

Background: Fungal cells require Ca2+ influx and signaling to survive some antifungal assaults.
Results: High affinity Ca2+ influx systems in yeasts required the Kch1 K+ transporters for full activation in response to ER stressors but not clinical antifungals.
Conclusion: Kch1 family proteins regulate Ca2+ influx in many yeast species.
Significance: Targeting fungal K+ transporters or Ca2+ channels may improve efficacy of existing antifungals in narrow situations.
Keywords: Calcineurin, Calcium Channels, Cell Death, ER Stress, Potassium Transport, Antifungals
Abstract
The activation of a high affinity Ca2+ influx system (HACS) in the plasma membrane is required for survival of yeast cells exposed to natural or synthetic inhibitors of essential processes (secretory protein folding or sterol biosynthesis) in the endoplasmic reticulum (ER). The mechanisms linking ER stress to HACS activation are not known. Here we show that Kch1, a recently identified low affinity K+ transporter in the plasma membrane of Saccharomyces cerevisiae, is up-regulated in response to several ER stressors and necessary for HACS activation. The activation of HACS required extracellular K+ and was also dependent on the high affinity K+ transporters Trk1 and Trk2. However, a paralog of Kch1 termed Kch2 was not expressed and not necessary for HACS activation in these conditions. The pathogenic yeast Candida albicans carries only one homolog of Kch1/Kch2, and homozygous knock-out mutants were similarly deficient in the activation of HACS during the responses to tunicamycin. However, the Kch1 homolog was not necessary for HACS activation or cell survival in response to several clinical antifungals (azoles, allylamines, echinocandins) that target the ER or cell wall. Thus, Kch1 family proteins represent a conserved linkage between HACS and only certain classes of ER stress in these yeasts.
Introduction
The endoplasmic reticulum (ER)2 is a membranous compartment consisting of tubules and flattened sacs involved in the biosynthesis of lipids and sterols and the processing, maturation, and transportation of secreted and membrane-bound proteins. ER stress can occur when any of these processes become limiting or taxed by environmental conditions such as nutrient starvation, hypoxia, calcium starvation, and toxins that inhibit the normal processing, folding, and assembly of secretory proteins. Chronic ER stress has been associated with several human pathologies including aging and neurodegenerative disease (1, 2). Similarly, tumor cells are thought to have chronic ER stress due to nutrient deprivation and hypoxic conditions, which if exacerbated can trigger apoptotic cell death, making the ER stress response a novel target for future therapies (3, 4).
Misfolded proteins in the ER can activate a well studied signaling network known as the unfolded protein response (UPR), which serves to relieve the stress by up-regulating molecular chaperones and coordinating protein trafficking pathways (5). The budding yeast Saccharomyces cerevisiae utilizes the transmembrane protein kinase and nuclease Ire1, the Hac1 transcription factor, and up-regulation of genes to help repair the damage caused by ER stressors (6–10). However, UPR-independent signaling networks also become activated and play crucial roles in cell survival. For example, Ire1 and Hac1 are not required for the survival of yeast cells during prolonged exposures to ER stressors, such as tunicamycin and dithiothreitol (11, 12). Instead, cell survival depends on the activation of a high affinity Ca2+ influx system (HACS), elevation of cytosolic free Ca2+ concentrations, and activation of the Ca2+/calmodulin-dependent protein phosphatase known as calcineurin (hereafter Cn) (13, 14). Although the essential targets of Cn in this process have not been identified, activated Cn somehow prevents limited rupture or permeabilization of the vacuolar membrane during prolonged responses to ER stress and thereby prevents subsequent nonapoptotic cell death (15). Inhibitors of Cn, such as FK506 and cyclosporine, dramatically convert tunicamycin, dithiothreitol, and other ER stressors from fungistats to fungicides. Similarly, Cn inhibitors convert the commonly used azole class of antifungal drugs, which target sterol biosynthesis in the ER of fungal cells, from fungistats to fungicides in a wide array of pathogenic fungi (13, 16–22). Inhibitors of HACS or its upstream regulators may be useful in antifungal therapies.
In S. cerevisiae, HACS is composed of Cch1, Mid1, and Ecm7 proteins that are homologous or analogous to the catalytic α-subunit, the regulatory α2δ-subunit, and the regulatory γ-subunit of voltage-gated Ca2+ channels present in animals (23–27). The hypothesis that HACS responds to membrane depolarization similar to its animal homologs is controversial for several reasons. Feedback inhibition of HACS by Cn suggests that protein kinases may stimulate HACS in response to certain stimuli (13, 28, 29). The four putative voltage-sensing S4 domains within Cch1 contain approximately half the number of the positively charged residues found in their mammalian counterparts (24), suggesting voltage insensitivity or perhaps tuning to the extremely low resting membrane potential of fungal cells (approximately −200 mV) (30–32). Finally, the Ca2+ currents generated through heterologous expression of Cch1 and Mid1 from the fungus Cryptococcus neoformans were largely insensitive to voltage (33). On the other hand, two putative K+ transporters, Kch1 and Kch2, were recently shown to regulate HACS through the transportation of extracellular K+, suggesting regulation through changes in the membrane potential (34). Expression of Kch1 and particularly Kch2 was strongly induced by the response to mating pheromones, which resulted in activated HACS, Cn, and cell survival mechanisms during prolonged mating responses (34).
Here we explore the possibility that Kch1 and Kch2 activate HACS during the response to ER stresses in S. cerevisiae and the pathogenic yeast Candida albicans. We also examine the individual roles of Kch1, HACS, and Cn in the responses to azole-class antifungals, other common antifungals, and additional classes of membrane stresses that were previously shown to activate HACS (13, 14, 27, 28). Remarkably, Kch1 family K+ transporters were induced or required for HACS activation by the endomembrane and ER stressors but not by the antifungals. Additional evidence suggests that K+ transporters in the Trk1 family represent alternative regulators of HACS. The findings strengthen the argument for HACS regulation by changes in transmembrane potential and suggest novel modes of antifungal intervention.
EXPERIMENTAL PROCEDURES
Yeast Strains, Plasmids, Culture Media, and Reagents
The S. cerevisiae strains used in this study (Table 1) were obtained from original sources or derived from parental strain W303-1A, by means of standard genetic crosses or PCR-based methods for introducing knock-out mutations and epitope tags (35). Yeast strains were cultured in rich YPD medium or synthetic SC medium (36) and shifted to alternative media as described below. Tunicamycin was purchased from Sigma-Aldrich and dissolved in DMSO. FK506 was obtained from Astellas Pharma and dissolved in DMSO. Aqueous 45CaCl2 was purchased from MP Biosciences or PerkinElmer.
TABLE 1.
Yeast strains used in this study
All strains are isogenic derivatives of S. cerevisiae strain W303-1A (MATa ade2-1 can1-100 his3-11,14 leu2-3,112 trp1-1 ura3-1) or C. albicans strain SC5314.
| Name | Description or genotype | Source or reference |
|---|---|---|
| K601 | Wild type | 41 |
| CS01 | kch2::G418 | 34 |
| CS02 | kch1::TRP1 | 34 |
| CS03 | kch1::TRP1 kch2::G418 | 34 |
| CS95 | cch1::HIS3 | 34 |
| CS34 | KCH2-MYC13::HIS3 | 34 |
| CS83 | KCH1-MYC13::HIS3 | 34 |
| CS166 | KCH1-MYC13::HIS3 ire1::TRP1 | This study |
| CS39 | pmr1::LEU2 | This study |
| CS40 | pmr1::LEU2 kch2::G418 | This study |
| CS41 | pmr1::LEU2 kch1::TRP1 | This study |
| CS42 | pmr1::LEU2 kch2::G418 kch1::TRP1 | This study |
| CS08 | trk1::LEU2 trk2::HIS3 | This study |
| CS11 | trk1::LEU2 trk2::HIS3 kch1::TRP1 kch2::G418 | This study |
| SC5314 | Candida albicans Wild-type | 69 |
| JLR48 | cch1-1::FRT/cch1-2::FRT | 70 |
| CS126 | kch1-1::FRT/kch1-2::FRT | This study |
| CS135 | cch1-1::FRT/cch1-2::FRT kch1-1::FRT/kch1-2::FRT | This study |
For C. albicans experiments, all deletion strains were generated in the SC5314 background. All primers used in strain construction are listed in Table 2. For disruption of the KCH1 gene, two ∼500-bp regions with homology to the 5′ promoter and 3′ terminator region were PCR-amplified and cloned into plasmid pSFS2A (38) with KpnI/XhoI, and NotI/SacI, respectively, generating plasmid pCS40. Plasmid pCS40 was digested with KpnI/SacI, and the disruption cassette was gel-purified. For transformations, 300 μl of saturated SC5314 and JLR48 cultures were allowed to recover in 10 ml of YPD, harvested, washed, and resuspended in 500 μl of LiOAc/Tris-EDTA solution. A standard transformation was then performed using 1 μg of linear DNA in 200 μl of resuspended cells. Cells were plated, and nourseothricin-resistant isolates selected on YPD + 200 μg/ml NAT were picked. Colonies were selected according to Ref. 38. Briefly, colonies were selected, allowed to grown overnight in YPD to allow the cassette to flip out, and then plated on 25 μg/ml nourseothricin plates. Small colonies representing strains in which one allele was excised (KCH1/kch1::FRT) were selected and confirmed by colony PCR. Confirmed colonies were then selected to undergo a second round of transformations. Double mutant strains (kch1::FRT/kch1::FRT) were confirmed by colony PCR.
TABLE 2.
Oligoneucleotides used in this study
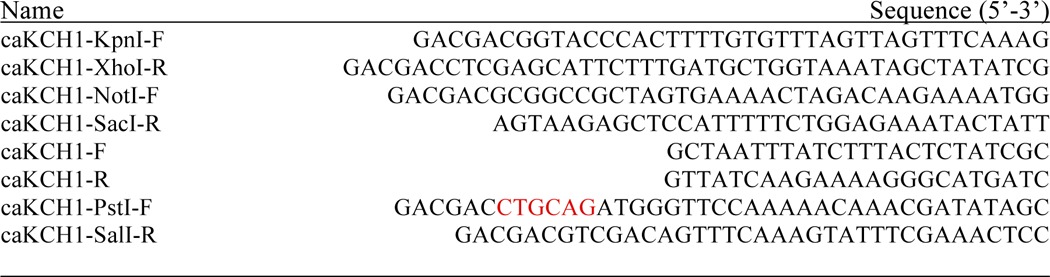
45Ca2+ Uptake Assays
Total cellular accumulation of Ca2+ was measured as described previously (39). Briefly, yeast cultures were grown to log phase in SC or YPD medium overnight, harvested, and resuspended in fresh SC-100 medium spiked with tracer 45CaCl2 (PerkinElmer) in the presence or absence of tunicamycin, N,N,N′,N″-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN), miconazole, terbinafine, or caspofungin with or without FK506 as indicated in the figures. SC-100 medium was similar to SC medium, except that the YNB component was replaced with yeast nitrogen base lacking calcium chloride, magnesium sulfate, sodium chloride, and potassium phosphate (Sunrise Scientific Products), supplemented with 100 mg/liter sodium phosphate, 0.5 g/liter MgSO4, and 100 μm CaCl2 and indicated concentrations of KCl. Cultures were then incubated at 30 °C in 96-well filtration plates (Millipore MultiScreen HTS plates), harvested by filtration, washed four times with ice-cold buffer A (10 mm CaCl2, 5 mm Na-HEPES, pH 6.5) on a vacuum filtration unit (Millipore), and dried at room temperature. The filters were covered with Microscint20 scintillation mixture (PerkinElmer), and counted using a TopCount NXT (Packard) or a Tri-Carb 2200 (Packard) liquid scintillation counter.
Western Blotting
Cultures were grown to log phase at 30 °C in YPD medium, adjusted to A600 ∼0.5 in standard SC medium, and split into five aliquots. Samples were treated with 1 μg/ml FK506, 2.5 μg/ml tunicamycin, 5 mm dithiothreitol (DTT) for 4.5 h at 30 °C before processing. Processing involved centrifuging one A600 unit of cells at 4 °C, lysis of cells with trichloroacetic acid, extraction of proteins with SDS sample buffer, then SDS-PAGE, and Western blotting as described previously (40). Blots were probed with anti-MYC monoclonal 9E10 antibodies (Covance). Protein standards were probed with anti-PGK1 (Abcam).
β-Galactosidase Assays
The activation of the UPR signaling pathway was monitored using plasmid pCZY1 (2μ URA3 UPRE-LacZ) (9). The activation of the calcium signaling pathway was monitored with plasmid pAMS366 (2μ URA3 4xCDRE-LacZ) (37). To measure the activation of the Ca2+ signaling cascade, cultures bearing the plasmid pCZY1 were grown overnight to log phase in SC medium lacking uracil. 2.5 μg/ml tunicamycin was added to 10 ml of log phase cultures and incubated at 24 °C with spinning and harvested every hour. To measure UPR activation, cultures bearing pAMS366 were grown overnight to log phase in SC medium lacking uracil. 5 μg/ml tunicamycin or 5 mm BAPTA was added to log phase cultures, and tubes were allowed to rotate at 24 °C with spinning for 3 and 3.5 h, respectively. Cultures were assayed for β-galactosidase activity as described previously (41).
Cell Death Measurements
S. cerevisiae cultures were grown overnight to log phase in SC medium at 30 °C. 0.2 A600 of cells was harvested, resuspended in standard SC medium, and mixed with 2-fold serial dilutions of tunicamycin. Cells were then incubated at room temperature for 10 h in a flat-bottom 96-well dish (BD Biosciences). For cell death of C. albicans strains in the presence of tunicamycin, log phase cultures grown in YPD overnight at 24 °C were added to 2-fold serial dilutions of tunicamycin in a flat-bottom 96-well dish and incubated at 30 °C for 7 h. For cell death measurements of C. albicans strains in the presence of antifungal drugs, cultures were grown overnight at 30 °C in SC medium to saturation, and saturated cells were diluted 1:4 into 2-fold dilutions of miconazole, terbinafine, or caspofungin in the presence or absence of 1 μg/ml FK506 in a flat-bottom 96-well dish and incubated at 30 °C for 24 h. In all experiments, the preincubated cells (20 μl) were mixed with 180 μl of PBS containing 1 μg/ml propidium iodide, and the live and dead cells were counted automatically using a 96-well flow cytometer FACSArray (BD Biosciences). At least 5,000 cells in each sample were counted.
Statistical Analyses
All statistical analysis was performed in the graphing software Prism. For all experiments, at least three biological replicates were analyzed in parallel and plotted as averages (±S.D.). Statistical significance was assessed by two-way analysis of variance on kch1 mutant strains relative to the isogenic control strains. Significant differences were marked in the figures as * (p < 0.05) or ** (p < 0.01).
RESULTS
Kch1 Regulates Ca2+ Uptake and CN Activation in S. cerevisiae Cells Exposed to Tunicamycin
The activation of HACS during the response to mating pheromones was recently shown to depend on the related fungus-specific proteins Kch1 and Kch2 (34). To test whether Kch1 and Kch2 are required for the activation of HACS during a very different kind of stress, ER stress, the uptake of 45Ca2+ was measured in kch1 kch2 double mutants and single mutants during a 4-h exposure to tunicamycin in the presence of FK506, an inhibitor of Cn that relieves the feedback inhibition of HACS. The kch2 single mutant cells exhibited wild-type levels of 45Ca2+ uptake, whereas the kch1 single mutant cells and the kch1 kch2 double mutant cells exhibited significantly lower levels of 45Ca2+ uptake during the response to tunicamycin (Fig. 1A). The defects observed in kch1 mutants and kch1 kch2 double mutants were not as severe as those of the cch1 mutants, suggesting residual HACS activation in the absence of Kch1 and Kch2. Similarly, when Cn was not inhibited (Fig. 1B), kch1 kch2 double mutants exhibited less 45Ca2+ accumulation than wild-type cells, although HACS activity was much lower in these conditions. To test whether Cn signaling also declines in kch1 mutants, expression of the CDRE-lacZ reporter gene was measured after exposure to tunicamycin. The kch1 mutant cells demonstrated significantly less CDRE-lacZ expression than wild-type cells and about the same as the HACS-deficient cch1 mutant cells and the cch1 kch1 double mutant cells (Fig. 1C). These results suggest that Kch1 functions in the same signaling pathway as HACS and Cn. To determine whether the defect in Cn signaling observed in kch1 mutant cells was extreme enough to prevent cell survival during the response to tunicamycin, cell death was measured after exposure to tunicamycin by staining with propidium iodide. Similar to Cn-deficient and HACS-deficient cells, the kch1 mutant cells exhibited enhanced cell death after prolonged exposure to tunicamycin (Fig. 1D). Collectively, these findings show that the activation of HACS, Cn, and essential cell survival pathways during the response to tunicamycin requires Kch1 but not Kch2.
FIGURE 1.

Kch1 regulates HACS-dependent Ca2+ accumulation in response to tunicamycin. A and B, 45Ca2+ uptake into cultures of S. cerevisiae strains that contain combinations of Kch1, Kch2, and Cch1 (strains K601, CS01, CS02, CS03, CS04) was measured after a 4-h incubation in SC-100 (10 mm KCl) medium in the presence or absence of 3 μg/ml tunicamycin plus 0.25 μg/ml FK506, as indicated. C, β-galactosidase activity of the above mentioned strains transformed with a CDRE-lacZ reporter gene (pAMS366) in SC medium in the presence of 2.5 μg/ml tunicamycin. Samples were collected at the indicated time points. D, wild type and kch1 mutants (K601 and CS02) were exposed to 2-fold dilutions of tunicamycin, incubated for 10 h, and stained with propidium iodide. Live and dead cells were counted by flow cytometry. In all panels, averages for three biological replicates (±S.D.) are shown, and significant differences between kch1 and KCH1 control cells are indicated (**; see “Experimental Procedures”).
DNA microarray experiments suggested that transcripts encoding Kch1, but not Kch2, were strongly induced during the exposure to tunicamycin via the canonical UPR signaling pathway (42). To test whether Kch1 and Kch2 protein levels were altered during the response to ER stress, Western blots were performed on cells expressing genomic variants of Kch1 and Kch2 containing the Myc13 tag at their C termini. Unlike the mating pheromone α-factor, tunicamycin and DTT exposures failed to induce expression of Kch2 (Fig. 2A). Both ER stressors strongly induced expression of Kch1 as well as a slight shift in mobility on SDS-PAGE that could not be blocked by FK506 (Fig. 2B). The up-regulation, but not the mobility shift, of Kch1 was dependent on Ire1 gene function (Fig. 2A). These findings explain why Kch2 was not required for HACS activation during the response to ER stressors and suggest that Kch1 up-regulation and post-translational modifications are associated with the effect.
FIGURE 2.
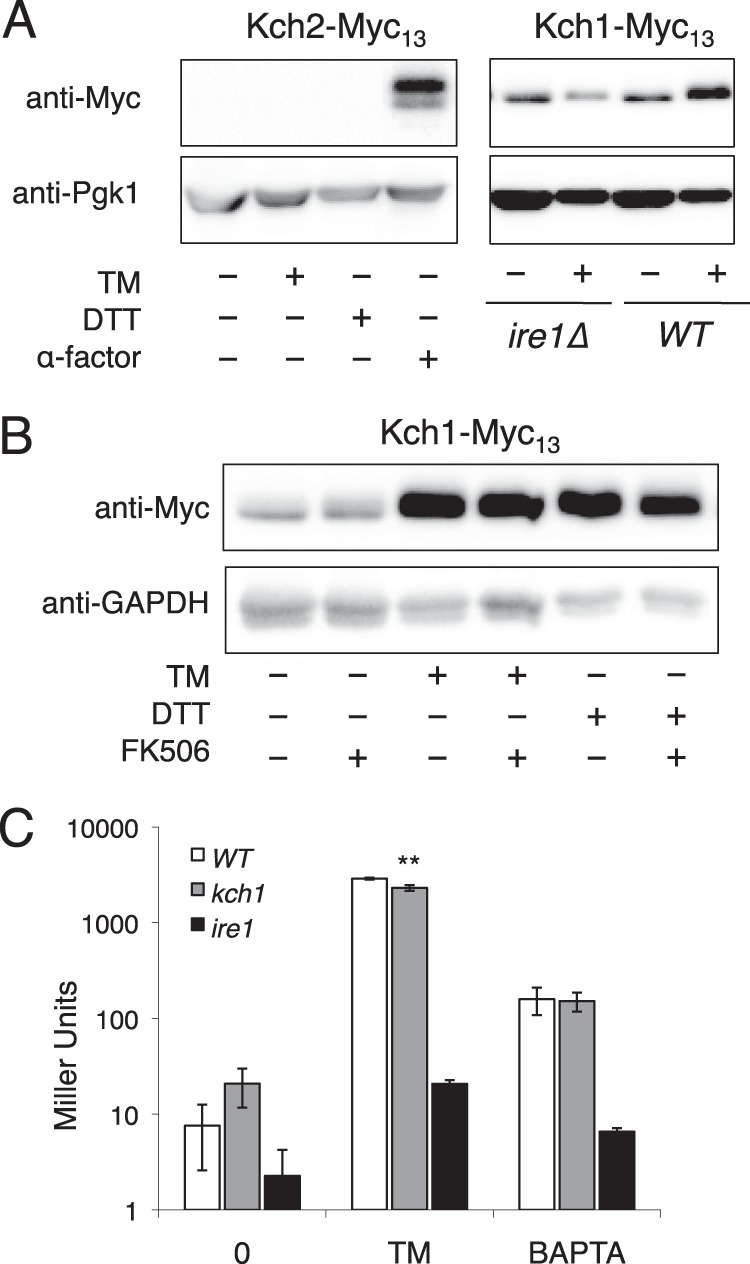
Altered expression and modification of Kch1 in response to tunicamycin and DTT. A and B, whole-cell lysates of cells expressing Kch2-Myc13 (A) or Kch1-Myc13 (A and B) were prepared after a 4-h exposure to tunicamycin (TM), DTT, α-factor, and/or FK506 as indicated. C, assays of β-galactosidase activity of wild type, kch1, and ire1 mutants (K601, CS03, DNY1048) transformed with a UPRE-lacZ reporter gene (pCZY1) in the presence or absence of tunicamycin or BAPTA after incubation for 3 or 3.5 h, respectively. Averages for three biological replicates (±S.D.) are shown, and significant differences between kch1 and KCH1 control cells are indicated (**; see “Experimental Procedures”).
To test whether Kch1 is necessary for proper signaling in the UPR pathway, UPRE-lacZ expression was measured in the presence of two ER-damaging agents, tunicamycin and BAPTA (Fig. 2B). Under these conditions, wild-type cells induced UPRE-lacZ expression more than 10-fold, whereas ire1 mutants exhibited greatly diminished responses. UPRE-lacZ expression in kch1 mutant cells was ∼80% of wild-type cells, suggesting that Kch1 is largely unnecessary for UPR signaling during ER stress.
Extracellular K+ Is Required for Ca2+ Influx in Response to Tunicamycin
The activation of HACS during the response to mating pheromones required extracellular K+ in addition to Kch1 or Kch2 (34). To test the role of extracellular K+ during the response to ER stress, kch1 kch2 double mutants and the control strain were exposed to tunicamycin plus FK506 in synthetic growth medium containing 45Ca2+ and varying amounts of dissolved KCl. Indeed, extracellular K+ promoted 45Ca2+ uptake in both strains in a dose-dependent fashion (Fig. 3A) with the wild-type strain exhibiting significantly stronger responses at almost every K+ concentration. The difference between the wild-type and kch1 kch2 double mutant strain was more pronounced when FK506 was omitted and the Cn-dependent feedback inhibition of HACS was restored (Fig. 3B). The residual 45Ca2+ uptake that occurred in the absence of Kch1 and Kch2 was likely due to residual HACS activation because it depended on extracellular K+ and it also depended on the high affinity K+ transporters Trk1 and Trk2 (Fig. 3, A and B). At high concentrations of K+, tunicamycin activated HACS in trk1 trk2 double mutant cells and, to a lesser degree, in the kch1 kch2 trk1 trk2 quadruple mutant cells. These experiments show that high affinity transporters Trk1 and Trk2 play important roles in the activation of HACS in low K+ conditions (less than 50 mm) that cannot be fulfilled by Kch1 (or Kch2). On the other hand, Kch1 contributed to HACS activation in high K+ conditions in the presence or absence of Trk1 and Trk2 and in low K+ conditions only when Trk1 and Trk2 were operational. The aggregate findings suggest that Kch1, Trk1, and Trk2 regulate HACS by controlling K+ uptake likely through their effects on the transmembrane electrical potential of the plasma membrane.
FIGURE 3.

Differential regulation of HACS by K+ ions through Kch1 and the K+ transporters Trk1 and Trk2 in response to tunicamycin and DTT. A–D, K+ dependent 45Ca2+ uptake was measured in wild type, kch1/2, trk1/2, and quadruple mutants (K601, CS03, CS08, CS11) after a 4-h incubation in SC-100 medium containing the indicated concentrations of KCl plus 2.5 μg/ml tunicamycin (TM) in the presence (A) or absence (B) of 0.25 μg/ml FK506 or DTT (C and D), respectively. Averages for four biological replicates (±S.E.) are shown, and significant differences between kch1 and KCH1 control cells are indicated (* or **; see “Experimental Procedures”).
Other ER Stressors Utilize Kch1 and Kch2 for Activation of HACS
The membrane permeable reducing agent DTT can suppress the formation of disulfide bonds in the endoplasmic reticulum, leading to protein misfolding and the activation of the UPR signaling pathway (13). DTT therefore elicits cellular responses that overlap with those of tunicamycin, an inhibitor of N- and O-glycosylation of secretory proteins (43). When DTT was used instead of tunicamycin to stimulate yeast cells in different K+ concentrations, the resulting uptake of 45Ca2+ was different in several ways. First, although the stimulatory effects of Kch1 remained evident, 45Ca2+ uptake into wild-type and kch1 kch2 double mutant cells in the presence of FK506 was several times lower in cells responding to DTT relative to the cells responding to tunicamycin (Fig. 3C). Second, the stimulatory effects of Kch1 and Kch2 on 45Ca2+ uptake disappeared in the trk1 trk2 double mutant cells (Fig. 3C) and in all the cells when FK506 was omitted (Fig. 3D). DTT and tunicamycin induced Kch1 expression and post-translational modifications to similar extents (Fig. 2B). It is possible that DTT secondarily inhibits HACS by reducing disulfide bridges in the regulatory subunits, Mid1 and Ecm7 (27).
Perturbation in ER Ca2+ homeostasis is known to result in ER stress and the activation of the UPR signaling pathway (13). The membrane-permeable metal chelator N,N,N′,N″-tetrakis-(2-pyridylmethyl) ethylenediamine (TPEN) has been shown to activate the UPR signaling pathway and HACS in yeast, presumably through an ability to chelate Ca2+ and other metal ions within the endoplasmic reticulum (13). Although TPEN did not induce expression or modification of Kch1 like DTT and tunicamycin (not shown), the elevated 45Ca2+ uptake observed in TPEN-exposed cells was dependent on Kch1 (Fig. 4B).
FIGURE 4.
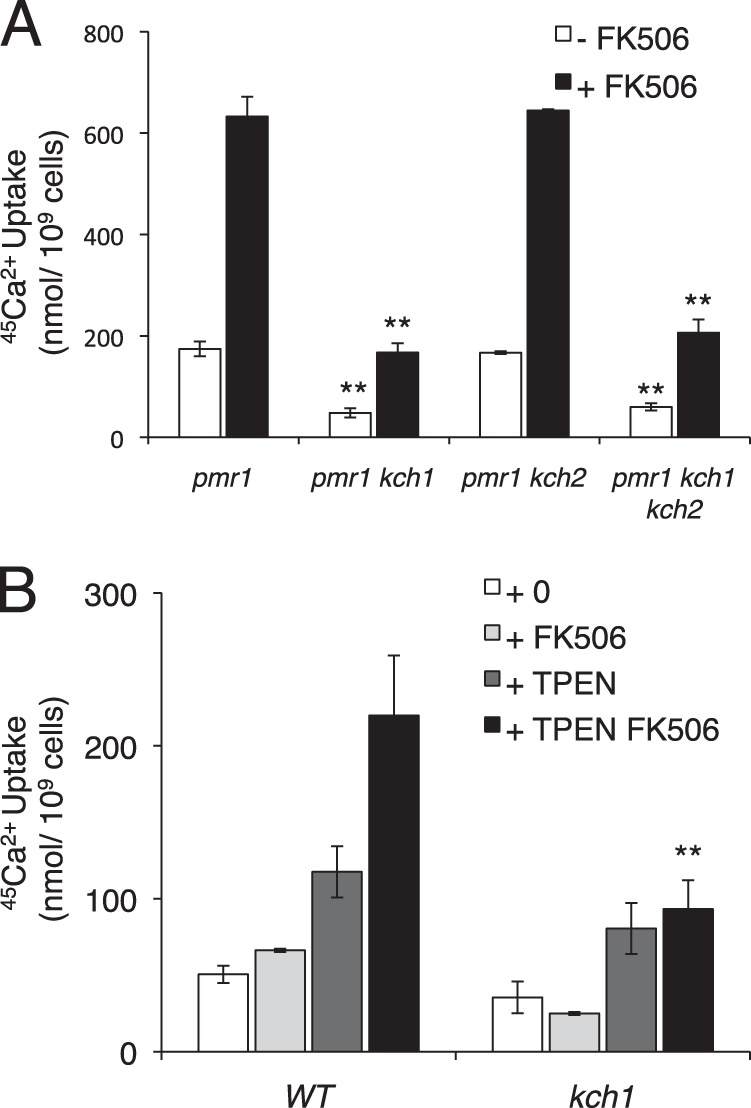
Defects in Ca2+ homeostasis result in a Kch1-dependent Ca2+ accumulation. A, 45Ca2+ uptake into pmr1 mutant backgrounds containing combinations of Kch1 and Kch2 (CS39, CS40, CS41, CS42) after 4 h of log-phase growth in SC-100 medium (5 mm KCl) in the presence or absence of 0.1 μg/ml FK506. B, 45Ca2+ uptake into wild type and kch1 mutants (K601, CS02) in YPD medium in the presence or absence of 200 μm TPEN and 1 μg/ml FK506 after incubation for 4 h. Averages for three biological replicates (±S.D.) are shown, and significant differences between kch1 and KCH1 control cells are indicated (**; see “Experimental Procedures”).
Yeast cells lack a homolog of the sarco/endoplasmic reticulum Ca2+ ATPase and instead utilize a homolog of the secretory pathway Ca2+ ATPase (SPCA) known as Pmr1 to supply Ca2+ to the endoplasmic reticulum and Golgi complex (44–46). Similarly to the addition of TPEN, pmr1 mutants also exhibit elevated 45Ca2+ uptake via activated HACS (13). The further loss of Kch1 in pmr1 mutants strongly diminished 45Ca2+ uptake, whereas the further loss of Kch2 had no significant effect (Fig. 4A). Therefore, Kch1 mediated the elevated HACS activity observed in the pmr1 mutant cells.
HACS Dependence on Kch1 Is Conserved in C. albicans
The opportunistic pathogen C. albicans utilizes HACS and Cn to survive assaults by tunicamycin (13) as well as antifungal compounds that target the ER (16–18, 20). To determine whether the sole homolog of Kch1 in C. albicans, termed caKch1, plays a role in these processes, both alleles of caKCH1 were deleted, and 45Ca2+ accumulation was measured in response to tunicamycin and FK506. Similar to S. cerevisiae, 45Ca2+ accumulation in the kch1−/− mutant strain was significantly lower than the parent strain and higher than both the cch1−/− mutant and the kch1−/− cch1−/− double mutant (Fig. 5A). To test whether this Ca2+ accumulation deficit resulted in cell death, strains were stained with propidium iodide after prolonged exposure to tunicamycin. The kch1−/− mutant exhibited a much higher level of cell death than the control strain, but somewhat less cell death than the kch1−/− cch1−/− double mutant strain (Fig. 5B). Thus, tunicamycin-stressed C. albicans depended on Kch1 and HACS for cell survival, similar to S. cerevisiae.
FIGURE 5.
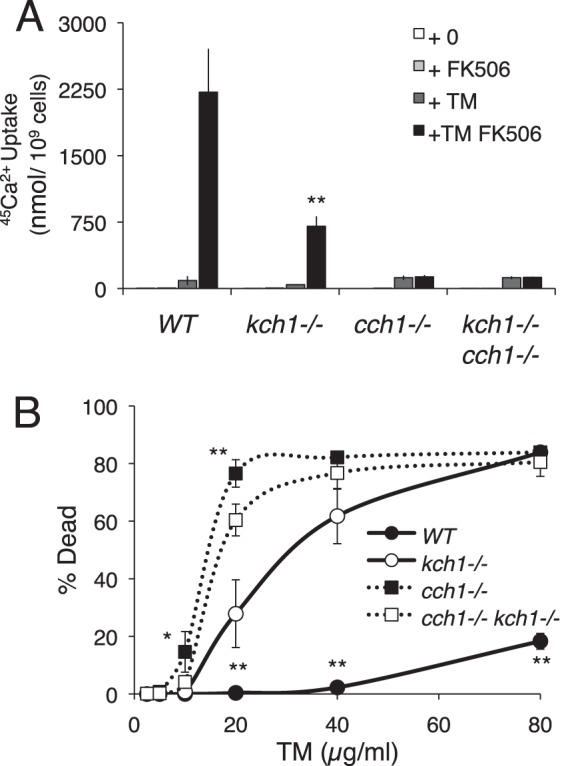
Kch1 function is conserved in the pathogenic fungi C. albicans. A, 45Ca2+ uptake into C. albicans wild type, kch1−/−, cch1−/−, and the quadruple mutant (SC5314, JLR48, CS126, CS135) in SC-100 media (10 mm KCl) in the presence or absence of 3 μg/ml tunicamycin and 1 μg/ml FK506 after a 4-h incubation. B, cell death measurements on log phase cultures of strains listed above in SC medium in the presence of indicated tunicamycin concentrations. Cells were stained with propidium iodide and measured by flow cytometry. Averages for three biological replicates (±S.D.) are shown, and significant differences between kch1 and KCH1 control cells are indicated (* or **; see “Experimental Procedures”).
Fig. 6 illustrates the roles of Kch1, HACS, and Cn in the responses of C. albicans to synthetic antifungal drugs that target different enzymes in the ER and plasma membrane. Miconazole, an inhibitor of cytochrome P-450 lanosterol 14α-demethylase during ergosterol biosynthesis in the ER (47), stimulated strong 45Ca2+ uptake similarly in wild-type cells and kch1−/− cells but not the cch1−/− and kch1−/− cch1−/− cells (Fig. 6A), suggesting that this compound activates HACS through a mechanism that does not depend on Kch1. Similarly, Kch1 was not required for HACS and Cn to suppress cell death after exposure to miconazole (Fig. 6B). Terbinafine, an inhibitor of squalene epoxidase (Erg1) early in the ergosterol biosynthesis pathway in the ER (20), produced 25-fold stronger 45Ca2+ uptake than miconazole in these conditions, and Kch1 was not detectably required for this process (Fig. 6C). Surprisingly, Kch1 promoted survival of cch1−/− cells in the presence or absence of FK506 and promoted the death of cells that were proficient in HACS and Cn (Fig. 6D), suggesting unique roles of Kch1 in this condition that were independent of HACS and Cn. Caspofungin, an inhibitor of the cell wall component β-1,3-d-glucan (48), generated a very small amount of 45Ca2+ uptake that was partially dependent on HACS and Kch1 (Fig. 6E). Unlike miconazole and terbinafine, caspofungin triggered cell death in the wild-type strain, and the effect was slightly increased in the mutants lacking Cch1 (Fig. 6F). Thus, these common antifungal compounds all activated HACS, Cn, and survival mechanisms independent of Kch1.
FIGURE 6.

Ca2+ accumulation and cell death in Ca2+ pathway mutants in response to commonly used antifungals. A, C, and E, 45Ca2+ uptake into strains listed in Fig. 5 in SC-100 medium (10 mm KCl) after exposure to 0.25 μg/ml miconazole (MIC) (A), 25 μg/ml terbinafine (TB) (C), or 25 μg/ml caspofungin (CAS) (E) in the presence or absence of 1 μg/ml FK506 after 4 h of incubation. Averages for three biological replicates (±S.D.) are shown, and significant differences between kch1 and KCH1 control cells are indicated (**; see “Experimental Procedures”). B, D, and F, cell death measurements on saturated cultures in SC media in the presence of indicated concentrations of miconazole (B), terbinafine (D), or caspofungin (F) in the presence or absence of 1 μg/ml FK506 after a 24-h incubation. Cells were stained with propidium iodide and measured by flow cytometry.
DISCUSSION
Regulation of HACS in Yeasts by Kch1 and K+
We report here that K+ regulates HACS in a broader variety of yeast species and conditions than just S. cerevisiae cells responding to mating pheromones (34). Specifically, the putative K+ transporters Kch1 in S. cerevisiae and its ortholog caKch1 in C. albicans participate in the activation of HACS during the response to ER stress. The ER stressors DTT and tunicamycin induced expression and post-translational modification of Kch1 in S. cerevisiae. Kch1 was necessary for most HACS-dependent Ca2+ uptake and Cn activation under these conditions, as indicated by diminished CDRE-lacZ expression and cell survival in kch1 knock-out mutants. Interestingly, full HACS activation still occurred in ire1 mutants responding to ER stress (13), suggesting that induction of Kch1 is not always necessary for HACS activation. Other ER stressors, such as TPEN and Pmr1 deficiency that both affect Ca2+ homeostasis in secretory organelles, failed to induce Kch1 expression (data not shown) and yet increased HACS activity through mechanisms that were only partially dependent on Kch1. These findings suggest that Kch1 may respond to ER stress through both transcriptional and post-translational mechanisms.
We show here that the physiological activation of HACS in response to tunicamycin also depended very strongly on Trk1 and Trk2, well characterized K+ transporters in yeasts (50–53), and was highly sensitive to the concentration of K+ in the environment. Trk1 and Trk2 lowered the requirements for extracellular K+ from ∼50 mm in trk1 trk2 double mutants to ∼1 mm in control strains that contained or lacked Kch1 (Fig. 3A). Therefore, HACS activation depended on the influx of K+ ions or intracellular K+ rather than extracellular K+ concentrations. Interestingly, Kch1 did not lower the requirements for extracellular K+ like Trk1 and Trk2 but instead increased the amplitude of HACS activation independent of Trk1 and Trk2 at the effective concentrations of K+. This finding could suggest a role for Kch1 on HACS activation that is either independent of K+ or dependent on some other K+ transporter; however, no such role was evident in trk1 trk2 mutant cells that were stressed with DTT instead of tunicamycin (Fig. 3C). Additionally, this finding is consistent with the possibility that Kch1 plays less of a role in K+ nutrition than Trk1 and Trk2 and more of a role in setting the transmembrane electrical potential, in much the same way that the Kir channels and NALCN channels help set the resting potentials of mammalian cells (54, 55). In fact, Kch1-dependent K+ currents were inwardly rectifying in both yeast and animal cells similar to Kir channels (34, 56). More work is necessary to discriminate whether Kch1 functions as a channel, transporter, or regulator of a channel or transporter.
The finding that HACS relies upon Trk1 family proteins in addition to Kch1 family proteins and their common substrate (K+) is further evidence that HACS senses changes to membrane voltage in yeasts, similar to its homologs in animals. More evidence comes from the rapid kinetics of HACS activation in response to sudden elevation of the environmental pH (27, 57). Because S. cerevisiae cells utilize an electrogenic H+ ATPase instead of an electrogenic Na+/K+ ATPase to energize their plasma membranes, the high pH shock is expected to depolarize the cell membrane (27, 57). The strongly negative resting potential of S. cerevisiae cells may necessitate fewer numbers of positively charged residues within the voltage-sensing S4 domains of Cch1 to achieve voltage sensitivity of HACS. Lastly, the apparent voltage insensitivity of a reconstituted HACS composed of Cch1 and Mid1 homologs from C. neoformans (33) may reflect the narrow voltage range examined (−100 mV and higher) and/or the absence of a γ-subunit homolog (Ecm7 in S. cerevisiae (27)). The effects of increasing or decreasing the number of charges in the S4 domains of HACS in these fungi has not been explored, but the results may be relevant to understanding the NALCN type of Na+ leak channels, which are the essential orthologs of Cch1 in animals (58) that contain a similarly reduced number of positive charges in their S4 domains.
Roles of Kch1, HACS, Ca2+, and Cn during Antifungal Assaults
Inhibitors of Cn, such as FK506 and cyclosporine, have long been known to convert azole-class antifungals, ER stressors, and natural mating pheromones from fungistats to fungicides (13, 16–22, 29, 49, 59–61), spurring research into the upstream regulators and downstream effectors of Cn in these phenomena that might be targeted for future antifungal therapies. The present study contributes several new findings to this ongoing enterprise. First, Kch1 family proteins played important roles in HACS activation, Cn activation, and cell survival during exposures to tunicamycin in both S. cerevisiae and C. albicans, suggesting a conservation of Kch1 function at least among these yeasts. However, the contributions of Kch1 were less important during exposures of S. cerevisiae to DTT and insignificant during exposures of C. albicans to miconazole, terbinafine, and, to a lesser extent, caspofungin, although HACS and Cn were strongly activated in all these conditions and important for cell survival. The findings therefore suggest that other cellular factors can sometimes regulate HACS independent of Kch1. The strong role of Trk1 and Trk2 in the regulation of HACS in tunicamycin-exposed S. cerevisiae suggests that other K+ transporters or channels or perhaps direct regulators of HACS itself may respond to common antifungals. Second, HACS and Cn occasionally promoted cell survival in an additive fashion, as if they functioned independently. The clearest example of this phenomenon was observed during the exposure of C. albicans to miconazole (Fig. 6A). In the absence of Cn, the uninhibited HACS may activate other Ca2+-sensitive survival factors such as Cmk2, which has been implicated already in the survival of S. cerevisiae cells exposed to mating pheromones and ER stressors (13, 14, 49). In the absence of HACS, alternative low affinity Ca2+ influx systems (29, 62) may contribute to the activation of Cn and Cmk2, resulting in cell survival. Therefore, functional redundancies may also exist at the levels of Ca2+ influx and Ca2+ effectors. Third, Kch1 uniquely promoted survival of C. albicans cells exposed to low doses of terbinafine in the absence of both HACS and Cn (Fig. 6D). Although more work would be required to understand this phenomenon, it hints at the possibility that K+ influx may influence the mechanism that governs vacuole rupture, which has been proposed as the key event that discriminates cell survival and eventual cell death (15). Rather than pinpointing a simple linear pathway that prevents vacuole rupture and promotes cell survival during antifungal assaults, this study suggests a network of interacting transporters and channels, Ca2+ influx systems, and Ca2+ effector proteins that contribute in varying degrees, under certain conditions and in differing species, to achieve cell survival.
The echinocandin class of antifungals, including caspofungin, is well known to have fungicidal activity on diverse species through blockade of cell wall biogenesis (63). Remarkably, HACS and Cn promoted survival of C. albicans cells that were exposed to sublethal doses of caspofungin (Fig. 6F). The increased cell death observed in HACS and Cn-deficient mutants of C. albicans may explain why these mutants exhibit hypersensitivity to caspofungin in standardized assays of culture growth (64). The abilities of HACS and Cn to suppress the lethal consequences of caspofungin, terbinafine, miconazole, tunicamycin, and dithiothreitol, which all inhibit different enzymes in the plasma membrane and ER, suggest that the calcium signaling network operates as a general defense system in fungal cells that responds to diverse membrane stressors.
Understanding how Cn defends fungal cells from antifungal assaults will be potentially useful in the development of new methods for controlling fungal pathogens in humans. Several prominent targets of Cn, including Crz1, Slm1/Slm2, Hph1/Hph2, and Rcn1/Rcn2, have been shown to be unnecessary for the survival-promoting activity of Cn in S. cerevisiae (14, 40, 65). Identifying the targets of Cn that promote cell survival or prevent cell death in each of the conditions will be important for determining whether the antifungals share a common mechanism of action that is normally thwarted by the activation of Cn.
Cn activation in chronic myelogenous leukemia cells has been shown recently to promote both cell survival in vitro and cancer progression in vivo after exposure to imatinib (Gleevec), which inhibits the Bcr-Abl oncogene that drives several cancers (66). Cn also prevents the death of normal cells of the proximal tubules of kidneys, thus explaining the nephrotoxic side effects of the immunosuppressants FK506 and cyclosporine (67, 68). Therefore, some of the cell death-defying functions of Cn may be conserved across the eukaryotic kingdoms and potentially exploited in the treatment of wide ranging diseases. More research in all these areas will be necessary to define the critical targets of Cn in all these circumstances.
Acknowledgments
We thank Joe Heitman (Duke University) for kind gifts of C. albicans strains SC5314 and JLR48 and for the disruption plasmid pSFS2A. We also thank Adam Kim (Johns Hopkins University) for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Research Awards NS057023 and NS074072 (to K. W. C.).
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- Cn
- calcineurin
- HACS
- high affinity calcium influx system
- SC
- synthetic complete
- TPEN
- N,N,N′,N″-tetrakis-(2-pyridylmethyl) ethylenediamine
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid.
REFERENCES
- 1. Brown M. K., Naidoo N. (2012) The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Macario A. J., Conway de Macario E. (2002) Sick chaperones and ageing: a perspective. Ageing Res. Rev. 1, 295–311 [DOI] [PubMed] [Google Scholar]
- 3. Kusio-Kobialka M., Podszywalow-Bartnicka P., Peidis P., Glodkowska-Mrowka E., Wolanin K., Leszak G., Seferynska I., Stoklosa T., Koromilas A. E., Piwocka K. (2012) The PERK-eIF2α phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 11, 4069–4078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schonthal A. H. (2012) Targeting endoplasmic reticulum stress for cancer therapy. Front. Biosci. (Schol. Ed.) 4, 412–431 [DOI] [PubMed] [Google Scholar]
- 5. Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 6. Harding H. P., Zhang Y., Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 [DOI] [PubMed] [Google Scholar]
- 7. Tirasophon W., Welihinda A. A., Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang X. Z., Harding H. P., Zhang Y., Jolicoeur E. M., Kuroda M., Ron D. (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 17, 5708–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mori K., Kawahara T., Yoshida H., Yanagi H., Yura T. (1996) Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1, 803–817 [DOI] [PubMed] [Google Scholar]
- 10. Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 [DOI] [PubMed] [Google Scholar]
- 11. Bonilla M., Cunningham K. W. (2002) Calcium release and influx in yeast: TRPC and VGCC rule another kingdom. Sci. STKE 2002, pe17. [DOI] [PubMed] [Google Scholar]
- 12. Chen Y., Feldman D. E., Deng C., Brown J. A., De Giacomo A. F., Gaw A. F., Shi G., Le Q. T., Brown J. M., Koong A. C. (2005) Identification of mitogen-activated protein kinase signaling pathways that confer resistance to endoplasmic reticulum stress in Saccharomyces cerevisiae. Mol. Cancer Res. 3, 669–677 [DOI] [PubMed] [Google Scholar]
- 13. Bonilla M., Nastase K. K., Cunningham K. W. (2002) Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 21, 2343–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dudgeon D. D., Zhang N., Ositelu O. O., Kim H., Cunningham K. W. (2008) Nonapoptotic death of Saccharomyces cerevisiae cells that is stimulated by Hsp90 and inhibited by calcineurin and Cmk2 in response to endoplasmic reticulum stresses. Eukaryot. Cell 7, 2037–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim H., Kim A., Cunningham K. W. (2012) Vacuolar H+-ATPase (V-ATPase) promotes vacuolar membrane permeabilization and nonapoptotic death in stressed yeast. J. Biol. Chem. 287, 19029–19039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchetti O., Entenza J. M., Sanglard D., Bille J., Glauser M. P., Moreillon P. (2000) Fluconazole plus cyclosporine: a fungicidal combination effective against experimental endocarditis due to Candida albicans. Antimicrob. Agents Chemother. 44, 2932–2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marchetti O., Moreillon P., Glauser M. P., Bille J., Sanglard D. (2000) Potent synergism of the combination of fluconazole and cyclosporine in Candida albicans. Antimicrob. Agents Chemother. 44, 2373–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cruz M. C., Goldstein A. L., Blankenship J. R., Del Poeta M., Davis D., Cardenas M. E., Perfect J. R., McCusker J. H., Heitman J. (2002) Calcineurin is essential for survival during membrane stress in Candida albicans. EMBO J. 21, 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edlind T., Smith L., Henry K., Katiyar S., Nickels J. (2002) Antifungal activity in Saccharomyces cerevisiae is modulated by calcium signalling. Mol. Microbiol. 46, 257–268 [DOI] [PubMed] [Google Scholar]
- 20. Onyewu C., Blankenship J. R., Del Poeta M., Heitman J. (2003) Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother. 47, 956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaur R., Castaño I., Cormack B. P. (2004) Functional genomic analysis of fluconazole susceptibility in the pathogenic yeast Candida glabrata: roles of calcium signaling and mitochondria. Antimicrob. Agents Chemother. 48, 1600–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinbach W. J., Schell W. A., Blankenship J. R., Onyewu C., Heitman J., Perfect J. R. (2004) In vitro interactions between antifungals and immunosuppressants against Aspergillus fumigatus. Antimicrob. Agents Chemother. 48, 1664–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer M., Schnell N., Chattaway J., Davies P., Dixon G., Sanders D. (1997) The Saccharomyces cerevisiae CCH1 gene is involved in calcium influx and mating. FEBS Lett. 419, 259–262 [DOI] [PubMed] [Google Scholar]
- 24. Paidhungat M., Garrett S. (1997) A homolog of mammalian, voltage-gated calcium channels mediates yeast pheromone-stimulated Ca2+ uptake and exacerbates the cdc1(Ts) growth defect. Mol. Cell. Biol. 17, 6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iida H., Nakamura H., Ono T., Okumura M. S., Anraku Y. (1994) MID1, a novel Saccharomyces cerevisiae gene encoding a plasma membrane protein, is required for Ca2+ influx and mating. Mol. Cell. Biol. 14, 8259–8271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iida K., Tada T., Iida H. (2004) Molecular cloning in yeast by in vivo homologous recombination of the yeast putative α1 subunit of the voltage-gated calcium channel. FEBS Lett. 576, 291–296 [DOI] [PubMed] [Google Scholar]
- 27. Martin D. C., Kim H., Mackin N. A., Maldonado-Báez L., Evangelista C. C., Jr., Beaudry V. G., Dudgeon D. D., Naiman D. Q., Erdman S. E., Cunningham K. W. (2011) New regulators of a high affinity Ca2+ influx system (HACS) revealed through a genome-wide screen in yeast. J. Biol. Chem. 286, 10744–10754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bonilla M., Cunningham K. W. (2003) MAP kinase stimulation of Ca2+ signaling is required for survival of endoplasmic reticulum stress in yeast. Mol. Biol. Cell 14, 4296–4305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muller E. M., Locke E. G., Cunningham K. W. (2001) Differential regulation of two Ca2+ influx systems by pheromone signaling in Saccharomyces cerevisiae. Genetics 159, 1527–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Slayman C. L. (1965) Electrical properties of Neurospora crassa. Effects of external cations on the intracellular potential. J. Gen. Physiol. 49, 69–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Slayman C. L., Long W. S., Lu C. Y. (1973) The relationship between ATP and an electrogenic pump in the plasma membrane of Neurospora crassa. J. Membr Biol. 14, 305–338 [DOI] [PubMed] [Google Scholar]
- 32. Peña A., Sánchez N. S., Calahorra M. (2010) Estimation of the electric plasma membrane potential difference in yeast with fluorescent dyes: comparative study of methods. J. Bioenerg. Biomembr. 42, 419–432 [DOI] [PubMed] [Google Scholar]
- 33. Hong M. P., Vu K., Bautos J., Gelli A. (2010) Cch1 restores intracellular Ca2+ in fungal cells during endoplasmic reticulum stress. J. Biol. Chem. 285, 10951–10958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stefan C. P., Zhang N., Sokabe T., Rivetta A., Slayman C. L., Montell C., Cunningham K. W. (2013) Activation of an essential calcium signaling pathway in Saccharomyces cerevisiae by Kch1 and Kch2, putative low-affinity potassium transporters. Eukaryot. Cell 12, 204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Longtine M. S., McKenzie A., 3rd, Demarini D. J., Shah N. G., Wach A., Brachat A., Philippsen P., Pringle J. R. (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 [DOI] [PubMed] [Google Scholar]
- 36. Sherman F., Hicks J. B., Fink G. R. (1986) Methods in Yeast Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 37. Stathopoulos A. M., Cyert M. S. (1997) Calcineurin acts through the CRZ1/TCN1 encoded transcription factor to regulate gene expression in yeast. Genes Dev. 11, 3432–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reuss O., Vik A., Kolter R., Morschhäuser J. (2004) The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341, 119–127 [DOI] [PubMed] [Google Scholar]
- 39. Cunningham K. W., Fink G. R. (1994) Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J. Cell Biol. 124, 351–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mehta S., Li H., Hogan P. G., Cunningham K. W. (2009) Domain architecture of the regulators of calcineurin (RCANs) and identification of a divergent RCAN in yeast. Mol. Cell. Biol. 29, 2777–2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cunningham K. W., Fink G. R. (1996) Calcineurin inhibits VCX1-dependent H+/Ca2+ exchange and induces Ca2+ ATPases in Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 2226–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Travers K. J., Patil C. K., Wodicka L., Lockhart D. J., Weissman J. S., Walter P. (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258 [DOI] [PubMed] [Google Scholar]
- 43. Lehle L., Tanner W. (1976) The specific site of tunicamycin inhibition in the formation of dolichol-bound N-acetylglucosamine derivatives. FEBS Lett. 72, 167–170 [DOI] [PubMed] [Google Scholar]
- 44. Rudolph H. K., Antebi A., Fink G. R., Buckley C. M., Dorman T. E., LeVitre J., Davidow L. S., Mao J. I., Moir D. T. (1989) The yeast secretory pathway is perturbed by mutations in PMR1, a member of a Ca2+ ATPase family. Cell 58, 133–145 [DOI] [PubMed] [Google Scholar]
- 45. Strayle J., Pozzan T., Rudolph H. K. (1999) Steady-state free Ca2+ in the yeast endoplasmic reticulum reaches only 10 μm and is mainly controlled by the secretory pathway pump Pmr1. EMBO J. 18, 4733–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Antebi A., Fink G. R. (1992) The yeast Ca2+-ATPase homologue, PMR1, is required for normal Golgi function and localizes in a novel Golgi-like distribution. Mol. Biol. Cell 3, 633–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Akins R. A. (2005) An update on antifungal targets and mechanisms of resistance in Candida albicans. Med. Mycol 43, 285–318 [DOI] [PubMed] [Google Scholar]
- 48. Clancy C. J., Huang H., Cheng S., Derendorf H., Nguyen M. H. (2006) Characterizing the effects of caspofungin on Candida albicans, Candida parapsilosis, and Candida glabrata isolates by simultaneous time-kill and postantifungal-effect experiments. Antimicrob. Agents Chemother. 50, 2569–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moser M. J., Geiser J. R., Davis T. N. (1996) Ca2+-calmodulin promotes survival of pheromone-induced growth arrest by activation of calcineurin and Ca2+-calmodulin-dependent protein kinase. Mol. Cell. Biol. 16, 4824–4831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gaber R. F., Styles C. A., Fink G. R. (1988) TRK1 encodes a plasma membrane protein required for high-affinity potassium transport in Saccharomyces cerevisiae. Mol. Cell. Biol. 8, 2848–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Haro R., Rodríguez-Navarro A. (2002) Molecular analysis of the mechanism of potassium uptake through the TRK1 transporter of Saccharomyces cerevisiae. Biochim. Biophys. Acta 1564, 114–122 [DOI] [PubMed] [Google Scholar]
- 52. Ko C. H., Buckley A. M., Gaber R. F. (1990) TRK2 is required for low affinity K+ transport in Saccharomyces cerevisiae. Genetics 125, 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ko C. H., Gaber R. F. (1991) TRK1 and TRK2 encode structurally related K+ transporters in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 4266–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 [DOI] [PubMed] [Google Scholar]
- 55. Ren D. (2011) Sodium leak channels in neuronal excitability and rhythmic behaviors. Neuron 72, 899–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bihler H., Slayman C. L., Bertl A. (1998) NSC1: a novel high-current inward rectifier for cations in the plasma membrane of Saccharomyces cerevisiae. FEBS Lett. 432, 59–64 [DOI] [PubMed] [Google Scholar]
- 57. Viladevall L., Serrano R., Ruiz A., Domenech G., Giraldo J., Barceló A., Ariño J. (2004) Characterization of the calcium-mediated response to alkaline stress in Saccharomyces cerevisiae. J. Biol. Chem. 279, 43614–43624 [DOI] [PubMed] [Google Scholar]
- 58. Liebeskind B. J., Hillis D. M., Zakon H. H. (2012) Phylogeny unites animal sodium leak channels with fungal calcium channels in an ancient, voltage-insensitive clade. Mol. Biol. Evol. 29, 3613–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Iida H., Yagawa Y., Anraku Y. (1990) Essential role for induced Ca2+ influx followed by [Ca2+]i rise in maintaining viability of yeast cells late in the mating pheromone response pathway: a study of [Ca2+]i in single Saccharomyces cerevisiae cells with imaging of fura-2. J. Biol. Chem. 265, 13391–13399 [PubMed] [Google Scholar]
- 60. Cyert M. S., Kunisawa R., Kaim D., Thorner J. (1991) Yeast has homologs (CNA1 and CNA2 gene products) of mammalian calcineurin, a calmodulin-regulated phosphoprotein phosphatase. Proc. Natl. Acad. Sci. U.S.A. 88, 7376–7380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cyert M. S., Thorner J. (1992) Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol. Cell. Biol. 12, 3460–3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Muller E. M., Mackin N. A., Erdman S. E., Cunningham K. W. (2003) Fig1p facilitates Ca2+ influx and cell fusion during mating of Saccharomyces cerevisiae. J. Biol. Chem. 278, 38461–38469 [DOI] [PubMed] [Google Scholar]
- 63. Morrison V. A. (2006) Echinocandin antifungals: review and update. Expert Rev. Anti. Infect. Ther. 4, 325–342 [DOI] [PubMed] [Google Scholar]
- 64. Del Poeta M., Cruz M. C., Cardenas M. E., Perfect J. R., Heitman J. (2000) Synergistic antifungal activities of bafilomycin A1, fluconazole, and the pneumocandin MK-0991/caspofungin acetate (L-743,873) with calcineurin inhibitors FK506 and L-685,818 against Cryptococcus neoformans. Antimicrob. Agents Chemother. 44, 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Heath V. L., Shaw S. L., Roy S., Cyert M. S. (2004) Hph1p and Hph2p, novel components of calcineurin-mediated stress responses in Saccharomyces cerevisiae. Eukaryot. Cell 3, 695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gregory M. A., Phang T. L., Neviani P., Alvarez-Calderon F., Eide C. A., O'Hare T., Zaberezhnyy V., Williams R. T., Druker B. J., Perrotti D., Degregori J. (2010) Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell 18, 74–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gijsen V. M., Madadi P., Dube M. P., Hesselink D. A., Koren G., de Wildt S. N. (2012) Tacrolimus-induced nephrotoxicity and genetic variability: a review. Ann. Transplant. 17, 111–121 [DOI] [PubMed] [Google Scholar]
- 68. Pallet N., Legendre C. (2010) Deciphering calcineurin inhibitor nephrotoxicity: a pharmacological approach. Pharmacogenomics 11, 1491–1501 [DOI] [PubMed] [Google Scholar]
- 69. Gillum A. M., Tsay E. Y., Kirsch D. R. (1984) Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol. Gen. Genet 198, 179–182 [DOI] [PubMed] [Google Scholar]
- 70. Reedy J. L., Filler S. G., Heitman J. (2010) Elucidating the Candida albicans calcineurin signaling cascade controlling stress response and virulence. Fungal Genet. Biol. 47, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]