα. NQO2 stabilization of C/EBPα was independent of NQO1, even though both interacted with the same C/EBPα domain. NQO2−/− mice, deficient in NQO2, failed to stabilize C/EBPα. This contributed to the development of γ radiation-induced myeloproliferative disease in NQO2−/− mice.
Introduction
Dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) is a flavoprotein that catalyzes the reductive metabolism of quinones (1). NQO2 belongs to a two-member quinone oxidoreductase gene family that also includes NQO1 (2). NQO2 utilizes NRH as an electron donor, and its activity is inhibited by benzo(a)pyrene (1). Analysis of the crystal structure of NQO2 revealed that NQO2 contains a specific metal binding site that is not present in NQO1 (3). NQO2 and/or NQO1 are stress-inducible proteins that are known to protect tumor suppressor p53, p33(INGIb), and p63 against 20 S proteasomal degradation (4–6).
NQO2−/− mice generated in our laboratory showed myeloid hyperplasia and significantly increased granulocyte levels in the peripheral blood (7). In addition, NQO2−/− mice demonstrated increased sensitivity to γ radiation-induced myeloproliferative disease and B cell lymphomas (7, 8). These studies suggest that NQO2 plays a significant role in protection against hematological disorders.
The transcription factors C/EBPα2 and PU.1 are known to regulate hematopoiesis (9, 10). C/EBPα is found predominantly in immature myeloid cells, whereas both lymphoid and myeloid cells express PU.1 (9, 10). Deregulation of C/EBPα has been found to be associated with myeloid transformation (11, 12). C/EBPα is known to regulate PU.1 gene expression (13).
In this report, we demonstrate that NQO2 protects myeloid differentiation factor C/EBPα against 20 S proteasomal degradation. NQO2 achieves this function by competing with 20 S proteasomes for binding with C/EBPα. Mutagenesis studies suggest that the C/EBPα region between amino acids Ser-268 to Val-279 is required for binding with both NQO2 and 20 S. Exposure of wild-type mice to γ radiation induced NQO2, which in turn led to increased NQO2 interaction and decreased 20 S interaction with C/EBPα. This resulted in the stabilization of C/EBPα, which affects myeloid differentiation. NQO2 stabilization of C/EBPα was independent of NQO1, even though both interacted with the same region in C/EBPα.
EXPERIMENTAL PROCEDURES
Materials
Purified 20 S fraction, FLAG antibody, Easyview Red anti-FLAG M2 affinity gel, NQO2 antibody for mouse cells, and MG132 were obtained from Sigma. 20 S core subunit and α5 subunit antibodies were from Calbiochem, and V5 antibody was from Invitrogen. NQO1 siRNA (catalog no. 4090) and NQO2 siRNA (catalog no. 9597) were from Ambion (Grand Island, NY). Antibodies against NQO2 (human cells) or C/EBPα and protein A/G-agarose were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). PU.1 antibody was from Cell Signaling Technology (Beverly, MA). Perfluoro-octanoic acid was from Oakwood Products (Columbia, SC). Nucleofector II and reagents were from Lonza (Cologne, Germany). Peptides were from Biosynthesis (Lewisville, TX).
Construction of Plasmids
All mouse C/EBPα truncated constructs were created by first performing a PCR to amplify the cDNA of interest using primers carrying the XbaI, EcoRI, or XhoI sites. By using XbaI (or EcoRI) and XhoI, the PCR products were digested and cloned into the pCMX vector containing a double FLAG tag, resulting in final expression constructs. To generate site-directed mutations of mouse C/EBPα cDNA, two PCRs were carried out to amplify the 5′ fragment of mouse C/EBPα cDNA (carrying the XbaI site) and the rest of the 3′ fragment of mouse C/EBPα cDNA (with site mutations and carrying the XhoI site). After blunt end ligation, the fragments that were ∼1080 base pairs long were digested with XbaI and XhoI and cloned into the pCMX vector containing a double FLAG tag. This approach was applied because C/EBPα is extremely GC-rich (72%), and site-directed mutagenesis kits (from Stratagene and Invitrogen) do not work. Double-strand DNA sequencing was used to verify all constructs.
FLAG-C/EBPα (1–162) and FLAG-C/EBPα (1–279) were engineered by PCR using the same forward primer (5′-TCTTTCTAGAATGGAGTCGGCCGACTTCTA-3′) and reverse primer (5′-ACCTGGGGATCCTCAGGGCTCTTGTTTGATCACCAG-3′ for construct 1–162 and 5′-ACCTGGGGATCCTCACACCGACTTCTTGGCTTTGCC-3′ for construct 1–279). FLAG-C/EBPα (157–359), FLAG-C/EBPα (268–359), and FLAG-C/EBPα(279–359) were generated by PCR, all using the same reverse primer (5′-CTTACTCGAGTCACGCGCAGTTGCCCATG-3′) and forward primers (5′-ACCTGGGAATTCGTGATCAAACAAGAGCCC-3′ for construct 157–359, 5′-ATTATTGAATTCAGCGGTGCCGGTGCGGGC-3′ for construct 268–359, and 5′-ACCTGGGAATTCGTGGACAAGAACAGCAACGAG-3′ for construct 279–359). FLAG-C/EBPαΔ268–279 was made by joining C/EBPα1–267 and C/EBPα269–359 with an EcoRI site.
All mouse NQO2-truncated constructs were created by blunt-ended cloning, and all truncated NQO2 cDNAs were subcloned into the pcDNA3.1/V5-His-TOPO vector for use. NQO2 (1–140)-V5 and NQO2 (1–165)-V5 were engineered by PCR using the same forward primer (5′-GCCACCATGGCAGGTAAGAAAGTGCT-3′) and the reverse primers 5′-ACCCTTGAGAAAACCAGAGTCATA-3′ and 5′-GAAATCTCCACTGACCCCATCTTT-3′, respectively; NQO2 (110–243)-V5 and NQO2 (141–243)-V5 were created by PCR using the forward primers 5′-GCCACCATGAAATTAGCTCTCCTTTCCTTA-3′ and 5′-GCCACCATGGCAATCCTAAAAGGTTGG-3′, respectively, and the same reverse primer (5′-CTCTTGGAAGTACCAAGGGGGTG-3′). The Invitrogen GeneArt site-directed mutagenesis system was used to produce NQO2Y104A, NQO2W105A, NQO2F106A, NQO2F126A, NQO2Y155A, NQO2F131A, NQO2Y132A, NQO2H173A, NQO2H177A, and NQO2C222A (all with a V5 tag).
Mice, Cells, and Cell Culture
The C57BL/6 wild-type and C57BL/6 NQO2−/− mice were produced in our laboratory (7). Human myelogenous leukemia HL-60 cells, U937 cells, and human CD34-positive KG-1 cells, purchased from the ATCC per its authentication, were cultured in RPMI/10% FBS/1% antibiotics. No further authentication of cells was performed. Bone marrow cells from mice femurs were accumulated and suspended in RPMI/10% FBS.
γ Radiation Treatment
Mice were either not irradiated or irradiated with 3 Gy of γ radiation. Mouse femur bone marrow cells were collected 48 h after irradiation, lysed, and analyzed. For ex vivo analysis, mice were euthanized, and their bone marrow cells were obtained in RPMI/10% FBS. Bone marrow cells were irradiated with 3 Gy of γ radiation and cultured for 48 h for further analysis.
Cell Lysate Preparation and Immunoprecipitation
Cells or bone marrow were lysed with cold lysis buffer (150 mm NaCl, 25 mm Tris (pH 7.4), 1 mm EDTA, and 1% perfluoro-octanoic acid) containing a protease inhibitor mixture (Roche) and centrifuged at 14,000 rpm for 10 min. The supernatant was precleared and immunoprecipitated overnight with the desired antibodies, followed by adsorption on protein A/G-agarose beads. The beads were washed and boiled for 5 min in 2× SDS loading buffer and analyzed by SDS-PAGE. Immunoprecipitation of FLAG-tagged proteins was done by Easyview Red anti-FLAG M2 affinity gel according to the protocol of the manufacturer.
Transfection
A Lonza cell line optimization nucleofector kit V was used to transfect HL-60, U937, or KG-1 cells in separate experiments with siRNA or plasmid DNA. 2 × 106 cells were centrifuged and suspended in 100 μl of nucleofector solution. Different concentrations of siRNA or plasmids were combined with the cell suspension, transferred into a cuvette, and transfected under Nucleofector Program T-019. Subsequently, 500 μl of the prewarmed RPMI 1640 was used to transfer the cell suspension into a 12-well plate. Cells were analyzed after 48 h.
In Vitro Translation of C/EBPα/Mutants and NQO1 and 20 S Proteasomal Degradation
1 μg of NQO2 (or mutants) or C/EBPα plasmid DNAs were in vitro-translated for 90 min at 30 °C. 2 μl of translated proteins were incubated with 20 S proteasome in degradation buffer (100 mm Tris-Cl (pH 7.5) containing 150 mm NaCl, 5 mm MgCl2, and 2 mm DTT) for various time periods and immunoblotted. In related experiments, in vitro-translated proteins were mixed with different reagents (NADH, NRH, benzo(a)pyrene, and omeprazole) for the same process.
Ubiquitination Assay
HL-60 cells were cotransfected with 0.5 μg of HA-tagged ubiquitin and 1 μg of FLAG-tagged wild-type C/EBPα or mutant C/EBPαK276A or C/EBPαK277A. 24 h later, the cells were lysed, and 1 mg of cell lysates was immunoprecipitated with anti-FLAG M2 affinity gel. After SDS-PAGE and Western blotting, the PVDF membrane was probed with anti-HA antibody.
Western Blot Analysis
Cell lysates were centrifuged at 10000 × g, and supernatant protein was quantified. SDS-PAGE analysis was performed as described previously (6).
Statistical Analysis
Microsoft Excel (2007) and Prism Graphpad (version 6) were used for statistical analysis. For statistical analysis, experiments were repeated three times, as shown in the figure legends. Differences were considered significant at p < 0.05.
RESULTS
NQO2 Protects C/EBPα against 20 S Proteasomal Degradation
WT and NQO2−/− mice were not irradiated or irradiated with 3 Gy of γ radiation to determine the short-term effect of radiation on the expression of NQO2, C/EBPα, and PU.1. The results are shown in Fig. 1A (left panel and bar graph). Bone marrow cells from non-irradiated NQO2−/− mice showed higher levels of C/EBPα and PU.1 as compared with wild-type mice, suggesting that increased C/EBPα and PU.1 were responsible for myeloid hyperplasia in NQO2−/− mice, as reported earlier (7). The mechanism of higher expression of C/EBPα and PU.1 in NQO2−/− bone marrow remains unknown. Interestingly, exposure to 3 Gy of γ radiation increased NQO2, C/EBPα, and PU.1 to varying levels in wild-type mice but failed to do so in NQO2−/− mouse bone marrow. These results indicated that NQO2−/− mice, deficient in NQO2, were incapable of inducing the myeloid differentiation factors C/EBPα and PU.1 upon exposure to γ radiation, as observed with wild-type mice expressing the NQO2 gene.
FIGURE 1.

NQO2 regulates C/EBPα and PU. 1. A, left panel and bar graph, lack of induction of C/EBPα and PU.1 in response to γ radiation. Mice were irradiated with 3 Gy of γ radiation, and bone marrow cells were immunoblotted. Right panel and bar graph, MG132 rescue of C/EBPα and PU.1 from γ radiation-induced degradation in NQO2−/− cells. Bone marrow cells were exposed to 3 Gy of γ radiation and left alone or treated with 20 μm MG132 for 10 h, lysed, and immunoblotted. * indicates statistically significant (p < 0.05) changes in protein levels between control and treatment groups. B, NQO2 regulates C/EBPα and PU.1. Left panel, HL60 cells immunoblotted for NQO2. Center panel and bar graph, HL-60 cells were transfected with increasing amounts of FLAG-NQO2, lysed, and immunoblotted. Right panel and bar graph, U937 cells were transfected with control or NQO2 siRNA and immunoblotted. Cntl, control. C, NQO2 protects C/EBPα against 20 S degradation. In vitro-translated C/EBPα and NQO2, purified 20 S proteasome, NRH (1 μm), and MG132 (20 μm) were mixed in combinations, as shown, for 1 h at 37 °C. 2 μl of mixtures from different treatments were immunoblotted.
Next, we performed ex vivo bone marrow studies to investigate the role of protein degradation in the lack of induction of C/EBPα and PU.1 because of exposure to γ radiation. Bone marrow cells from wild-type and NQO2−/− mice were exposed to 3 Gy of γ radiation in the absence and presence of the proteasome inhibitor MG132 and analyzed for C/EBPα, PU.1, NQO2, and actin. MG132 treatment protected C/EBPα in NQO2−/− mouse bone marrow cells from proteasomal degradation (Fig. 1A, right panel and bar graph). This was evident from increased levels of C/EBPα in bone marrow cells from NQO2−/− mice exposed to γ radiation in the presence of MG132. The results, together, suggest that exposure to γ radiation leads to proteasomal degradation of C/EBPα in the absence of NQO2. In other words, NQO2 is required for the stabilization and induction of C/EBPα in response to γ radiation.
Next, HL-60 and U937 cells were immunoblotted to compare NQO2 levels. The results showed that U937 cells express 2-fold higher levels of NQO2 as compared with HL-60 cells (Fig. 1B, left panel). These cells were used to further strengthen our observation that NQO2 controls C/EBPα. HL-60 cells transfected with the FLAG-NQO2 plasmid (Fig. 1B, center panel and bar graph) and U937 cells transfected with NQO2 siRNA (right panel and bar graph) were analyzed for alterations in NQO2 and its effect on C/EBPα and PU.1. The results revealed that overexpression of NQO2 in HL-60 cells led to the up-regulation of both C/EBPα and PU.1, whereas siRNA-mediated inhibition of NQO2 in U937 cells led to the down-regulation of both C/EBPα and PU.1. The inhibition of NQO2 followed by down-regulation of C/EBPα and Pu.1 was observed with two different NQO2 siRNA (Fig. 1B, right panel and bar graph). These results demonstrated and strengthened our earlier observation that NQO2 controls C/EBPα stability, followed by increased PU.1 expression.
We performed in vitro assays to investigate the role of the 20 S proteasome in C/EBPα degradation and the role of NQO2 protection against 20 S degradation of C/EBPα. Incubation of in vitro-translated C/EBPα with the purified 20 S proteasome showed significant degradation of C/EBPα within 1 h (Fig. 1C). Inclusion of in vitro-translated NQO2 in the incubation mixture partially protected C/EBPα. Interestingly, NQO2, in combination with its cofactor NRH in the same experiment, provided complete protection of C/EBPα against 20 S proteasomal degradation (Fig. 1C). The results collectively indicate that NQO2 stabilizes C/EBPα against 20 S proteasomal degradation.
NQO2 and 20 S Both Interact with C/EBPα but Not with PU.1
Immunoprecipitation assays were performed to investigate the role of NQO2 and 20 S interaction with C/EBPα in the inhibition of 20 S degradation of C/EBPα. Bone marrow from wild-type and NQO2−/− mice, HL-60 cells, and HL-60 cells transfected with NQO2-V5 and FLAG-C/EBPα plasmids were analyzed for interactions among C/EBPα, 20 S, and NQO2 (Fig. 2A). The results showed that NQO2 and 20 S interact with C/EBPα in mouse bone marrow (Fig. 2A, left panels), HL-60 cells (center panel), and transfected HL-60 cells (right panel). The results also showed that NQO2 and 20 S did not interact with each other (Fig. 2A). The results further indicated that C/EBPα interacted with 20 S in the absence of NQO2 in NQO2−/− mouse bone marrow. In similar experiments, as described above for C/EBPα in Fig. 2A, immunoprecipitation assays failed to show PU.1 interaction with either NQO2 or 20 S proteasomes (Fig. 2B). This confirmed that NQO2 controls PU.1 through C/EBPα, which regulates the transcription of PU.1.
FIGURE 2.

NQO2 and 20 S both interact with C/EBPα but not PU.1. A, NQO2 and 20 S both interact with C/EBPα. Bone marrow from wild-type and NQO2−/− mice (left panel), HL-60 cells (center panel) and HL-60 cells transfected with FLAG-C/EBPα and NQO2-V5 (right panel) were lysed; immunoprecipitated (IP) with IgG (control), C/EBPα, 20 S, and NQO2 antibodies; and immunoblotted. B, PU.1 does not interact with C/EBPα, NQO2, or 20 S. Mouse bone marrow (left two panels) and HL-60 cell (right two panels) lysates were immunoprecipitated with IgG (control), PU.1, C/EBPα, NQO2, and 20 S antibodies and immunoblotted.
Amino Acids Ser-268 to Val-279 of C/EBPα Are Required for Its Interaction with Both NQO2 and 20 S Proteasome, and Lysines 276/277 of C/EBPα Are Essentially Required for Its Interaction with NQO2
FLAG-tagged C/EBPα deletions were generated to identify protein domain(s) that interact with NQO2 and 20 S. The various deletions produced expected sizes of truncated C/EBPα bands upon transfection in HL-60 cells (Fig. 3A, left panel). Transfection and immunoprecipitation assays revealed that all deletions, except 1–162, 279–359, and Δ268–279, interacted with NQO2 and 20 S (Fig. 3A, right panel). The results, together, clearly indicate that the C/EBPα protein region amino acid 268–279 is essential for interaction with both NQO2 and 20 S.
FIGURE 3.

The C/EBPα region between Ser-268 and Val-279 is required for interaction with both NQO2 and 20 S. Lysines 276 and 277 of C/EBPα are required for its interaction with NQO2. A, the C/EBPα region between Ser-268 and Val-279 is required for interaction with both NQO2 and 20 S. Left top panel, schematic domain structure of C/EBPα mutants and input for coimmunoprecipitation. DBD, DNA binding domain; LZD, leucine zipper region. Right panel, HL-60 cells were cotransfected with full-length or truncated mutant FLAG-C/EBPα and NQO2-V5. Cell lysates were immunoprecipitated (IP) and immunoblotted with the indicated antibodies. WB, Western blot. B, mutation of C/EBPα Lys-276 and Lys-277 to alanine abrogates the interaction with NQO2 but not with 20 S. HL-60 cells were cotransfected with FLAG-C/EBPα or mutant FLAG-C/EBPα and NQO2-V5, lysed, immunoprecipitated, and immunoblotted. C, His-tagged-C/EBPα but not His-tagged-C/EBPα-Δ268–279 pulls down NQO2 and 20 S, whereas the C/EBPαK276A and C/EBPαK277A mutants pull down 20 S but not NQO2. HL-60 cells were cotransfected with the FLAG-NQO2 and C/EBPα-V5-His or C/EBPα mutant-V5-His plasmids. 1 mg of cell lysates was pulled down using protein AG or nickel-nitrilotriacetic acid beads and immunoblotted with anti-V5, FLAG, or 20 S antibodies. Lane 1, FLAG-NQO2 + C/EBPα-V5-His6; lane 2, FLAG-NQO2 + C/EBPαΔ268–279-V5-His6; lane 3, FLAG-NQO2 + C/EBPαK276A-V5-His6; lane 4, FLAG-NQO2 + mutant C/EBPαK277A-V5-His6. D, Lys-276 and Lys-277 are not the ubiquitination sites of C/EBPα. HL-60 cells were cotransfected with HA-Ub and FLAG-tagged WT C/EBPα or C/EBPαK276A or C/EBPαK277A. 1 mg of cell lysates was immunoprecipitated with anti-FLAG M2 affinity gel and probed with anti-HA antibody.
Alignment of the mouse C/EBPα protein region between amino acids 268–279 with the complementary human and rat showed conserved residues Lys-274, Lys-276, and Lys-277; Gly-269, Gly-271, and Gly-273; Ala-272 and Ala-275; Ser-278; and Val-279. Site-directed mutagenesis was used to individually mutate Lys-274, Lys-276, and Lys-277 to generate the FLAG-tagged C/EBPα K274A, K276A, and K277A mutant plasmids. Transfection and immunoprecipitation assays demonstrated that mutation of Lys-276 and Lys-277 to alanine resulted in the loss of interaction of C/EBPα with NQO2, whereas the C/EBPα K274A mutant still interacted with NQO2 (Fig. 3B). These results suggest that Lys-276/Lys-277 of C/EBPα is required for its interaction with NQO2. Interestingly, all four C/EBPα mutants (K274A, K276A, K277A, and the double mutant K276A/K277A) still interacted with 20 S (Fig. 3B). These results indicated that mutations in individual amino acids between Ser-268 and Val-279 had no effect on C/EBPα interaction with 20 S, suggesting the requirement of all of the amino acids in the 268–279 domain for the interaction of C/EBPα with 20 S.
Histidine pull-down assays showed same results as those observed in the immunoprecipitation assays (Fig. 3C). Both NQO2 and 20 S were pulled down with C/EBPα-V5-His6 but not with C/EBPαΔ268–279-V5-His6. In related pull-down assays, 20 S, but not NQO2, was pulled down with both mutant C/EBPαK276A-V5-His6 and C/EBPαK277A-V5-His6. Ubiquitination assays results showed that C/EBPα and the mutants were all ubiquitinated to the same extent (Fig. 3D). This indicated that neither Lys-276 nor Lys-277 are the ubiquitination sites of C/EBPα.
NQO2 Deletions Fail to Interact with C/EBPα
V5-tagged deletions of NQO2 were generated to identify the domain(s) required for interaction with C/EBPα (Fig. 4A). Transfection of these mutants in HL-60 cells produced the expected size of truncated NQO2-V5 proteins (Fig. 4B, top panel). Transfection and immunoprecipitation assays revealed that all deletions failed to interact with C/EBPα (Fig. 4B). This suggests that a full-length NQO2 protein is required to interact with C/EBPα. In similar experiments, the mutations in FAD binding sites (Y104A, W105A, F106A, and Y155A), catalytic sites (F126A, F131A, and Y132A), and metal binding sites (H173A, H177A, and C222A) of NQO2 also failed to block C/EBPα protection against 20 S degradation (Fig. 4C). In related experiments, three C/EBPα peptides (Ser-268 to Val-279 (full-length), Ser-268 to Gly-273 (N-terminal half), and Lys-274 to Val-279 (C-terminal half)) were synthesized and used in in vitro assays to determine their effects on C/EBPα interaction with 20 S (Fig. 4D). All three peptides competed with C/EBPα for binding with 20 S and degradation. Interestingly, C-terminal half peptides containing basic lysine residues were significantly more efficient in competing with C/EBPα against 20 S degradation as compared with the N-terminal half containing small/neutral amino acids.
FIGURE 4.

NQO2 with terminal deletions fails to interact with C/EBPα. A, schematic of C- and N-terminal NQO2 deletions. B, HL-60 cells were cotransfected with V5-tagged, full-length NQO2 (NQO2-FL) or NQO2 deletion mutants and FLAG-C/EBPα, immunoprecipitated (IP), and immunoblotted. WB, Western blot. C, in vitro 20 S degradation assay using NQO2 site mutations. In vitro-translated C/EBPα and NQO2 site mutations, purified 20 S proteasome, and NRH were mixed in combinations for 1 h at 37 °C. 2 μl of mixtures from different treatments were immunoblotted. D, in vitro-translated, wild-type C/EBPα was incubated with purified 20 S proteasomes in the absence or presence of the synthesized peptides C/EBPα (Ser-268 to Val-279, Ser-268 to Gly-273, and Lys-274 to Val-279) for 1 h and analyzed by immunoblotting.
Benzo(a)pyrene Inhibits NQO2 Protection of C/EBPα against 20 S Degradation, and γ Radiation Leads to an Increase in NQO2:C/EBPα Interaction and a Decrease in 20 S:C/EBPα Interaction
We performed in vitro assays to determine the effect of benzo(a)pyrene inhibition of NQO2 on its capacity to protect C/EBPα against 20 S degradation. Benzo(a)pyrene treatment significantly reduced NQO2 protection of C/EBPα against 20 S degradation in the presence or absence of 50 μm CYP1A1 inhibitor omeprazole (Fig. 5A, top panel). Omeprazole at 50 μm concentration inhibited 70–80% CYP1A1 activity in Hepa-1 cells and rabbit reticulocyte lysate (Fig. 5A, bottom panel). These results suggest that both NQO2 and cofactor NRH are required for protection of C/EBPα against 20 S degradation and blocking of benzo(a)pyrene inhibition. The results also demonstrate that the benzo(a)pyrene metabolism is not required for inhibition of NQO2 activity. Next, we used in vitro assays to determine direct competition between NQO2 and 20 S for C/EBPα. In vitro-translated C/EBPα and purified 20 S were mixed with increasing concentrations of NQO2 alone or NQO2 + NRH, and coimmunoprecipitation assays were performed to determine C/EBPα interactions with NQO2 and 20 S. The results indicated that NQO2 and 20 S proteasome competed to interact with C/EBPα. Increasing NQO2 concentration in the absence of NRH provided an NQO2 concentration dependent increase in NQO2:C/EBPα interaction and a decrease in 20 S:C/EBPα that was clearly visible but not highly significant (Fig. 5B, left two panels). In related experiments, increasing the concentration of NRH reverted back the inhibitory effect of benzo(a)pyrene on NQO2 protection of C/EBPα against 20 S degradation, and increasing the NRH concentration in the presence of NQO2 led to NRH concentration-dependent, highly significant increases in NQO2:C/EBPα interaction and decreases in 20 S:C/EBPα interaction (Fig. 5B, right two panels). These results suggest that cofactor NRH, in association with NQO2, controls the interactions between NQO2:C/EBPα and 20 S:C/EBPα.
FIGURE 5.

Alterations in C/EBPα interaction with NQO2 and 20 S. A, top panel, benzo(a)pyrene (B(a)p) inhibits NQO2 protection of C/EBPα against 20 S degradation. In vitro-translated C/EBPα and NQO2, purified 20 S proteasome, and 1 μm NRH were mixed with or without 2 μm benzo(a)pyrene in the absence and presence of 50 μm CYP1A1 inhibitor omeprazole for 1 h at 37 °C and immunoblotted. Bottom panel, 7-ethoxy-resorufin-O-deethylase (EROD) assays. Hepa-1 cells or reticulocyte cell lysates were plated in 96-well plates and treated with without omeprazole (OME, 50 μm). EROD activities were determined by adding 7-ethoxyresorufin (40 mm final concentration) in PBS to each well and measuring in a TECAN fluorometer at a wavelength of 344 nm with a reference filter set at 590 nm at room temperature. Each sample was analyzed in triplicate, and the mean ± S.E. were used for the bar graph. B, in vitro 20 S degradation of C/EBPα in the presence of different amounts of NQO2 (left two panels) and varying NRH concentrations (right two panels). The FLAG-C/EBPα and NQO1-V5 plasmids were in vitro-translated and mixed with different amounts of in vitro-translated NQO2-V5 and different concentrations of NRH. The mixtures were immunoprecipitated (IP) and probed with antibodies. C, NQO2 competes with the 20 S proteasome for C/EBPα binding, leading to stabilization of C/EBPα. HL-60 cells were either not irradiated or irradiated with 3 Gy of γ radiation, lysed, immunoprecipitated, and immunoblotted. The band density was measured and normalized by densitometry. The ratios of interactions are the mean of three independent experiments. D, exposure of mice to γ radiation increased NQO2:C/EBPα and decreased 20 S:C/EBPα interactions. WT mice were either not irradiated or irradiated with 3 Gy of γ radiation. Bone marrow cells were collected, lysed, immunoprecipitated, and immunoblotted. * indicates statistically significant (p < 0.05) difference between control and irradiated samples.
HL-60 cells, not irradiated or irradiated with γ radiation in three independent experiments, were analyzed for C/EBPα, NQO2, and 20 S interactions, and a densitometry analysis was performed (Fig. 5C). The results demonstrated a 90% increase in NQO2 interaction with C/EBPα in response to γ radiation. The results also revealed that γ radiation induced a 40% decrease in the 20 S interaction with C/EBPα (Fig. 5C). In similar experiments, WT mice were exposed to 3 Gy of γ radiation. Bone marrow cells were collected and analyzed for C/EBPα, NQO2, and 20 S interaction. A significant increase in NQO2 interaction with C/EBPα and a nearly 2-fold decrease in 20 S interaction with C/EBPα were observed (Fig. 5D). The results, together, suggested that exposure to γ-radiation led to a significantly greater interaction of C/EBPα with NQO2 and reduced its interaction with 20 S proteasomes, leading to the stabilization of C/EBPα and the up-regulation of downstream gene expression.
NQO2 and NQO1 Independently Protect C/EBPα
The current report on NQO2 and a report published previously on NQO1 (14) suggest that both protect C/EBPα against 20 S proteasomal degradation during stress. Therefore, we performed in vitro and in vivo experiments to compare similarities and differences between NQO2 and NQO1 protection of C/EBPα.
In Vitro Studies
NQO1 cofactor NADH or NQO2 cofactor NRH alone were ineffective in blocking 20 S degradation of C/EBPα (Fig. 6A, top panel). However, NADH in the presence of NQO1 protected C/EBPα against 20 S degradation, and the protective effect is positively proportional to the NADH concentration from 1–10 μm. In the meantime, NRH (1–10 μm) in the presence of NQO1 was unable to provide more protection of C/EBPα than NQO1 alone (Fig. 6A, center left panel and bottom left bar graph). In a similar experiment, NRH in the presence of NQO2 protected C/EBPα against 20 S degradation, and the protective effect is positively proportional to the NRH concentration from 1–10 μm. Although NADH (1–10 μm) in the presence of NQO2 was unable to provide more protection of C/EBPα than NQO2 alone (Fig. 6A, center right panel and bottom right bar graph). In related experiments, the NQO1 inhibitor dicoumarol blocked NQO1 + NADH stabilization of C/EBPα, but NQO2 inhibitor benzo(a)pyrene was unable to do so (Fig. 6B, top panel). In a similar experiment, the NQO2 inhibitor benzo(a)pyrene abolished NQO2 + NRH protection of C/EBPα, but the NQO1 inhibitor dicoumarol was unable to do so (Fig. 6B, bottom panel). The above results collectively suggested that NQO1+NADH and NQO2+NRH independently protected C/EBPα against 20 S proteasomal degradation. The results also suggested that NQO1 could not complement NQO2 and vice versa because of differences in cofactor requirements.
FIGURE 6.

NQO1 or NQO2 independently protect C/EBPα in in vitro assays. A, different concentrations of NADH and NRH (micromolar) in the absence and presence of in vitro-translated NQO1 and NQO2 were used in a 20 S degradation of C/EBPα assay and analyzed. B, in vitro-translated C/EBPα and NQO1 or NQO2 were used for in vitro 20 S proteasome degradation in the presence or absence of dicoumarol or benzo(a)pyrene and analyzed.
In Vivo Studies
HL-60 cells were treated with either 10 μm NQO1 inhibitor dicoumarol or 50 μm NQO2 inhibitor benzo(a)pyrene for 24 h, not irradiated or irradiated with γ radiation, and analyzed for C/EBPα, NQO1, NQO2, and 20 S interactions. Dicoumarol treatment abrogated C/EBPα interaction with NQO1, but not with NQO2, in both control and γ radiation-exposed cells (Fig. 7A). The results also demonstrated the increase in NQO2 interaction with C/EBPα in response to γ radiation and the decrease in 20 S interaction with C/EBPα (Fig. 7A). The benzo(a)pyrene treatment in a similar experiment blocked C/EBPα interaction with NQO2 but not with NQO1 in non-irradiated and irradiated cells (Fig. 7B). Benzo(a)pyrene also increased NQO2 interaction with C/EBPα in response to γ radiation and decreased 20 S interaction with C/EBPα (Fig. 7B). The results, together, suggested that exposure to γ radiation led to a significantly greater interaction of C/EBPα with NQO1 or NQO2 and reduced its interaction with 20 S proteasomes, leading to the stabilization of C/EBPα and the up-regulation of downstream gene expression. In related experiments, HL-60 cells were transfected with either control siRNA, NQO1 siRNA, NQO2 siRNA, or a combination of NQO1 and NQO2 siRNAs, exposed to 3 Gy of γ radiation, and analyzed. The results revealed that siRNA-mediated inhibition of NQO1 or NQO2 both significantly reduced C/EBPα levels in HL-60 cells (Fig. 7C). Interestingly, the combined inhibition of NQO1 and NQO2 led to a complete loss of C/EBPα in γ radiation-exposed cells (Fig. 7C). The same results were observed in bone marrow cells from wild-type, NQO1−/−, NQO2−/−, and double knockout mice. The deletion of NQO1 or NQO2 led to a significant decrease in C/EBPα levels (Fig. 7D). In addition, the combined deletion of NQO1 and NQO2 led to a complete loss of C/EBPα in radiation-exposed cells (Fig. 7D). Collectively, the in vivo results supported in vitro results and led to the conclusion that NQO1 and NQO2 independently control the stability of C/EBPα against 20 S proteasomal degradation.
FIGURE 7.

NQO1 and NQO2 independently protect C/EBPα in HL-60 cells and mice. A, HL-60 cells treated with dicoumarol were either not irradiated or irradiated with 3 Gy of γ radiation. 24 h later, cells were lysed, immunoprecipitated (IP), and immunoblotted. B, HL-60 cells treated with benzo(a)pyrene were either not irradiated or irradiated with 3 Gy of γ radiation. 24 h later, cells were lysed, immunoprecipitated, and immunoblotted. C, HL-60 cells transfected with control, NQO1, NQO2, and NQO1/NQO2 siRNA were either not irradiated or irradiated with 3 Gy of γ radiation, lysed, immunoprecipitated, and immunoblotted. The band density was measured and normalized by densitometry. D, wild-type, NQO1−/−, NQO2−/−, or double knockout (DKO) mice were either not irradiated or irradiated with 3 Gy of γ radiation. Bone marrow cells were collected, lysed, immunoprecipitated, and immunoblotted.
DISCUSSION
Human gene polymorphism and mouse studies have shown a direct association between NQO1 and its protection against myeloproliferative diseases, including leukemia (15–16). NQO2, structurally related to NQO1, also exhibited protection against myeloid hyperplasia (7) and myeloproliferative disease (8). This raised intriguing questions regarding the mechanism of NQO2 protection against radiation-induced myeloproliferative diseases. Disruption of C/EBPα and PU.1 is known to be associated with the development of myeloid leukemia (17). This study investigated NQO2 control of C/EBPα and PU.1 as a mechanism contributing to the protection against myeloproliferative diseases. NQO2−/− mice exposed to γ radiation showed a lack of induction of C/EBPα and PU.1. Wild-type mice, upon exposure to γ radiation, showed a significant induction of NQO2, C/EBPα, and PU.1 (this work) and the absence of myeloproliferative diseases (7, 8). Therefore, the lack of induction of C/EBPα and PU.1 contributed to myeloproliferative diseases in NQO2−/− mice. The lack of induction of C/EBPα was due to the rapid degradation by 20 S proteasomes in the absence of NQO2. NQO2 directly interacted with C/EBPα and prevented its degradation by the 20 S proteasomes. This led to the stabilization of C/EBPα, normal differentiation of myeloid progenitor cells, and protection against myeloproliferative diseases. NQO2 indirectly regulates PU.1 because PU.1 does not directly interact with NQO2 or 20 S proteasomes. C/EBPα is known to up-regulate PU.1 gene transcription (16). Therefore, NQO2-mediated stabilization or 20 S degradation of C/EBPα up- or down-regulated PU.1 transcription, respectively.
NQO2 and 20 S proteasomes competed for the same C/EBPα region between amino acids Ser-268 and Val-279. Internal deletion of this in C/EBPα resulted in the loss of interaction with both NQO2 and 20 S. The NQO2 interaction with C/EBPα was further narrowed down to two lysine residues at positions 276 and 277. Mutations of one or both of these lysine residues abolished C/EBPα interaction with NQO2 but not with 20 S. Interestingly, single mutations in the 20 S-interacting domain had no effect on CEBPα interaction with 20 S. This indicated that the complete domain was required for C/EBPα and 20 S interaction. The NQO2/20 S-interacting domain in mouse C/EBPα was highly conserved in humans and rats. Deletions in NQO2 protein had deleterious effects on NQO2 interaction with C/EBPα. This indicates that secondary/tertiary/folding structures are required for NQO2 interaction with C/EBPα. This study also revealed that NQO2 requires its cofactor NRH to protect C/EBPα against 20 S degradation. The role of NRH in NQO2 stabilization of C/EBPα requires further investigation.
A similar mechanism as that described above for NQO2 is also reported by us for NQO1 stabilization of C/EBPα against 20 S degradation (14). Therefore, we investigated similarities/differences and complementarity between NQO2 and NQO1 protection of C/EBPα. The similarities included NQO2 and NQO1 competition with 20 S proteasome for interaction with the C/EBPα domain S(268)GAGAGKAKKSV(279); the requirement of full-length NQO2 and NQO1 proteins to interact with C/EBPα; and stress-induced, increased C/EBPα interaction with NQO2 and NQO1 and decreased interaction with 20 S proteasome, leading to the stabilization of C/EBPα. The differences included the requirement of different cofactors (NADH for NQO1 and NRH for NQO2), the lack of complementarity between NQO2 and NQO1 with respect to C/EBPα stabilization because of different cofactor requirements, and the inability of the NQO1 inhibitor dicoumarol to inhibit C/EBPα:NQO2 interaction and of the NQO2 inhibitor benzo(a)pyrene to inhibit C/EBPα:NQO1 interaction. These differences led to the conclusion that the NQO2 + NRH and NQO1 + NADH stabilization of C/EBPα against 20 S degradation are independent of each other. This is supported by the observation that combined siRNA inhibition of NQO1 and NQO2 in HL-60 cells and double NQO1 and NQO2 deletion in mice both demonstrated complete degradation of C/EBPα by 20 S proteasomes. Individual deletions of NQO1 and NQO2 reduced, but did not eliminate, C/EBPα stabilization.
The expression of the NQO1 and NQO2 genes is coordinately activated in response to radiation and chemicals and is critical in the protection against myeloproliferative diseases (7, 8, 18). The induction of NQO2 enhances interaction with C/EBPα and reduces 20 S interaction with C/EBPα, leading to the protection and stabilization of C/EBPα. NQO2-mediated protection of C/EBPα allows the accumulation of C/EBPα and downstream PU.1 that is required for differentiation of myeloid cells. NQO2−/− mice and, possibly, human individuals lacking or deficient in NQO2 fail to accumulate C/EBPα, which leads to myeloproliferative diseases. It is noteworthy that human NQO2 gene promoter polymorphism is known to lead to lower expression of NQO2 in a significant portion of the population (19, 20).
A hypothetical model of NQO2 protection of C/EBPα against 20 S degradation is shown in Fig. 8. Under normal conditions, both 20 S and NQO2 interact with C/EBPα and control the physiological levels of C/EBPα, PU.1, and myeloid differentiation. Exposure to γ radiation leads to increased NQO2 levels and increased NQO2 interaction with C/EBPα. Increased NQO2 interaction with C/EBPα and decreased interaction of C/EBPα with 20 S lead to the stabilization of C/EBPα, increased expression of PU.1, and differentiation of myeloid cells and protection against γ radiation. On the contrary, the loss of NQO2 leads to increased 20 S interaction with C/EBPα, degradation of C/EBPα, down-regulation of PU.1, lack of myeloid cell differentiation, and myeloproliferative diseases, including leukemia/lymphoma.
FIGURE 8.
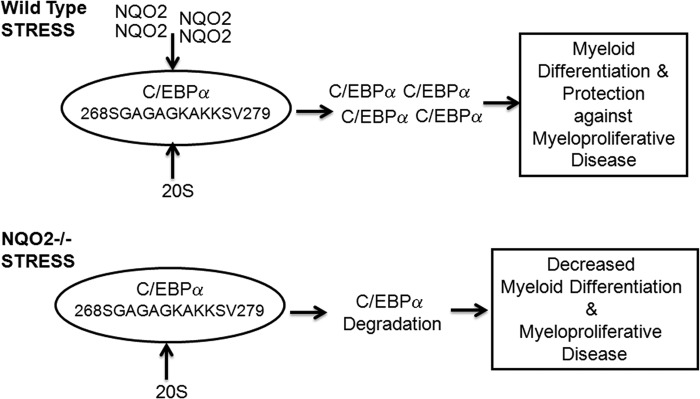
Model to demonstrate NQO2 protection against stress-induced myeloproliferative diseases.
Acknowledgments
We thank Dr. Richard Knox (UK) for NRH. We also thank Lubna Refai for help with manuscript preparation.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 ES007943 (to A. K. J.).
- C/EBPα
- CCAAT enhancer-binding protein α
- Gy
- gray
- NRH
- dihydronicotinamide riboside.
REFERENCES
- 1. Zhao Q., Yang X. L., Holtzclaw W. D., Talalay P. (1997) Unexpected genetic and structural relationships of a long-forgotten flavoenzyme to NAD(P)H:quinone reductase (DT-diaphorase). Proc. Natl. Acad. Sci. U.S.A. 94, 1669–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen S., Wu K., Knox R. (2000) Structure-function studies of DT-diaphorase (NQO1) and NRH:quinone oxidoreductase (NQO2). Free Radic. Biol. Med. 3–4, 276–284 [DOI] [PubMed] [Google Scholar]
- 3. Bianchet M. A., Foster C., Faig M., Talalay P., Amzel L. M. (1999) Structure and mechanism of cytosolic quinone reductases. Biochem. Soc. Trans. 27, 610–615 [DOI] [PubMed] [Google Scholar]
- 4. Gong X., Kole L., Iskander K., Jaiswal A. K. (2007) NRH:quinone oxidoreductase 2 and NAD(P)H:quinone oxidoreductase 1 protect tumor suppressor p53 against 20 S proteasomal degradation leading to stabilization and activation of p53. Cancer Res. 67, 5380–5388 [DOI] [PubMed] [Google Scholar]
- 5. Garate M., Wong R. P., Campos E. I., Wang Y., Li G. (2008) NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of the tumour suppressor p33(ING1b). EMBO Rep. 9, 576–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Patrick B. A., Gong X., Jaiswal A. K. (2011) Disruption of NAD(P)H:quinone oxidoreductase 1 gene in mice leads to 20 S proteasomal degradation of p63 resulting in thinning of epithelium and chemical-induced skin cancer. Oncogene 30, 1098–1107 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7. Long D. J., 2nd, Iskander K., Gaikwad A., Arin M., Roop D. R., Knox R., Barrios R., Jaiswal A.K. (2002) Disruption of dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) leads to myeloid hyperplasia of bone marrow and decreased sensitivity to menadione toxicity. J. Biol. Chem. 277, 46131–46139 [DOI] [PubMed] [Google Scholar]
- 8. Iskander K., Barrios R. J., Jaiswal A. K. (2009) NRH:quinone oxidoreductase 2-deficient mice are highly susceptible to radiation-induced B-cell lymphomas. Clin. Cancer Res. 15, 1534–1542 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9. Scott L. M., Civin C. I., Rorth P., Friedman A. D. (1992) A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood 80, 1725–1735 [PubMed] [Google Scholar]
- 10. Klemsz M. J., McKercher S. R., Celada A., Van Beveren C., Maki R. A. (1990) The macrophage and B cell-specific transcription factor PU. 1 is related to the ets oncogene. Cell 61, 113–124 [DOI] [PubMed] [Google Scholar]
- 11. Pabst T., Mueller B. U., Zhang P., Radomska H. S., Narravula S., Schnittger S., Behre G., Hiddemann W., Tenen D. G. (2001) Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat. Genet. 27, 263–270 [DOI] [PubMed] [Google Scholar]
- 12. Fröhling S., Schlenk R. F., Stolze I., Bihlmayr J., Benner A., Kreitmeier S., Tobis K., Döhner H., Döhner K. (2004) CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics. Prognostic relevance and analysis of cooperating mutations. J. Clin. Oncol. 22, 624–633 [DOI] [PubMed] [Google Scholar]
- 13. Yeamans C., Wang D., Paz-Priel I., Torbett B. E., Tenen D. G., Friedman A. D. (2007) C/EBPα binds and activates the PU. 1 distal enhancer to induce monocyte lineage commitment. Blood 110, 3136–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu J., Jaiswal A. K. (2012) NAD(P)H:quinone oxidoreductase 1 (NQO1) competes with 20 S proteasome for binding with C/EBPα leading to its stabilization and protection against radiation-induced myeloproliferative disease. J. Biol. Chem. 287, 41608–41618 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15. Iskander K., Barrios R. J., Jaiswal A. K. (2008) Disruption of NAD(P)H:quinone oxidoreductase 1 gene in mice leads to radiation-induced myeloproliferative disease. Cancer Res. 68, 7915–7922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith M.T., Wang Y., Skibola C. F., Slater D. J., Lo Nigro L., Nowell P. C., Lange B. J., Felix C. A. (2002) Low NAD(P)H:quinone oxidoreductase activity is associated with increased risk of leukemia with MLL translocations in infants and children. Blood 100, 4590–4593 [DOI] [PubMed] [Google Scholar]
- 17. Rosenbauer F., Wagner K., Kutok J. L., Iwasaki H., Le Beau M. M., Okuno Y., Akashi K., Fiering S., Tenen D. G. (2004) Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU. 1. Nat. Genet. 36, 624–630 [DOI] [PubMed] [Google Scholar]
- 18. Kaspar J. W., Niture S. K., Jaiswal A. K. (2009) Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47, 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang W., Jaiswal A. K. (2004) Sp3 repression of polymorphic human NRH:quinone oxidoreductase 2 gene promoter. Free Radic. Biol. Med. 37, 1231–1243 [DOI] [PubMed] [Google Scholar]
- 20. Wang W., Le W. D., Pan T., Stringer J. L., Jaiswal A. K. (2008) Association of NRH:quinone oxidoreductase 2 gene promoter polymorphism with higher gene expression and increased susceptibility to Parkinson's disease. J. Gerontol. A Biol. Sci. Med. Sci. 63, 127–134 [DOI] [PubMed] [Google Scholar]