

Background: Elastin degradation is a key event in various pathologies of the skin, lung, and the vascular system.
Results: Elastin degradation by cathepsin V requires two exosites.
Conclusion: Cathepsin exosites support the recognition of and binding to insoluble elastin.
Significance: Exosites may represent novel target sites for the selective inhibition of the elastase activity of cathepsins.
Keywords: Cysteine Protease, Elastin, Extracellular Matrix Proteins, Protein Degradation, Site-directed Mutagenesis, Elastase, Cathepsin V, Exosite, Substrate Specificity
Abstract
Cathepsin V is a highly effective elastase and has been implicated in physiological and pathological extracellular matrix degradation. However, its mechanism of action remains elusive. Whereas human cathepsin V exhibits a potent elastolytic activity, the structurally homologous cathepsin L, which shares a 78% amino acid sequence, has only a minimal proteolytic activity toward insoluble elastin. This suggests that there are distinct structural domains that play an important role in elastinolysis. In this study, a total of 11 chimeras of cathepsins V and L were generated to identify elastin-binding domains in cathepsin V. Evaluation of these chimeras revealed two exosites contributing to the elastolytic activity of cathepsin V that are distant from the active cleft of the protease and are located in surface loop regions. Replacement of exosite 1 or 2 with analogous residues from cathepsin L led to a 75 and 43% loss in the elastolytic activity, respectively. Replacement of both exosites yielded a non-elastase variant similar to that of cathepsin L. Identification of these exosites may contribute to the design of inhibitors that will only affect the elastolytic activity of cysteine cathepsins without interfering with other physiological protease functions.
Introduction
Elastin is a major structural extracellular matrix protein that provides elasticity and tensile strength to tissues such as skin, blood vessels, and lungs (1). It consists of numerous cross-linked chains of tropoelastin with an α-helical conformation (2). Cross-linking and hydrophobicity of elastin monomers result in the formation of an insoluble elastin meshwork that allows connective tissues and organs to stretch and recoil back to their original state (3). Because of the lack of de novo synthesis of elastin after reaching adulthood, its degradation during aging is irreversible and can lead to various symptoms and diseases (4). These include wrinkled skin, the weakening and rupture of blood vessels, and the destruction of alveoli in lung emphysema (5–7).
However, elastin is highly resistant to proteolytic degradation, and only very few selected serine, metallo-, and cysteine proteases are capable of hydrolyzing insoluble elastin (8–12). The commonality of these proteases is that they prefer the binding of small hydrophobic amino acids such as alanine and valine in their substrate-binding sites. When compared with known mammalian elastases, such as serine elastases, matrix metalloproteinases, and several lysosomal cysteine proteases (cathepsin S and K), cathepsin V revealed an exceptionally high elastolytic activity (9, 13, 14). Because of the insoluble nature of elastin, elastolytic activity of an enzyme is frequently measured as the release of soluble fragments from the insoluble core protein. These fragments are typically attached or associated with a chromophore, fluorophore, or radioactive label for easy detection. Interestingly, human cathepsin L, which shares a 78% amino acid sequence identity and a highly similar three-dimensional structure with cathepsin V (15, 16), exhibits minimal cleavage activity toward insoluble elastin even though its specific peptidolytic efficacy (kcat/Km) is higher than that of cathepsin V (14, 15). The difference in their elastase activities appears unrelated to the nature of the classical subsite-defined binding areas in both proteases. Substrate profiling using a diversified combinatorial peptide library revealed qualitatively similar subsite specificities between cathepsins V and L (17), thus requiring other factors to explain the elastase activity of cathepsin V.
A recent study has suggested a two-step mechanism for elastin degradation by cathepsins as follows: a noncatalytic adsorption onto the elastin surface followed by the formation of a catalytically competent structure prior to desorption (18). This implies cathepsins may utilize certain surface structures in addition to the traditional substrate-binding sites in facilitating substrate binding. Recent NMR and bioinformatics studies followed by mutagenesis experiments have identified two exosites in MMP-12, which are critically contributing to elastin degradation (19, 20). However, no distinct elastin-binding sites have been identified in cathepsins.
In this study, using cathepsin V and cathepsin L as model enzymes, a series of chimeras were generated to identify noncatalytic regions that are responsible for the potent elastolytic activity of cathepsin V. Two exosites were identified, which may contribute to the elastolytic activity of cathepsin V. Both exosites are distant from the active site of the protease and form a triangle-like “docking station” with the subsite-binding area for elastin. The replacement of either exosite 1 or 2 with the analogous residues from cathepsin L led to a 75 or 43% loss in the elastolytic activity. The replacement of both exosites resulted in a poor elastolytically active variant despite retaining its full peptidolytic ability. The identification of exosites may contribute to the design of inhibitors that will primarily affect the elastolytic activity of cathepsin V without interfering with the normal protease functions of the enzyme.
EXPERIMENTAL PROCEDURES
Materials
Benzyloxycarbonyl-Phe-Arg-7-amido-4-methylcoumarin (Z-FR-MCA)3 was purchased from Enzyme System Products (Dublin, CA). The cathepsin inhibitor E-64 (l-3-carboxy-trans-2–3-epoxypropionyl-leucylamido-(4-guanidino)-butane) and elastin-rhodamine conjugate were purchased from BioBasic Inc. (Markham, Ontario, Canada). Bovine neck elastin was purchased from EPC (Owensville, MO). The complete diverse tetrapeptide-7-amino-4-carbamoylmethylcoumarin positional scanning-synthetic combinatorial library was synthesized as described previously (17).
Construction of Chimera Cathepsin V-L Expression Vectors
Previously generated human wild-type cathepsin V and cathepsin L expression pPIC9 vectors (Invitrogen) (17, 21) served as templates for constructing all chimera vectors. The cDNAs were generated by PCR using Pfu-polymerase (Fermentas, Burlington, Ontario, Canada) with the primers outlined in Table 1. Based on the amino acid sequence alignment, the primers were designed to allow the incorporation of different parts of the cathepsin V and cathepsin L sequences. The resulting chimera cDNAs were digested with XhoI and NotI restriction enzymes and ligated into the Pichia pastoris expression vector pPIC9 (Invitrogen) with the extracellular secretion signal α-factor replacing the endogenous signal peptides of the cathepsins. Subsequently, the vectors were linearized with SacI and electroporated into the P. pastoris strain, GS115, using a standard protocol provided by Invitrogen.
TABLE 1.
PCR primers for the generation of chimera cathepsin V-L variants
| Mutants | Primer sequences |
|---|---|
| Mutant 1 (M1) | |
| Forward | 5′-TCA CTG AGC GAG CAG AAT CTG GTG-3′ |
| Reverse | 5′-CAC CAG ATT CTG CTC GCT CAG TGA-3′ |
| Mutant 2 (M2) | |
| Forward | 5′-GGC CTG GAC TCT GAG GAA TCC TAT CC-3′ |
| Reverse | 5′-GGA TAG GAT TCC TCA GAG TCC AGG CC-3′ |
| Mutant 3 (M3) | |
| Forward | 5′-TCT GTT GCT AAT GAC ACC GGC TTT-3′ |
| Reverse | 5′-AAA GCC GGT GTC ATT AGC AAC AGA-3′ |
| Mutant 4 (M4) | |
| Forward | 5′-GAG AAG GCC CTG ATG AAA GCA-3′ |
| Reverse | 5′-TGC TTT CAT CAG GGC CTT CTC-3′ |
| Mutant 5 (M5) | |
| Forward | 5′-GGT GGT CTG GTG GTT GGC TAC GGC-3′ |
| Reverse | 5′-GCC GTA GCC AAC CAC CAG ACC ACC-3′ |
| Mutant 6 (M6) | |
| Forward | 5′-CCT ATC CAT ATG AAG CAA CG G AAG AAA GCT GTA AGT AC-3′ |
| Reverse | 5′-GTA CTT ACA GCT TTC TTC CGT TGC TTC ATA TGG ATA GG-3′ |
| Mutant 7 (M7) | |
| Forward | 5′-TGT AAG TAC AAC CCT AAG TAT TCT GTT GCT-3′ |
| Reverse | 5′-AGC AAC AGA ATA CTT AGG GTT GTA CTT ACA-3′ |
| Mutant 8 (M8) using M4 as the template | |
| Forward | Same primer as M5 |
| Reverse | Same primer as M5 |
| *Mutant 9 (M9) | |
| Forward | 5′-ACT GGC TTC GTG GAC ATC CCT AAG GAG AAG GCC-3′ |
| Reverse | 5′-GGC CTT CTC CTT AGG GAT GTC CAC GAA GCC AGT-3′ |
| Mutant 10 (M10) | |
| Forward | 5′-ACA GTG GTC GCA CCT AAG GAG-3′ |
| Reverse | 5′-CTC CTT AGG TGC GAC CAC TGT-3′ |
| Mutant 11 (M11) using M6 as the template | |
| Forward | Same primer as M9 |
| Reverse | Same primer as M9 |
| Flanking primers | |
| Forward | 5′-GAC TGG TTC CAA TTG ACA AGC-3′ |
| Reverse | 5′-GCA AAT GGC ATT CTG ACA TCC-3′ |
P. pastoris Protein Expression
GS115 yeast clones transformed with cathepsin V-L chimera cDNAs were screened and selected using minimal dextrose (dextrose 2% v/v) and minimal methanol (methanol 0.5% v/v) agar plates. The resulting His+ Mut+ clones were screened for maximal cathepsin activity toward the substrate Z-FR-MCA. The clones were briefly incubated in 5 ml of BMGY media ((1% w/v) yeast extract, 2% (w/v) peptone, 100 mm potassium phosphate, pH 6.0, 1.34% (w/v) yeast nitrogen base, 4× 10–15% (w/v) biotin, 1% (v/v) glycerol) at 30 °C until the culture reached an A600 of 2.0–6.0. The cells were then transferred into 3 ml of BMMY media with 0.5% (v/v) methanol to induce protein expression. During growth, the induction was maintained by adding 0.5% (v/v) methanol every 24 h. Cells were incubated at 30 °C for 4 days to reach the approximate maximum protein production, and aliquots were tested for activity after processing of the proenzymes by pepsin as described previously (21).
For large scale production, the clone expressing the highest amount of chimera protein was first inoculated in 5 ml of minimal dextrose media at 30 °C for 24 h and then followed by further inoculation in 50 ml of BMGY media for 12 h. The cells were then pelleted and resuspended into 500 ml of BMGY and then allowed to grow until an A600 of 6.0. Afterward, the cells were transferred into 1.5 liters of BMMY media with induction of 0.5% (v/v) methanol every 24 h, and aliquots of the supernatant were withdrawn every day to monitor enzyme activity and contamination. The cells were harvested on the 4th day of induction. The supernatant was concentrated 50-fold using an Amicon Ultrafiltration membrane (EMD Millipore, Darmstadt, Germany) to 30 ml at 4 °C.
Protein Activation and Purification
The concentrated yeast culture supernatant containing the chimera proenzyme was adjusted to pH 4.0 using diluted glacial acetic acid (Fisher) and activated with 0.6 mg/ml pepsin (Sigma). The mixture was incubated at 37 °C and monitored for activity using Z-FR-MCA in 100 mm sodium acetate buffer (pH 5.5, containing 2.5 mm EDTA and 2.5 mm dithiothreitol (DTT)). Activation was stopped by increasing the pH to 5.5. The activated enzyme was supplemented with ammonium sulfate to a final concentration of 2 m. After centrifugation at 4,000 × g for 20 min at 4 °C, the cleared supernatant containing the activated enzyme was loaded onto an N-butyl-Sepharose column (GE Healthcare) and washed thoroughly with the loading buffer (100 mm sodium acetate, pH 5.0, 2 m ammonium sulfate, 0.5 mm EDTA, and 0.5 mm DTT). The protein was eluted from the column using a linear gradient with reducing ammonium sulfate concentration. The presence of the chimera protein was monitored using Z-FR-MCA as a substrate. Active fractions were combined and further concentrated 10-fold using an Amicon ultra-concentrator (EMD Millipore) to about 1.5 ml and 3× buffer exchange with 100 mm sodium acetate buffer (pH 5.5, supplemented with 0.5 mm EDTA and 0.5 mm DTT). The final solution was divided into 100-μl aliquots for storage at −80 °C. The homogeneity of the proteins was assessed by SDS-PAGE.
Enzyme Titration and Kinetic Studies
Enzyme concentrations were determined by active site titration using E-64 and steady state kinetics, which were performed with the fluorogenic substrate Z-FR-MCA, as described previously (22). The excitation and emission wavelengths used for the assays were set at 380 and 460 nm, respectively, and the rates were recorded using the spectrofluorometer LB50 (PerkinElmer Life Sciences). Kinetic parameters, Km, and kcat values were determined using nonlinear regression analysis. All enzymes were assayed at room temperature at fixed enzyme concentrations (1–5 nm) and variable substrate concentrations (1–20 μm) in 100 mm sodium acetate buffer (pH 5.5, containing 2.5 mm DTT and 2.5 mm EDTA).
The substrate specificity of the S1 to S4 subsites of variant, M11, and wild-type cathepsin V was determined using four positional scanning fluorogenic 4-mer substrate combinatorial libraries, as described (17). The P1 diverse library consisted of 20 sublibraries in which only the P1 position was systematically held constant with one of the 20 proteinogenic amino acids, omitting cysteine and including norleucine. The P2, P3, and P4 positions were randomized with an equimolar mixture of the 19 amino acids, for a total of 6,859 substrate sequences per well. The extended P4 to P2 substrate specificity of proteases was profiled with the tetrapeptide P1-Arg fixed library in which the P1 position was held constant as Arg, which was determined to be the most favored amino acid at the P1 site by the P1 diverse library. An aliquot of the P1 diverse library was added to 20 wells (6,859 compounds per well), and assays were carried out in a 96-well Microfluor plate reader (Dynex Technologies, Chantilly, VA) for a final concentration of 7.3 nm for each compound. To determine the P2, P3, and P4 specificities of both cathepsins, a P1-Arg fixed tetrapeptide library comprised of P2, P3, and P4 sublibraries was used. This library had Arg at the P1 position and the P2, P3, and P4 positions were spatially addressed with 19 amino acids (excluding cysteine and including norleucine). Approximately 9 × 10−9 mol aliquots of each sublibrary of the P1-Arg fixed library were added to 57 wells (361 compounds per well) of a 96-well plate for a final concentration of 0.25 μm. The hydrolysis of the substrate libraries was measured in the presence of 5 nm of cathepsin V and 5 nm of M12 by monitoring the fluorescence released at wavelengths of 380 nm for excitation and of 460 nm for emission. Assays were performed at 25 °C in a buffer containing 100 mm sodium acetate (pH 5.5, 100 mm NaCl, 10 mm DTT, 1 mm EDTA, and 0.01% Brij-35).
Elastin-Rhodamine Conjugate Degradation Assay and HPLC Degradation Profile
Reaction mixture containing 1 μm enzyme and 10 mg/ml elastin-rhodamine conjugate for a final volume of 1 ml of 100 mm sodium acetate buffer (pH 5.5, containing 2.5 mm EDTA and 2.5 mm DTT) was incubated at 37 °C at 200 rpm on a MaxQ 5000 floor shaker (Geneq Inc, Montreal, Quebec, Canada). Samples were withdrawn at time points 0, 30, 60, 90, and 120 min, and mixed with E-64 to stop the reaction. Samples were then centrifuged at top speed using a bench-top AccuSpin microcentrifuge (Fisher) to sediment all undigested elastin particles. The fluorescence of the supernatants was recorded using a 96-well plate format at excitation and emission wavelengths of 570 and 590 nm, respectively, using a Gemini plate reader (Molecular Devices, Sunnyvale, CA). All measurements were carried out in triplicate. Rates for the hydrolysis of rhodamine elastin slowly decreased over the 2-h reaction time for both wild-type proteases and mutant variants. All proteases retained ∼40–50% residual activity after 2 h and thus revealed similar protein stability. The 2-h values were used for the comparison of the protease variants and normalized to 100% for wild-type cathepsin V. The residual cathepsin activity was recorded at the same time points prior to the addition of E-64 using the substrate Z-FR-MCA as described above.
The HPLC degradation profile of bovine neck elastin used 1 μm cathepsin (final concentration) mixed with 10 mg/ml bovine neck elastin (EPC, Owensville, MO) in 1 ml of 100 mm sodium acetate buffer (pH 5.5, containing 2.5 mm EDTA and 2.5 mm DTT) at 37 °C and 200 rpm for 18 h. The reaction was stopped with E-64; samples were centrifuged, and the supernatants were loaded onto a reversed phase C-18 analytical column (Phenomenex, Torrance, CA) on a System Gold© 126 HPLC (Beckman-Coulter, Brea, CA) and eluted with a linear gradient starting with 0.1% trifluoric acid and ending with 90% acetonitrile supplemented with 0.1% trifluoric acid.
Binding Assay for Cathepsins to Elastin
Aliquots of 1 mg of elastin-Congo Red conjugate (Sigma) was separately incubated with E-64-inhibited 1 μm cathepsin V, cathepsin L, and M11 for 2 h in 50 mm sodium acetate buffer (pH 5.5, containing 2.5 mm DTT and EDTA) at 600 rpm and 37 °C. Assays were stopped by centrifugation at top speed using a bench-top AccuSpin microcentrifuge to remove the insoluble contents. The soluble protein contents were then visualized and quantified on Coomassie Blue-stained SDS-polyacrylamide gel.
Electron Scanning Microscopy of Elastin Digests
Aliquots of 10 mg of bovine neck elastin (EPC, Owensville, MO) in a final volume of 1 ml of 100 mm sodium acetate buffer (pH 5.5, containing 2.5 mm EDTA and 2.5 mm DTT) were separately incubated with 1 μm cathepsin V, cathepsin L, and M11 at 37 °C and 200 rpm on a MaxQ 5000 floor shaker (Geneq Inc, Montréal, Quebec, Canada) overnight. The elastin specimens were washed and pelleted at 1,500 rpm for 30 s at room temperature. The pellets were subsequently resuspended evenly in 20 μl of distilled water. 1 μl of the suspension was air-dried on a nickel-copper EM grid and sputter-coated with gold palladium for examining with a Cambridge 260 Stereoscan SE at 6–8 kV (Imaging Facility, Dentistry, University of British Columbia).
Statistics
Results are expressed as mean ± S.D. The significance of differences of the mean values was calculated using one-way analysis of variance (t test). A p value of less than 0.05 was considered significant.
RESULTS
Mapping of Potential Elastin-binding Regions in Cathepsin V
There is a 22% amino acid sequence variation between wild-type human cathepsin V and cathepsin L (15). The highest incidence of sequence differences is found in the middle region of both proteases (Fig. 1A). To identify potential sites that are important for the elastolytic activity of cathepsin V, a series of chimeras were generated by sequentially replacing parts of cathepsin V with its analogous sequence from cathepsin L. Five chimeras, M1, M2, M3, M4, and M5, were constructed, each of which contained various proportions of cathepsin V and cathepsin L (Fig. 1B). The integrity of the active sites of the chimeras was determined by kinetic analysis using the synthetic substrate Z-FR-MCA (Table 2). Chimeras M1 to M5 all possessed a functional active site, which allowed the effective cleavage of the peptide substrate. The kcat values for both wild-type cathepsins and their chimeras were similar. The Km values, however, decreased, and thus the kcat/Km values increased from M1 to M5. The kcat/Km values were also in line with their parent enzymes depending on the sequence contributions of cathepsins V and L in each chimera. Chimera M2, with approximately one-third of its the N-terminal section replaced with that of cathepsin L, revealed a Km value much closer to the value of cathepsin L. This is not surprising as the main substrate specificity determining subsite S2 is greatly swayed by the amino acid residue at position 69 (phenylanine in cathepsin V and a leucine in cathepsin L) (16, 23). M2, which like cathepsin L has a leucine at this position, would therefore exhibit a preference for bulky aromatic residues in the P2 position for substrates like Z-FR-MCA.
FIGURE 1.

Mapping of exosites in cathepsin V. A, amino acid sequence alignment of the mature wild-type cathepsin V (catV) and cathepsin L (catL). Exosite residues from catV are boxed, and their analogues residues in catL are underlined. The “–” is inserted to allow proper alignment. Glycine 118 residue after the proline 117 in exosite 2 is highlighted in green. B, schematic diagram of the wild-type cathepsins and all chimeras. Cathepsin V is presented in red, and cathepsin L is presented in blue, whereas the chimeras were generated with portions taken from cathepsin V and cathepsin L. Cys25, His (residue 163 in cathepsin V and 162 in cathepsin L), and Asn (residue 188 in cathepsin V and 187 in cathepsin L) are the three signature active site amino acid residues in cathepsins. Exosite 1 spans from Val92 to Asn104, respectively (A, Val92 to Ile97; B, Arg101 to Asn104), and exosite 2 contains amino acid residues Thr113 to Lys119. The “.” in M10 indicates glycine 118 deletion.
TABLE 2.
Enzyme kinetics of wild-type cathepsins and chimeras
The chimera mutants are ordered with decreasing portions of cathepsin V to show the changing trends in the kcat, Km, and kcat/Km values. The values for each enzyme was determined based on the synthetic substrate Z-FR-AMC using GraphPad Prism program through a nonlinear regression algorithm.
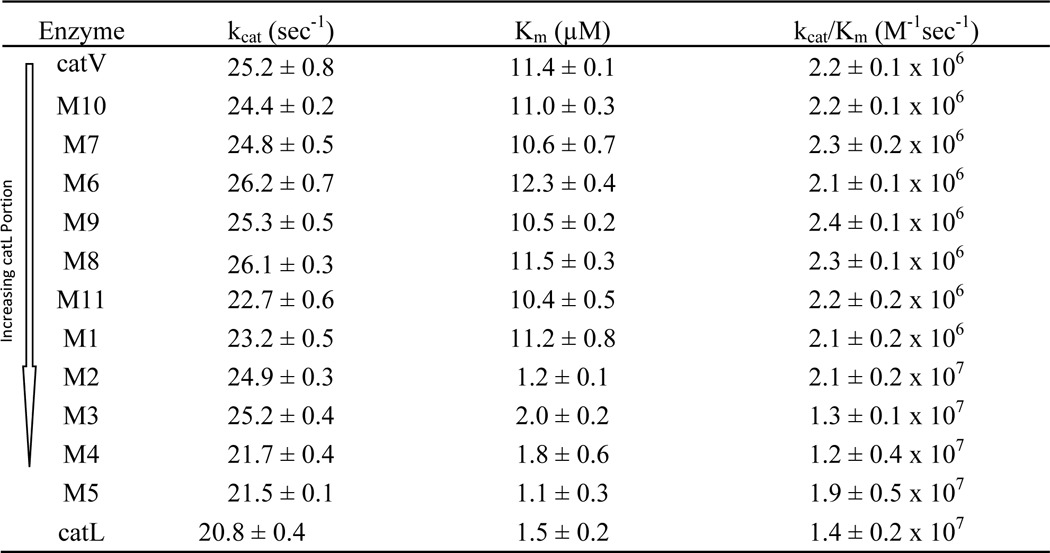
The elastolytic potencies of the chimeras were evaluated using elastin-rhodamine conjugate. Upon cleavage of insoluble elastin into small soluble fragments, rhodamine is released into the aqueous phase and can be monitored at excitation and emission wavelengths of 570 and 590 nm, respectively. As shown in Fig. 2A, chimeras M1 and M2 showed comparable activities to wild-type cathepsin V, whereas M3, M4, and M5 lost 41, 64, and 85% of the elastolytic activities, respectively. The loss in activity of chimeras M3 to M5 was not due to an impairment of the active site as shown by their increasing kcat/Km values. For example, M5 revealed an ∼9-fold increase in the kcat/Km value from cathepsin V for the cleavage of Z-FR-MCA (Table 2).
FIGURE 2.
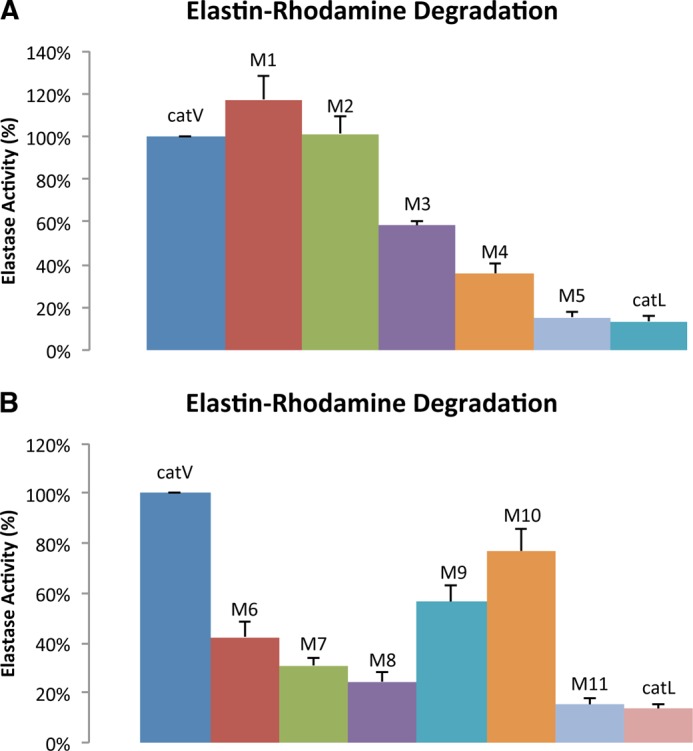
Elastolytic activity of wild-type cathepsins V and L and chimeras using elastin-rhodamine as substrate. A, elastase activity of M1 to M5. B, elastase activities of M6 to M11. Assays were performed at 37 °C with 1 μm of enzyme at 200 rpm in the activity buffer containing 100 mm sodium acetate, 2.5 mm EDTA, and 2.5 mm dithiothreitol at pH 5.5 for 120 min. Fluorescence measurement was determined by the amount of rhodamine released with the excitation and emission wavelengths at 570 and 590 nm, respectively. All measurements were done in triplicate.
Identification of Exosites 1 and 2 within Amino Acid Region Pro90 and Glu120 on Cathepsin V
Chimeras M1 to M5 provided important information for narrowing down the precise locations of elastin-binding exosites in cathepsin V. As M2, which contained the N-terminal 90 amino acid residues of cathepsin L, preserved the same level of elastolytic activity as cathepsin V, it was unlikely that potential exosites resided within the amino acid sequence of Leu1 to Pro90 (Fig. 1B). However, M5, which had nearly 80% of its C-terminal sequence replaced with that of cathepsin L, lost 85% of the elastolytic activity. This excluded residue His163 to Val221 from being crucial in binding elastin. Together, M3 and M4, which diminished 41 and 64% of the activity, conveyed the finding that amino acid residues Pro90 to Glu120 were most likely accountable for the elastolytic activity of cathepsin V (Figs. 1B and 2A). From these 31 amino acid residues, 12 residues were different between cathepsins V and L and formed two clusters separated by a cathepsin V/L-identical octapeptide, 105SVANDTGF112. Four more chimeras were designed to further dissect this area. M6 was generated from substitution of 92VAVDEI97 of cathepsin V with 92EATEES97 of cathepsin L; M7 replaced 101RPEN104 with 101NPKY104; M8 contained both substitutions of M6 and M7; and finally M9 swapped 113TVVAPGK119 with 113VDIPKQ118 from cathepsin L (Fig. 1B). Chimeras M7 to M9 showed kcat, Km, and kcat/Km values comparable with those of the wild-type cathepsins V and L, indicating the presence of fully functional active sites (Table 2). However, similar to chimeras M3 and M4, they demonstrated up to 75% loss of elastolytic activity. M6, M7, and M8 lost 57, 69, and 75%, respectively, of the elastolytic activity compared with cathepsin V (Fig. 2B). It was noted that the reduction of activities was not simply additive as M8 exhibited an activity that did not correspond to the total reduction sum of M6 and M7. This suggested a synergistic relationship between motifs 92VAVDEI97 and 101RPEN104. Based on this observation and the fact that the three amino acid residues, 98CKY100, were also present in cathepsin L, motifs 92VAVDEI97 and 101RPEN104 were combined together and termed exosite 1. In addition, M9 showed a 43% reduction in digesting elastin implying that motif 113TVVAPGK119 was important and was thus denoted as exosite 2 (Fig. 1).
To obtain more information about the cleavage specificities of the chimeras, an HPLC profile for each chimera was obtained by analyzing the solubilized peptide fragments from digesting unlabeled bovine neck elastin. Comparison of the HPLC profiles suggested that M1 to M5 generated a highly similar cleavage pattern as cathepsin V with only a few noticeable variations (Fig. 3, A and B). As chimeras gained more cathepsin L-like characteristics, significant reductions in HPLC peak sizes of proteolytic fragments were observed. M1 showed nearly the same intensities as cathepsin V at peaks 1 and 2 but displayed apparent losses at the rest of the peaks (Fig. 3A). Continuing this pattern, the HPLC profiles of M2, M3, M4, and M5 became increasingly similar to that of cathepsin L. The base line of M5 nearly overlapped the elution profile of cathepsin L with some minor spikes that were unique (Fig. 3B). Chimeras M8 and M9 also demonstrated reduced degradation efficacies, which were in line with the results of their elastin-rhodamine assays. M8 produced less degradation products compared with M9, but both contained the characteristic elution peaks of cathepsin V with significantly less intensities (Fig. 3, C and D). To further evaluate if exosites 1 and 2 were fully responsible for elastin binding, M11 was generated by integrating cathepsin L sequences into exosites 1 and 2 of cathepsin V (Figs. 1 and 2). As shown in Fig. 2B, M11 exerted 15% residual activity and thus was nearly identical to the minimal elastase activity exhibited by cathepsin L. In addition, the HPLC elastin degradation profile of M11 was very similar to that of cathepsin L (Fig. 3D). The only discernable difference was the presence of peak 2 that is dominant in the elastolytic HPLC profile of cathepsin V, although it is significantly reduced. Fig. 3E displays the quantification of all HPLC profiles.
FIGURE 3.

HPLC degradation profile of bovine neck elastin with wild-type cathepsins and chimera cathepsins. A, overlay of wild-type enzymes with those of M1, M2, and M3 chimera HPLC profiles. B, overlay of wild-type cathepsin, M4, and M5 profiles. C, overlay of wild-type cathepsins, M8, and M9 profiles. D, overlay of wild-type cathepsins and M10 and M11 profiles. E, quantification of area below the elution profiles for the individual enzyme variants and wild-type cathepsins V and L. In general, 1 μm of each enzyme was mixed with 10 mg/ml bovine neck elastin in assay buffer at 37 °C at 200 rpm for 18 h. The reaction was stopped with E-64, and the supernatant was load onto an analytical reserved phase C-18 column on HPLC to generate the above elution profiles. A linear gradient starting with 0.1% trifluoroacetic acid and ending with 90% acetonitrile supplemented with 0.1% trifluoroacetic acid was used to elude the digestion products. Major peaks are labeled with numbers for easier comparison.
Two methods were used to verify that the substitutions of both exosites do not affect the integrity of the active site and the classical subsite-binding area: (i) Michaelis-Menten kinetics revealed that M11 had a very similar kcat/Km value to that of cathepsin V for the cleavage of Z-FR-MCA (Table 2), and (ii) substrate specificity profiling analysis using a complete combinatorial library containing 160,000 7-amino-4-carbamoylmethylcoumarin-tagged tetrapeptides revealed no major differences between M11 and cathepsin V in subsites S1, S2, and S4. Observable differences were noted in S3 with a drastically increased acceptability of threonine by M11 when compared with cathepsin V (Fig. 4). Furthermore, substrates with bulky aromatic residues such as tryptophan, tyrosine, and phenylalanine in the P3 position were more efficiently hydrolyzed, and the overall selectivity of this subsite to individual amino acid residues dropped. Altogether, the kinetic and subsite specificity studies indicated that exosites 1, 2, and 4 do not significantly affect the overall subsite specificity of the variant protein with the exception of a certain specificity loss in the S3-binding site.
FIGURE 4.

S4 to S1 subsite specificity determination of wild-type cathepsin V and M11 using a complete diverse substrate library. All assays were carried out in triplicate. P4, P3, P2, and P1 denote the four substrate amino acid residues binding to subsites S4 to S1. The x axis indicates a total of 20 amino acid residues. The y axis represents the picomolar fluorophore produced per s.
Involvement of Glycine 118 in Cathepsin V
Sequence alignments of the known cathepsin elastases (cathepsins K, S, and V) indicated that proline 117 from exosite 2 (based on the numbering of cathepsin V) was followed by a glycine residue that is missing in cathepsin L (Fig. 1A). The comparison of the crystal structures of cathepsins V and L further revealed that lysine 119 adjoining glycine 118 seems to position its side chain outward, away from the active cleft, whereas its orientation is opposite in cathepsin L (Fig. 5) (16, 24). This structural difference within exosite 2 led to the speculation that glycine 118 in cathepsin V might be involved in orienting the adjacent lysine residue to create an elastin interacting point, as well as opening up the entrance for elastin into the active site area. To test this hypothesis, M10 was created by deleting glycine 118 from cathepsin V (Fig. 1B). The results depicted that M10 retained nearly all active site characteristics of cathepsin V, including the corresponding kcat, Km, and kcat/Km values for the Z-FR-MCA substrate. However, the degradation of elastin-rhodamine showed a 23% reduction in cleavage efficacy (Fig. 2B). The effect of the glycine 118 residue was also supported by the HPLC analysis demonstrating a reduction in elastin degradation product intensities to that of wild-type cathepsin V (Fig. 3D). These results support that glycine 118 plays a role in exosite 2.
FIGURE 5.

Superimposition of cathepsin V and cathepsin L. Cathepsin V is colored in cyan (lighter blue) with its exosite 1, shown as sticks and colored in orange, and exosite 2, colored in purple. Cathepsin L is colored in dark blue with its exosite analogous shown as sticks. Active site residues Cys25 and His163 are colored in yellow. Lys119 of cathepsin V is highlighted in red and Gly118 in beige, whereas Lys118 of cathepsin L is highlighted in green. Crystal images are obtained from the Protein Data Bank. Cathepsin V, 1FH0, and cathepsin L, 2XU3.
Binding of Cathepsin V, Cathepsin L, and M11 to Insoluble Elastin
The ability to exit the soluble phase and adhere to the insoluble substrates might be necessary for an effective elastase to carry out proteolysis. This implies that both exosites of cathepsin V serve as initial anchor sites to elastin. To verify this hypothesis, an elastin adsorption assay was developed. Equimolar cathepsin V, cathepsin L, and M11 were incubated with insoluble elastin Congo Red. The proteases were E-64-inhibited to prevent elastinolysis. Results showed that cathepsin L and M11 did not bind to elastin as indicated by the amount of soluble protease before and after the addition of insoluble elastin on SDS-PAGE (Fig. 6A). In contrast, cathepsin V revealed a 52% drop in soluble enzyme concentration, indicating that more than half of the cathepsin V molecules were bound to elastin. When the reaction buffer was supplemented with 300 mm NaCl, all cathepsin V was recovered in the soluble phase (Fig. 6B) indicating that NaCl completely abolishes the absorption of cathepsin V to elastin.
FIGURE 6.

Protein absorption assay onto insoluble elastin-Congo Red. 1 μm E-64-inhibited enzymes (wild-type cathepsin V (catV), cathepsin L (catL), and M11) were inoculated with 10 mg of insoluble elastin-Congo Red in 1 ml of activity buffer for 2 h at 600 rpm at 37 °C. Samples prior to contacting and after contacting elastin-rhodamine were compared and quantified on Coomassie Blue-stained SDS-polyacrylamide gel (A and B).
As elastin is highly hydrophobic, we determined the GRAVY scores of the exosites. GRAVY scores measure the average degree of hydrophobicity in peptide sequences (25). For example, replacement of exosite 1 with its analogous residues from cathepsin L reduced hydrophobicity by 187% from a GRAVY score of −0.62 to −1.78, and in the case of exosite 2, the GRAVY score changed from 0.51 to −0.63, which is a 224% decrease (Table 3). This clearly indicates that the exosites in cathepsin V are highly hydrophobic when compared with the appropriate regions in cathepsin L and thus are likely involved in hydrophobic interactions with insoluble elastin. The GRAVY scores for the analogous mouse cathepsin L sequences lie between those of human cathepsins V and L, which reflect the intermediate elastase activity of the mouse orthologue (14).
TABLE 3.
Comparison of hydrophobicity between exosites of cathepsin V and their analogues in cathepsin L and mouse cathepsin L
Exosite 1 of cathepsin V is divided into exosite 1a and exosite 1b. GS stands for GRAVY score, which is used to measure the hydrophobicity of peptide sequences. GRAVY scores were calculated using the on-line calculator provided by Dr. Stephan Fuchs (University of Greifswald, Department of Microbiology, Greifswald, Germany).
| Exosite 1a | GS | Exosite 1b | GS | Exosite 1 | GS | Exosite 2 | GS | |
|---|---|---|---|---|---|---|---|---|
| Human cathepsin V | VAVDEI | 1.28 | RPEN | −3.28 | VAVDEICKYRPEN | −0.62 | TVVAPGK | 0.51 |
| Human cathepsin L | EATEES | −1.70 | NPKY | −2.57 | EATEESCKYNPKY | −1.78 | VDIPKQ | −0.63 |
| Mouse cathepsin L | EAKDGS | −1.72 | RAEF | −0.85 | EAKDGSCKYRAEF | −1.26 | VDIPQQ | −0.57 |
Electron Microscopy Imaging of Cathepsin-digested Elastin
Morphological changes were observed on bovine neck elastin digested with cathepsin V. Particles lost their surface structure and appeared to fuse into each other with indefinable surface morphology (Fig. 7b). This was noticeably different from the control in the absence of any protease where elastin particles exhibited cylinder-like shapes with sharp surface contours and larger particle size (Fig. 7a). The images of elastin particles incubated with cathepsin L and M11 (Fig. 7, c and d) were very similar to the undigested sample and thus coincided with their lack of significant elastolytic activities.
FIGURE 7.

Scanning electron microscopy images of bovine neck elastin powder incubated in acetate buffer, pH 5. 5 (a), with cathepsin V (b), with M11 variant (c), and with cathepsin L (d). All cathepsin concentrations were 1 μm. The incubation time was done overnight at room temperature.
DISCUSSION
Sharing 78% amino acid sequence identity, cathepsins V and L are very similar cysteine proteases. Moreover, their three-dimensional crystal structures are highly superimposable (Fig. 5) (16, 24). The active site cleft, even with a few differences in some residues in the S2 and S3 subsites (16), demonstrates very similar substrate preferences in subsites S1 to S4 as shown previously (17). Despite these similarities, their elastin-degrading efficacies are dramatically different. Cathepsin V is described as a highly potent elastase, whereas cathepsin L demonstrates only minimal elastolytic activity. This implies the existence of exosite interactions with elastin in cathepsin V. To pinpoint the locations of these exosites, a total of 11 chimeras were produced by swapping analogous sequences of cathepsin L into cathepsin V (Fig. 1B). Evaluation of their elastinolysis revealed two exosites in cathepsin V. Exosite 1 spans through the region of 92VAVDEICKYRPEN104, and exosite 2 contains amino acid residues 113TVVAPGK119. Neither region contains any amino acid residues that are part of the catalytic site nor of the classical substrate binding pockets, S4 to S2′ (16). As shown in Fig. 8, exosite 1 resides on the L-domain and contains 54% hydrophobic and 46% neutral amino acid residues. The analogous loop in cathepsin L has 54% hydrophobic, 31% neutral, and 15% hydrophilic amino acid residues, respectively. This is also supported by GRAVY scores, which provide a more direct understanding on the shifts of hydrophobicity in this case (25). As shown in Table 3, the hydrophobicity of both exosites increases when going from the appropriate sequences present in cathepsin L to cathepsin V (187% increase for exosite 1 and 224% increase for exosite 2). This indicates that the hydrophobicity of the exosites is a critical matter for the elastolytic activity of cathepsin V. Elastin mainly consists of neutral and hydrophobic amino acid residues (26), and thus a higher hydrophobicity in the exosites of cathepsin V can serve as a better binding interface for elastin via van der Waals interactions (27). This rationale is supported by the dramatic reduction in the elastinolytic activity of M11, implying a weakening of the enzyme-substrate interaction by cathepsin L analogues (Figs. 2B and 3E). Interestingly, mouse cathepsin L, which represents an intermediate between human cathepsins L and V based on its amino acid sequence and some of its biological functions (15, 28, 29), also has GRAVY scores between those determined for human cathepsins L and V (Table 3). This may also support the intermediate elastin activity of mouse cathepsin L when compared with the human cathepsin L and V orthologues (14).
FIGURE 8.
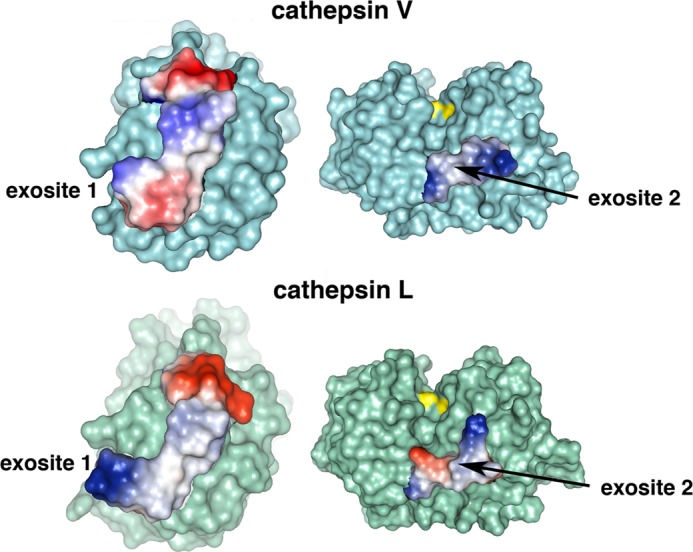
Locations and electrostatics of exosites of cathepsin V. Upper panel, locations of exosites on cathepsin V. Exosite 1, colored in orange, contains residues 92VAVDEICKYRPEN104 (left). Exosite 2, colored in purple, includes residues 113TVVAPGK119 (right). Active site residue Cys25, colored in yellow, is bound to the LHVS inhibitor. Crystal images are modified with PyMOL from the original Protein Data Bank code 1FH0. Electrostatics of exosites of cathepsin V. Lower panel, electrostatics of analogous sequences in cathepsin L (Protein Data Bank code 2XU3). Positive electrostatics are shown in red; negative is shown in blue; neutral is shown in white.
Both exosites clearly contribute to the elastase activity of cathepsin V but not in an additive manner. Exosite 1 is responsible for 75% and exosite 2 for 42% as shown with the M8 and M9 chimeras. Both exosites together (M11 chimera) account for 85% of the elastolytic activity that is in line with the elastolytic activity of cathepsin L of about 14% (when compared with cathepsin V). This may imply a cooperative mode of enzyme-substrate interaction. As the M8 chimera, deprived of exosite 1, causes a more severe reduction in the elastolytic activity of cathepsin V, it is possible that this exosite provides the initial contact site to elastin, which then leads to the interaction with exosite 2. Exosite 2 is located adjacent to the classical subsite binding region (S1 to S3) of cathepsin V. The binding of elastin to both exosites may thus maneuver susceptible peptide bonds into the active site. A recent study by Novinec et al. (18) supports this scenario. The authors proposed that cysteine cathepsins might degrade elastin through cycles of dynamic adsorption and desorption. Such a process involves the reorientation of the enzyme from a nonproductive manner to a catalytically active form upon adsorbing to elastin. Interestingly and in contrast to cathepsins K and S, cathepsin L was found to be poorly absorbed onto insoluble elastin (18). This finding is in agreement with our data obtained from the elastin Congo Red adsorption assay (Fig. 6). Overall, both exosites and the active cleft form a triangular arrangement on the enzyme surface of cathepsin V and act like a docking station for elastin. More importantly, such a nonlinear arrangement may even force the cross-linked tropoelastin to bend and unfold itself for cleavage. Here, it would be interesting to explore in a future study whether the introduction of cathepsin V exosites into human cathepsin L results in a potent elastase. If yes, then the simple docking of elastin to the exosites would be sufficient to direct a putative scissile substrate bond into the active site. If not, the specific orientation of the active site to the two docking sites would likely be critical as well. Nevertheless, the principle of triangularization between exosite elastin-binding sites and the active site seem to represent a general mechanism as this was recently also shown for the elastolytic activity of MMP-12 (19).
In addition, the identification of cathepsin V exosites provides an explanation to the underlying mechanism where they function as docking points to allow the enzyme in an aqueous phase to come in contact with elastin in the insoluble phase. On the contrary, cathepsin L, lacking both exosites, may not generate an absorption strong enough to allow the subsequent hydrolysis to occur. It is important to point out that once elastin loses its insoluble nature due to breakdown of the cross-linkages between tropoelastin monomers, cathepsin L was reported to demonstrate a potent activity against soluble elastin such as ETNA-elastin (18). Although this observation has earned cathepsin L the designation of an elastase in some studies, our studies show that cathepsin L is incapable of digesting insoluble elastin in vitro.
In addition to the hydrophobicity of the exosites, the location and configuration of exosite 2 appear to be crucial. Exosite 2, which resembles a bridge-like structure connecting the L- and R-domains of cathepsin V, sits beneath the S2 subsite pocket of the active cleft (Fig. 6). Common with the other elastolytic cathepsins K and S, and in contrast with cathepsin L, exosite 2 has a glycine residue in position 118. The additional glycine makes the exosite 2 bridge somewhat more flexible, which may be crucial to escort the elastin to the catalytic site for hydrolysis. Moreover, the glycine in cathepsin V directs the side chain of adjacent lysine 119 outwardly away from the active site cleft (Fig. 5) (16). In contrast, the side chain of lysine 117 in cathepsin L is oriented toward the cleft between its L- and R-domain, which may impair a large substrate from entering the active cleft (Fig. 5) (24). The M10 variant with the single point deletion at glycine 118 supports this model. This mutant showed a 23% decrease in its elastolytic activity (Fig. 2B).
The substrate specificity analysis of M11 revealed similar S1, S2, and S4 subsite preferences to its parent wild-type cathepsin V but with some specificity shifts in the S3 subsite. Preferences for individual amino acids in this binding site were lost suggesting a shallower binding site. It should be noted that none of the exosite 1- and 2-forming residues constitute a part of the S3 subsite. The structural nature why these exosite substitutions led to the observed S3P3 specificity shifts remains elusive. The S3 subsite-forming residues are between 20 and 30 amino acid residues in the N-terminal direction from exosite 1-forming residues. Significant alteration in the hydrophobicity of the exosite 1 residues may have conformational consequences in the S3 subsite. However, this can only be clarified if a structure of the M11 becomes available.
The identification of elastin-binding exosites in cathepsin V may be exploited for target-specific inhibitor design. Besides functioning as a potent elastase, cathepsin V is involved in many physiological activities such as the generation of antigen-presentable αβ-CLIP complexes in the immune system and in angiogenesis by releasing endostatin from collagen XVIII (14, 29, 30). Selective cathepsin V exosite inhibitors may therefore specially block the matrix degradation by this protease without interfering with its other proteolytic activities. Ongoing studies will verify whether cathepsin V-like exosites are also required for the elastolytic activities of cathepsins K and S.
This work supported by Canadian Institutes of Health Research Grant MOP89974 (to D. B.) and National Institutes of Health Grant R01 CA128765 (to C. S. C.).
- Z-FR-MCA
- benzyloxycarbonyl-Phe-Arg-7-amido-4-methylcoumarin
- E-64
- l-3-carboxy-trans-2–3-epoxypropionyl-leucylamido-(4-guanidino)-butane.
REFERENCES
- 1. Debelle L., Tamburro A. M. (1999) Elastin: molecular description and function. Int. J. Biochem. Cell Biol. 31, 261–272 [DOI] [PubMed] [Google Scholar]
- 2. Tamburro A. M., Pepe A., Bochicchio B. (2006) Localizing α-helices in human tropoelastin: assembly of the elastin “puzzle”. Biochemistry 45, 9518–9530 [DOI] [PubMed] [Google Scholar]
- 3. Shapiro S. D., Endicott S. K., Province M. A., Pierce J. A., Campbell E. J. (1991) Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of d-aspartate and nuclear weapons-related radiocarbon. J. Clin. Invest. 87, 1828–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robert L., Robert A. M., Fülöp T. (2008) Rapid increase in human life expectancy: will it soon be limited by the aging of elastin? Biogerontology 9, 119–133 [DOI] [PubMed] [Google Scholar]
- 5. Harman D. (2001) Aging: overview. Ann. N. Y. Acad. Sci. 928, 1–21 [DOI] [PubMed] [Google Scholar]
- 6. Makrantonaki E., Zouboulis C. C. (2007) William J. Cunliffe Scientific Awards. Characteristics and pathomechanisms of endogenously aged skin. Dermatology 214, 352–360 [DOI] [PubMed] [Google Scholar]
- 7. Robert L., Robert A. M., Jacotot B. (1998) Elastin-elastase-atherosclerosis revisited. Atherosclerosis 140, 281–295 [DOI] [PubMed] [Google Scholar]
- 8. Liu J., Sukhova G. K., Sun J. S., Xu W. H., Libby P., Shi G. P. (2004) Lysosomal cysteine proteases in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 1359–1366 [DOI] [PubMed] [Google Scholar]
- 9. Shapiro S. D. (1998) Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr. Opin. Cell Biol. 10, 602–608 [DOI] [PubMed] [Google Scholar]
- 10. Turk D., Guncar G. (2003) Lysosomal cysteine proteases (cathepsins): promising drug targets. Acta Crystallogr. D Biol. Crystallogr. 59, 203–213 [DOI] [PubMed] [Google Scholar]
- 11. Wiedow O., Schröder J. M., Gregory H., Young J. A., Christophers E. (1990) Elafin: an elastase-specific inhibitor of human skin. Purification, characterization, and complete amino acid sequence. J. Biol. Chem. 265, 14791–14795 [PubMed] [Google Scholar]
- 12. Hornebeck W., Emonard H. (2011) The cell-elastin-elastase(s) interacting triade directs elastolysis. Front. Biosci. 16, 707–722 [DOI] [PubMed] [Google Scholar]
- 13. Matsumoto S., Kobayashi T., Katoh M., Saito S., Ikeda Y., Kobori M., Masuho Y., Watanabe T. (1998) Expression and localization of matrix metalloproteinase-12 in the aorta of cholesterol-fed rabbits: relationship to lesion development. Am. J. Pathol. 153, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yasuda Y., Li Z., Greenbaum D., Bogyo M., Weber E., Brömme D. (2004) Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem. 279, 36761–36770 [DOI] [PubMed] [Google Scholar]
- 15. Brömme D., Li Z., Barnes M., Mehler E. (1999) Human cathepsin V functional expression, tissue distribution, electrostatic surface potential, enzymatic characterization, and chromosomal localization. Biochemistry 38, 2377–2385 [DOI] [PubMed] [Google Scholar]
- 16. Somoza J. R., Zhan H., Bowman K. K., Yu L., Mortara K. D., Palmer J. T., Clark J. M., McGrath M. E. (2000) Crystal structure of human cathepsin V. Biochemistry 39, 12543–12551 [DOI] [PubMed] [Google Scholar]
- 17. Choe Y., Leonetti F., Greenbaum D. C., Lecaille F., Bogyo M., Brömme D., Ellman J. A., Craik C. S. (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 281, 12824–12832 [DOI] [PubMed] [Google Scholar]
- 18. Novinec M., Grass R. N., Stark W. J., Turk V., Baici A., Lenarcic B. (2007) Interaction between human cathepsins K, L, and S and elastins: mechanism of elastinolysis and inhibition by macromolecular inhibitors. J. Biol. Chem. 282, 7893–7902 [DOI] [PubMed] [Google Scholar]
- 19. Fulcher Y. G., Van Doren S. R. (2011) Remote exosites of the catalytic domain of matrix metalloproteinase-12 enhance elastin degradation. Biochemistry 50, 9488–9499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Palmier M. O., Fulcher Y. G., Bhaskaran R., Duong V. Q., Fields G. B., Van Doren S. R. (2010) NMR and bioinformatics discovery of exosites that tune metalloelastase specificity for solubilized elastin and collagen triple helices. J. Biol. Chem. 285, 30918–30930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brömme D., Okamoto K. (1995) Human cathepsin O2, a novel cysteine protease highly expressed in osteoclastomas and ovary molecular cloning, sequencing and tissue distribution. Biol. Chem. Hoppe-Seyler 376, 379–384 [DOI] [PubMed] [Google Scholar]
- 22. Lecaille F., Kaleta J., Brömme D. (2002) Human and parasitic papain-like cysteine proteases: their role in physiology and pathology and recent developments in inhibitor design. Chem. Rev. 102, 4459–4488 [DOI] [PubMed] [Google Scholar]
- 23. Puzer L., Cotrin S. S., Alves M. F., Egborge T., Araújo M. S., Juliano M. A., Juliano L., Brömme D., Carmona A. K. (2004) Comparative substrate specificity analysis of recombinant human cathepsin V and cathepsin L. Arch. Biochem. Biophys. 430, 274–283 [DOI] [PubMed] [Google Scholar]
- 24. Hardegger L. A., Kuhn B., Spinnler B., Anselm L., Ecabert R., Stihle M., Gsell B., Thoma R., Diez J., Benz J., Plancher J. M., Hartmann G., Banner D. W., Haap W., Diederich F. (2011) Systematic investigation of halogen bonding in protein-ligand interactions. Angewandte Chemie 50, 314–318 [DOI] [PubMed] [Google Scholar]
- 25. Kyte J., Doolittle R. F. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 26. He D., Chung M., Chan E., Alleyne T., Ha K. C., Miao M., Stahl R. J., Keeley F. W., Parkinson J. (2007) Comparative genomics of elastin: Sequence analysis of a highly repetitive protein. Matrix Biol. 26, 524–540 [DOI] [PubMed] [Google Scholar]
- 27. Le Brun A. P., Chow J., Bax D. V., Nelson A., Weiss A. S., James M. (2012) Molecular orientation of tropoelastin is determined by surface hydrophobicity. Biomacromolecules 13, 379–386 [DOI] [PubMed] [Google Scholar]
- 28. Nakagawa T., Roth W., Wong P., Nelson A., Farr A., Deussing J., Villadangos J. A., Ploegh H., Peters C., Rudensky A. Y. (1998) Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science 280, 450–453 [DOI] [PubMed] [Google Scholar]
- 29. Tolosa E., Li W., Yasuda Y., Wienhold W., Denzin L. K., Lautwein A., Driessen C., Schnorrer P., Weber E., Stevanovic S., Kurek R., Melms A., Bromme D. (2003) Cathepsin V is involved in the degradation of invariant chain in human thymus and is overexpressed in myasthenia gravis. J. Clin. Invest. 112, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma D. H., Yao J. Y., Kuo M. T., See L. C., Lin K. Y., Chen S. C., Chen J. K., Chao A. S., Wang S. F., Lin K. K. (2007) Generation of endostatin by matrix metalloproteinase and cathepsin from human limbocorneal epithelial cells cultivated on amniotic membrane. Invest. Ophthalmol. Vis. Sci. 48, 644–651 [DOI] [PubMed] [Google Scholar]