

Background: Prolactin regulates the activity of nociceptors in pain conditions.
Results: Prolactin regulation of sensory neurons is acute and mediated via PI3K and PKCϵ following activation of prolactin receptor short isoform. Prolactin receptor short isoform actions are inhibited by the long isoform.
Conclusion: Prolactin receptor short isoform mediates transient sensitization of nociceptors.
Significance: The proposed mechanism could underlie prolactin involvement in hyperalgesia/pain.
Keywords: Neuroendocrinology, Pain, PI3-kinase (PI3K), Prolactin, Protein Kinase C (PKC)
Abstract
Prolactin (PRL) regulates activity of nociceptors and causes hyperalgesia in pain conditions. PRL enhances nociceptive responses by rapidly modulating channels in nociceptors. The molecular mechanisms underlying PRL-induced transient signaling in neurons are not well understood. Here we use a variety of cell biology and pharmacological approaches to show that PRL transiently enhanced capsaicin-evoked responses involve protein kinase C ϵ (PKCϵ) or phosphatidylinositol 3-kinase (PI3K) pathways in female rat trigeminal (TG) neurons. We next reconstituted PRL-induced signaling in a heterologous expression system and TG neurons from PRL receptor (PRLR)-null mutant mice by expressing rat PRLR-long isoform (PRLR-L), PRLR-short isoform (PRLR-S), or a mix of both. Results show that PRLR-S, but not PRLR-L, is capable of mediating PRL-induced transient enhancement of capsaicin responses in both male and female TG neurons. However, co-expression of PRLR-L with PRLR-S (1:1 ratio) leads to the inhibition of the transient PRL actions. Co-expression of PRLR-L deletion mutants with PRLR-S indicated that the cytoplasmic site adjacent to the trans-membrane domain of PRLR-L was responsible for inhibitory effects of PRLR-L. Furthermore, in situ hybridization and immunohistochemistry data indicate that in normal conditions, PRLR-L is expressed mainly in glia with little expression in rat sensory neurons (3–5%) and human nerves. The predominant PRLR form in TG neurons/nerves from rats and humans is PRLR-S. Altogether, PRL-induced transient signaling in sensory neurons is governed by PI3K or PKCϵ, mediated via the PRLR-S isoform, and transient effects mediated by PRLR-S are inhibited by presence of PRLR-L in these cells.
Introduction
Prolactin (PRL)3 acts as an endocrine hormone, a growth factor, a neurotransmitter, and an immune modulator (1). PRL is produced by the anterior pituitary gland and in other tissues (extrapituitary PRL) in a variety of pain conditions (2–6). The release of PRL from both the pituitary and extrapituitary tissues is sex-dependent and correlates with estrogen levels in serum (2, 4, 7). The systemic and local release of PRL can modulate the activity of a wide variety of cell types, including peripheral sensory neurons in pain conditions where this modulation contributes to hyperalgesic responses (2–4, 8, 9). Even though the molecular mechanisms responsible for PRL-directed modulation of nociception are unknown, one possible mechanism is the PRL regulation of sensory neuronal channels, including transient receptor potential (TRP) channels that play critical roles in hyperalgesia/pain (2, 7, 10, 11). Because PRL can differentially activate nociceptors in a sex-dependent manner (2), this modulation may involve short lasting (transient) effects that could vary in males compared with females. Accordingly, one of the main goals of this study is to define mechanisms underlying transient effects of PRL in female and male sensory neurons because this transient modulation by PRL may represent an important mechanism that contributes to analgesia and pain.
The actions of PRL are mediated by the PRL receptor (PRLR) belonging to the class 1 cytokine receptor family (12). Although the PRLR is encoded by a single gene, alternative splicing of the PRLR gene generates a variety of isoforms that differ by length and amino acid sequence at their cytoplasmic tail. In contrast, the extracellular PRL-binding domain is identical for all PRLR isoforms (13, 14). In rats and humans, there are three PRLR isoforms: short and long, as well as a seldom expressed intermediate variant (15). The neuronal expression of PRLR-L and PRLR-S can vary depending on neuron location, sex of animals, and other systemic conditions. For example, PRLR-S mRNA was either undetectable or present at low levels in several hypothalamic regions of diestrous rats, but was significantly up-regulated in lactating rats (16). Even though a role for PRLR in peripheral sensory neurons appears likely, because PRL can modulate sensory neuron activities, information on the relative expression and functions of PRLR isoforms in sensory neurons is mostly lacking (7). Hence, another goal of the present study was to define the roles of PRLR isoforms in rapid sensitizing effect of PRL in female and male sensory neurons.
Variances in the cytoplasmic part of PRLR isoforms implicate different transducer pathways for each of the PRLR isoforms. All isoforms contain the box-1 domain, which is required for Janus kinase 2 (JAK2) binding, as well as the membrane-proximal region responsible for activation of Fyn and mitogen-activated protein (MAP) kinases (15). Importantly, only the PRLR-L triggers activation of the signal transducer and activator of transcription 3 and 5 (STAT3 and STAT5, respectively) pathways, because it contains the required box-2 domain (1). PRL is also capable of producing rapid action in neurons (7, 17). However, it is not clear which intracellular pathways govern the rapid actions of PRL in sensory neurons. To address this question, we examined the role of different kinases in mediating the PRL-evoked sensitization of female sensory neurons.
EXPERIMENTAL PROCEDURES
Animals
The use of animals in all experiments was approved by IACUC protocols. Sprague-Dawley male and female rats were 45–60 days old (Charles River). Adult female and male PRLR-null mutant (PRLR KO) and corresponding littermate wild-type (WT) mice were obtained from The Jackson Laboratory. PRLR KO mice are viable and normal in size and do not display any gross physical or behavioral abnormalities. However, male and female homozygous PRLR KO mice are completely sterile. PRLR KO mice were produced by creating an in-frame stop codon in exon 5 (18). The lack of functional PRLR in homozygous mutant animals was confirmed using Northern, Western, and binding assays, and all demonstrated the lack of a functional receptor (18). PRLR KO mice were produced in C57BL/6J line. Because PRLR KO mice have irregular estrous cycles, trigeminal ganglia (TG) were removed from WT and PRLR KO females only at the estrous phase. The reproductive stage of cycling females was determined by vaginal lavage as described previously (19).
Human Samples
This study was approved by the Human Subjects Institutional Review Board at the University of Texas Health Science Center at San Antonio. The inclusion criteria consisted of female patients seeking dental therapy for extraction of a normal healthy third molar tooth (wisdom tooth) with fully formed apices and lacking a past history of pain and pathology. The pulpal tissue from 10 teeth (one tooth each from 10 different female patients) was evaluated in the anatomical studies.
Chinese Hamster Ovary (CHO) Cells and Sensory Neuron Culture
We used the following expression constructs: enhanced green fluorescent protein (pEGFP-N1 from Clontech); and rat TRPV1 (accession number NM031982), rat STAT5b (kindly provided by Dr. Rotwein, Oregon Health and Science University, Portland, OR); rat short PRLR (NM001034111.1) and rat long PRLR (NM012630.1) in pcDNA3 (Invitrogen). The expression constructs were delivered into CHO cells using PolyFect (Qiagen) or FuGENE (Promega) according to the manufacturers' protocols. CHO cells were subjected to experimental procedures within 24–48 h after transfection. Expression constructs were delivered into TG sensory neurons using the Amaxa nucleofector according to the manufacturer's protocol. In brief, plasmids were mixed with the provided transfection solution and dispersed sensory neurons and then electroporated at the G013 setting on the nucleofector (20). TG sensory neurons were maintained in DMEM supplemented with 2% FBS at low density on poly-d-lysine/laminin-coated coverslips (Clontech) as described previously (21). Recordings were performed within 16–24 h after plating.
In Situ Hybridization and Immunohistochemistry
The removed rat TG and human dental pulp were fixed with 4% paraformaldehyde, cryoprotected with 30% sucrose in phosphate buffer, and embedded in Neg 50 (Richard Allan Scientific, Kalamazoo, MI), and 30-μm longitudinal and transverse cryosections were generated. In situ hybridization was performed on cryosections of rat TG that had been permeabilized with 0.5% Triton X-100, acylated in acetic anhydride, dehydrated in alcohol, and delipidated in chloroform. Sections were hybridized at 55 °C with DIG-cRNA (containing digoxigenin-UTP) probe specific against rat PRLR-L (position 1037–2005; GenBank NM012630.1). After RNase treatment, slides/coverslips were washed with decreasing concentration of SSC buffer (final wash, 0.1× SSC at 55 °C), and hybridization was detected using standard alkaline phosphate-based reaction (substrates 5-bromo-4-chloro-3-indolyl phosphate nitro blue tetrazolium; Roche Applied Science). Negative controls for in situ hybridization were hybridization with sense PRLR-L probe (position 1037–2005). Immunohistochemistry (IHC) was carried out as described previously (22). Control IHC was performed on tissue sections processed as described but either lacking primary antibodies or lacking primary and secondary antibodies. The following previously characterized primary antibodies were used: (i) PRLR mouse monoclonal 1A2B1 clone (Invitrogen) used at 1:100; (ii) PRLR mouse monoclonal U5 clone (Affinity BioReagents, Golden, CO) used at 1:33; (iii) human-specific PRLR-L (LF), PRLR-S (SF)1a, and PRLR-S (SF)1b rabbit polyclonal used at 1:300, 1:100, and 1:100, respectively (23); (iv) TRPV1 guinea pig polyclonal from Neuromics (Bloomington, MN; GP14100) used at 1:2000; (v) neurofilament heavy (NFH) chicken polyclonal from Abcam (AB4680) used at 1:1000; and (vi) myelin basic protein (MBP) rat monoclonal (12 clone) from Millipore (MAB386) used at 1:500. Sections were incubated in species appropriate Alexa Fluor secondary antibodies (1:200; Molecular Probes). Nuclei were stained with either TO-PRO-3 iodide (TO-PRO, Invitrogen; 1:5000 dilution) added to the secondary antibodies or with DAPI in Vectashield (Vector Laboratories) used as mounting medium. Experiments evaluating PKCϵ trafficking in cultured TG neurons were conducted by double IHC using the above mentioned TRPV1 antibody and an already characterized mouse monoclonal antibody against PKCϵ (Molecular Probes) used at 1:1000 (24). Images were acquired using a Nikon Eclipse 90i microscope (Melville, NY) equipped with a C1si laser scanning confocal imaging system.
Western Blotting and pSTAT5 ELISA
Western blotting and ELISA were performed as described previously (3, 21). Transfected CHO cells were homogenized by 20 strokes in a Potter-Elvehjem homogenizer in solution provided by ELISA kit (InstantOne ELISA for pSTAT5; eBioscience, San Diego, CA) and supplemented with protease inhibitors aprotinin (1 μg/ml; Sigma-Aldrich), leupeptin (1 μg/ml; Sigma-Aldrich), pepstatin (1 μg/ml; Sigma-Aldrich), and phenylmethylsulfonyl fluoride (PMSF, 100 nm; Sigma-Aldrich). Cell extract was incubated on ice for 15 min and then centrifuged at 500 × g for 1 min at 4 °C. Supernatants were used for protein amount measurement, ELISA, and Western blotting. Protein quantification of crude plasma membrane homogenates was completed using the Bradford method (25) as recommended by the manufacturer (Thermo Scientific). ELISA for pSTAT5 was performed according to the manufacturer's protocol. Equal amounts of protein extracts (≈4 μg) were resolved via 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinyl difluoride membrane (Millipore). Western blots were blocked in 5% nonfat milk in Tris-buffered saline/Tween 20 (Fisher Scientific), labeled using monoclonal anti-rat PRLR antibodies (1:1000; U5 clone; Affinity BioReagents) followed by appropriate horseradish peroxidase-conjugated secondary antisera (GE Healthcare) and enhanced chemiluminescence detection following the manufacturer's instructions (GE Healthcare).
Ca2+ Imaging
The Ca2+ imaging experiments and ratiometric data conversion were basically performed as described previously (21). Fluorescence was detected by a Nikon TE 2000U microscope fitted with a 20×/0.9 NA Fluor objective. Data were collected and analyzed with MetaFluor Software (MetaMorph; Universal Imaging Corporation). The experiments were performed in standard external solution (see under “Electrophysiology”). The calcium-sensitive dye was Fura-2/AM (2 μm; Molecular Probes). The net changes in Ca2+ influx were calculated by subtracting the basal [Ca2+]i (mean value collected for 60 s prior to agonist addition) from the peak [Ca2+]i value achieved after exposure to the agonists. Increases in [Ca2+]i above 50 nm were considered positive. This minimal threshold criterion was established by application of 0.1% dimethyl sulfoxide as a vehicle. Ratiometric data were converted to [Ca2+]i (in nm) as described previously (26).
Electrophysiology
Recordings were made in whole cell voltage clamp (Vh = −60 mV) mode at 22–24 °C from the somata of small-to-medium TG rat and mouse neurons (20–45 picofarads) as described (21). Data were acquired and analyzed using an Axopatch 200B amplifier and pCLAMP9.0 software (Molecular Devices). Recording data were filtered at 0.5 kHz and sampled at 2 kHz. Borosilicate pipettes (Sutter, Novato, CA) were polished to resistances of 4–7 megohms in whole cell pipette solution. Access resistance (Rs) was compensated (40–80%) when appropriate up to the value of 10–15 megohms. Currents were considered positive when their amplitudes were 5-fold bigger than displayed noise (in root mean square). Standard external solution contained 140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm d-glucose, and 10 mm HEPES, pH 7.4. The standard pipette solution for the whole cell configurations contained 140 mm KCl, 1 mm MgCl2, 1 mm CaCl2, 10 mm EGTA, 10 mm d-glucose, 10 mm HEPES, 2 mm GTP, 5 mm ATP, pH 7.3. Ca2+-free conditions were constituted with standard external solution without Ca2+, and standard pipette solution without Ca2+, whereas 10 mm EGTA was replaced with 10 mm BAPTA. Drugs were applied using a fast, pressure-driven and computer-controlled 8-channel system (ValveLink8; AutoMate Scientific, San Francisco, CA).
Immunoreactive Calcitonin Gene-related Peptide (iCGRP) Release Assay
All release assays were performed on 5–7-day TG neuronal cultures, at 37 °C, using modified Hanks' (Invitrogen) buffer (10.9 mm HEPES, 4.2 mm sodium bicarbonate,10 mm dextrose, and 0.1% bovine serum albumin were added to 1× Hanks') as described previously (27). After two initial washes, a 15-min base-line sample was collected. The cells then were pretreated with either vehicle or kinase antagonists and then exposed to vehicle, PRL (1 μg/ml), or mixes of PRL and kinase antagonists for 15 min. Finally, capsaicin (30 nm)-evoked iCGRP release was performed for 10 min, and the supernatants were collected for analysis of iCGRP content by radioimmunoassay. Radioimmunoassay was performed as described in detail previously (28). The basal release was typically 6–8 fmol/well.
Data Analysis
Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad, San Diego, CA) as specified in legends to figures. The data in figures are given as means ± S.E. with the value of n referring to the number of analyzed cells or trials for each group. Experiments were performed at least in duplicate. Significant differences between groups were assessed by one-way or two-way analysis of variance (ANOVA) with Bonferroni's multiple comparison post hoc test (compares all pairs of columns). Two conditions were compared using unpaired t test. A difference was accepted as significant when p < 0.05.
RESULTS
Transient Signaling Pathways Involved in PRL-induced Sensitization of TRPV1 Channel in Female Rat TG Neurons
PRL is able to exert transient effects in a variety of cell types (13) including neurons where the acute application (3–10 min) of PRL (0.1 μg/ml) regulates TRPV1, TRPA1, and TRPM8 channel activities in female, but not male rat sensory neurons (2, 7). Signaling pathways underlying this transient action of PRL on sensory neurons are unknown. PRL-induced enhancement of TRP responses is most likely mediated by kinases because we have detected direct phosphorylation of the TRPV1 channel by acute (5-min) application of PRL (1 μg/ml) to TG neurons from OVX-E female rats (7). Activation of several kinases, including protein kinase C (PKC (29)), protein kinase A (PKA (30, 31)), and phosphatidylinositol-3 kinase (PI3K (32)), can lead to phosphorylation of TRPV1. Accordingly, we employed pharmacological and cell biology approaches in combination with Ca2+ imaging, iCGRP release assays, and whole cell voltage clamp recording to investigate the contributions of several acute signaling pathways in regulating capsaicin (specific TRPV1 agonist) responses by PRL in female rat TG sensory neurons.
TG neurons were pretreated for 5 min with vehicle or either selective intracellular pathway inhibitors followed by co-treatment for 5 min with PRL or vehicle as indicated on Fig. 1B. The utilized kinase inhibitors had no effect on their own for altering either basal Ca2+ levels or CAP (50 nm)-evoked intracellular Ca2+ ([Ca2+]i) rise in TG neurons (Fig. 1, A, C, and D). PRL (1 μg/ml)-induced sensitization of CAP-evoked [Ca2+]i accumulation was substantially reversed by pan-PKC inhibitor bisindolylmaleimide I (BIS; 0.5 μm; Fig. 1, A and B). Similarly, PRL sensitization effects were also inhibited by the blockade of the PI3K pathway with LY294002 (20 μm; Fig. 1C (32). However, pretreatment of TG neurons with PKA inhibitor KT-5720 (0.2 μm) had no effect on PRL augmentation of CAP-evoked [Ca2+]i rise, even though it did inhibit prostaglandin E2 (1 μm)-induced enhancement of CAP responses (Fig. 1D).
FIGURE 1.

A, intracellular calcium ([Ca2+]i) accumulation by capsaicin (Cap; 50 nm) in TG neurons from female rats pretreated for 5 min with vehicle (Veh), PRL (40 nm (1 μg/ml); BIS (0. 5 μm), or PRL+BIS. CAP was applied for 1 min. Statistical analysis was performed using a one-way ANOVA with Bonferroni's post hoc test (*, p < 0.05; **, p < 0.01; n.s., nonsignificant; n = 40–60). Error bars, S.E. B, representative Ca2+ imaging traces from TG neurons responsive to CAP after pretreatment for 5 min with vehicle, PRL, or PRL+BIS. Durations of CAP and PRL applications and concentrations are indicated. C, CAP-evoked [Ca2+]i rise in TG neurons pretreated for 5 min with vehicle, PRL, LY294002 (LY; 20 μm), or PRL+LY. CAP was applied for 1 min. One-way ANOVA with Bonferroni's post hoc test was used (*, p < 0.05; **, p < 0.01; n.s., nonsignificant; n = 40–60). D, CAP-evoked [Ca2+]i rise in TG neurons pretreated for 5 min with vehicle, PRL, KT-5720 (KT; 0.2 μm), or PRL+KT. The positive control was regulation of CAP-evoked [Ca2+]i rise by prostaglandin E2 (PGE2; 1 μm), and blockade of this effect by KT. CAP was applied for 1 min. One-way ANOVA with Bonferroni's post hoc test was used (**,p < 0.01; n.s., nonsignificant; n = 30–60).
PRL may contribute to the initiation of local inflammation (4). This effect could be either via activation of immune cells or due to PRL-driven neurogenic inflammation (7). Therefore, to test whether PKC and PI3K mediate PRL-induced sensitization of CAP-evoked exocytosis, we evaluated actions of kinase inhibitors on PRL-sensitized CAP-evoked iCGRP release as a measure for neurogenic inflammation. Fig. 2, A and B, demonstrates that pretreatment (15 min) and co-treatment (15 min) of cultured TG neurons with PKC inhibitor BIS (1 μm) and PI3K inhibitor LY294002 (20 μm) completely abolished the previously observed PRL (1 μg/ml)-induced augmentation of CAP (30 nm)-evoked iCGRP release from cultured TG neurons.
FIGURE 2.
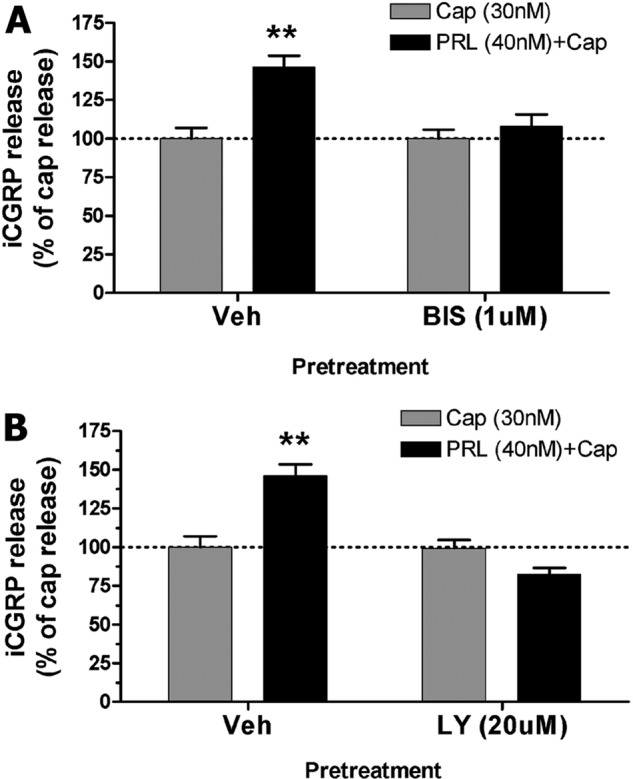
iCGRP release experiments were performed on 5-day cultures of TG neurons isolated from female rats. The cultures were supplemented with 100 ng/ml NGF and estradiol (50 nm). A, iCGRP release was evoked by CAP (30 nm) in TG neurons pretreated for 15 min with vehicle (Veh), PRL (1 μg/ml), BIS (1 μm), or PRL + BIS. CAP was applied for 10 min. Statistical analysis was performed using two-way ANOVA with Bonferroni's post hoc test (**, p < 0.01; n = 4/group). Error bars, S.E. B, CAP-evoked iCGRP release in TG neurons pretreated for 15 min with vehicle, PRL, LY294002 (LY; 20 μm), or PRL + LY. Two-way ANOVA with Bonferroni's post hoc test was used (**, p < 0.01; n = 4/group).
We next examined PRL enhancement of CAP responses with alternative kinase inhibitors using whole cell patch voltage clamp recording. Pretreatment with kinase inhibitors was performed as for Ca2+ imaging. PRL (0.1 μg/ml) sensitization of CAP (50 nm)-gated current (ICAP) was significantly reversed by pretreatment with the potent and selective PKC inhibitor, GF 109203X (0.1 μm; Fig. 3, A and B). Selective PI3Kγ inhibitor, AS 605240 (0.1 μm), which displays 30-fold selectivity over PI3Kδ and PI3Kβ and 7.5-fold selectivity over PI3Kα (33), also blocked sensitization of ICAP by PRL (Fig. 3C). Unlike PKC and PI3Kγ blockers, herbimycin A (0.5 μm) antibiotic, which antagonizes a Src family of kinases including BCR-ABL tyrosine kinases (34), did not produce an inhibitory effect on PRL-evoked potentiation of ICAP (Fig. 3D).
FIGURE 3.

A, enhancement of magnitude of ICAP (50 nm) by PRL (0.1 μg/ml) is blocked by GF 109203X (GF; 0.1 μm) in TG sensory neurons from female rats. Statistical analysis was performed using a one-way ANOVA with Bonferroni's post hoc test (*, p < 0.05; **, p < 0.01; n = 7–12; error bars, S.E.). B, representative ICAP traces from TG neurons pretreated with vehicle (Veh), PRL, or PRL+GF. Duration of CAP application (40 s) is indicated. C, ICAP in TG neurons pretreated with vehicle, PRL, AS 605240 (AS; 0.1 μm) or PRL+AS. CAP was applied for 40 s. One-way ANOVA with Bonferroni's post hoc test was used (*, p < 0.05; **, p < 0.01; n = 6–11). D, ICAP in TG neurons pretreated with vehicle, PRL, herbimycin A (HerbA; 0.5 μm) or PRL+HerbA. CAP was applied for 40 s. One-way ANOVA with Bonferroni's post hoc test was used (*,p < 0.05; NS, nonsignificant; n = 7–10). All pretreatment times for vehicle and PRL are 5 min.
The PKC inhibitors used in this study are known to affect several PKC isoforms with different affinities (35). Because the exact isoform or combination of isoforms that mediate PRL sensitization of TG neurons is largely unknown, we carried out experiments to identify the isoforms involved. There are two classes of PKC isoforms: [Ca2+]i-dependent and -independent (36). We found that PRL (0.1 μg/ml) is able to sensitize CAP responses in TG neurons in presence and absence of extracellular and intracellular Ca2+ in recording solutions (Fig. 4A). This result implies that a transient PRL effect in sensory neurons is probably not mediated by PKCα and PKCβ isoforms (37). In the next experiments, PKCδ, PKCϵ, and PKCζ isoforms were blocked with selective peptide-based inhibitors (38–40). PKCδ and PKCϵ were inhibited with cell-permeable N-myristoylated peptides δPKC (8–17) and ϵ-V1-2 PKC (both made by AnaSpec), respectively. Inhibitor of PKCζ is a pseudosubstrate peptide, which is attached to cell permeabilization Antennapedia domain vector peptide (Tocris). PRL (1 μg/ml)-evoked sensitization of CAP responses was effectively blocked by the PKCϵ, but not PKCζ inhibitors (Fig. 4B), whereas PKCδ had a partial effect (Fig. 4B). These results are in accordance with a previous publication that demonstrated an important role for PKCϵ in the regulation of a heat-gated channel (now known as TRPV1) in sensory neurons (24).
FIGURE 4.
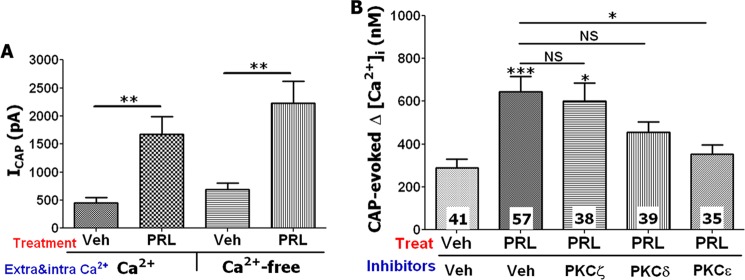
Inhibition of transient PRL effects with PKC isoform-specific inhibitors. A, pretreatment (5 min) with PRL (0.1 μg/ml) increased ICAP (50 nm) in TG sensory neurons from female rats at both physiological concentrations of extracellular (2 mm) and intracellular (≈150 nm) Ca2+ and Ca2+-free (extracellular and intracellular) conditions. Unpaired t test was used (**, p < 0.01; n = 7–9; error bars, S.E.). Veh, vehicle. B, effect of PKCϵ (ϵ-V1-2 PKC, 1 μm), PKCδ (δPKC (8–17), 1 μm), and PKCζ (ζPKC(113–129) of PKCζ, 1 μm) inhibitors on sensitization of CAP (50 nm)-evoked Ca2+ influx by PRL (1 μg/ml) in TG sensory neurons from female rats is shown. Cells were pretreated for 10 min with inhibitors and then 5 min with inhibitors + PRL. CAP was applied for 1 min. Numbers of responding cells are indicated within bars. One-way ANOVA with Bonferroni's post hoc test was used (*, p < 0.05; ***, p < 0.001; NS, nonsignificant); error bars, S.E.
PKCϵ is an important isoform of the PKC family that is expressed in sensory neurons and plays a key role in sensitization of TRPV1 by inflammatory mediators (24, 41). Moreover, PKCϵ is translocated to the plasma membrane of TG neurons upon activation (24, 42), providing a convenient measure of kinase activities. Thus, we next evaluated the possible PRL activation of PKCϵ as detected by the translocation of this kinase to the plasma membrane. We freshly isolated female rat TG neurons and cultured them up to 6 h. The cultures were supplemented with 100 ng/ml NGF and estradiol (50 nm). TG neurons were treated for 5 min at 37 °C with vehicle, PRL (1 μg/ml), or PMA (0.5 μm). Following treatments, TG neurons were fixed with 4% formalin and processed for IHC with antibodies against PKCϵ and TRPV1. PKCϵ translocation was measured only in TRPV1-positive neurons. Fig. 5 illustrates that PRL (1 μg/ml; panel B) pretreatment, unlike vehicle (panel A), triggers translocation of PKCϵ to the plasma membrane of TRPV1-positive TG neurons (Fig. 5, D and E). Moreover, PRL-induced translocation of PKCϵ was mimicked by treatment of TG neurons with PMA (0.5 μm), a direct activator of several isoforms of PKC (Fig. 5, C–E, and Refs. 24, 42). Altogether, the results obtained with Ca2+ imaging, whole cell recording, iCGRP release, and PKC translocation assays combined with pharmacological tools demonstrated that PRL is able to induce acute effects in female rat TG sensory neurons by activating PKCϵ and PI3K.
FIGURE 5.

A–C, PRL evokes the translocation of PKCϵ to the plasma membrane in cultured TRPV1-positive TG neurons from female rats. Confocal image of TG neurons was taken at 120× magnification. TG neurons were treated for 5 min at 37 °C with vehicle (Veh), PRL (1 μg/ml), or PMA (0.5 μm). Following treatments, TG neurons were fixed with 4% formalin and processed for IHC with antibodies against PKCϵ and TRPV1. Treatment with vehicle produced no translocation of PKCϵ (A). PRL (B) and PMA (C) induced translocation of PKCϵ to the plasma membrane in a subset of TRPV1-positive TG neurons. D and E, quantification of translocation (f/f0 at 470 nm) for PKCϵ is presented for cytoplasmic (D) and plasma membrane regions (E). Number of analyzed cells are indicated within bars. One-way ANOVA with Bonferroni's post hoc test was used (***, p < 0.001). Error bars, S.E.
Roles of Short and Long Forms of Prolactin Receptor in Transient Actions of PRL in TG Neurons
It is well documented that PRL exerts long term effects via the JAK/STAT pathway (1). This pathway is triggered by binding of PRL to PRLR-L, but not PRLR-S isoforms (43). We found that a transient action of PRL in TG sensory neurons is mediated by PKCϵ and PI3K (Figs. 1–5). However, it is unclear which PRLR isoforms mediate this effect. To answer this question, we have co-transfected CHO cells with GFP, TRPV1, and rat PRLR-L or PRLR-S. GFP transfection was used for visual identification of CHO cells co-transfected with either TRPV1 and PRLR-L or TRPV1 and PRLR-S. Cells co-transfected with TRPV1 and PRLR-L did not exhibit any sensitization of TRPV1 after pretreatment (5 min) of cells with mouse PRL (0.1 μg/ml; Fig. 6, A, right two columns, and C). In contrast, ICAP was sensitized by PRL (0.1 μg/ml) pretreatment of CHO cells containing TRPV1 and PRLR-S isoform (Fig. 6, A, left two columns, and B).
FIGURE 6.

A, enhancement of magnitude of ICAP (50 nm) by PRL (0.1 μg/ml) in CHO cells co-expressing GFP (for visualization of transfected cells), rat TRPV1, and rat PRLR-L or PRLR-S isoforms. Treatments by PRL were for 5 min in all experiments. CAP was applied for ≈40 s. Statistical analysis was unpaired t test (NS, nonsignificant; **, p < 0.01; n = 7–10; error bars, S.E.). B, representative ICAP traces from CHO cells transfected with GFP, TRPV1, and PRLR-S and pretreated with vehicle (Veh) or PRL. C, representative ICAP traces from CHO cells transfected with GFP, TRPV1, and PRLR-L, and pretreated with vehicle or PRL. Durations of CAP applications are indicated.
Biochemistries of CHO cells and sensory neurons are principally different (21); hence, we evaluated whether PRLR is involved in acute PRL signaling in TG neurons. PRLR-L and PRLR-S were reconstituted in TG neurons derived from PRLR-null mutant mice (PRLR KO). The reconstitution was performed in both male and female TG sensory neurons because PRL action in sensory neurons is critically sex-dependent (2). Fig. 7A and representative traces (Fig. 7B) illustrate that, as expected, PRL (0.1 μg/ml) does not sensitize ICAP in WT male mice transfected with GFP. However, reconstitution of PRLR-S, but not PRLR-L, in TG neurons from male PRLR KO mice restores acute action of PRL on TRPV1 (Fig. 7A). In TG neurons from WT female mice, PRL was capable of sensitizing CAP responses (Fig. 7C and Ref. 2). This effect was ablated in TG neurons from PRLR KO mice, indicating that PRL effects are indeed arbitrated by PRLR (Fig. 7C). Reconstitution experiments revealed that like that seen in male TG neurons, PRLR-S alone can restore transient PRL actions on ICAP in female PRLR KO TG neurons (Fig. 7C). Importantly, the PRLR-S isoform can heterodimerize with PRLR-L, and this heteromer inhibits PRLR-L function (44–46). This finding implies that tissue-specific relative expression of PRLR-L and PRLR-S isoforms may alter the physiological effect mediated by PRL. Here, we investigated the role of PRLR-L and PRLR-S co-expression on PRL-induced transient signaling in sensory neurons. Using the above described experimental approach, PRLR-S and PRLR-L were co-transfected at a molar ratio of 1:1 into TG neurons from male or female PRLR KO mice, and CAP responses were assessed after pretreatment with vehicle or PRL (0.1 μg/ml). Fig. 7D illustrates that the co-presence of PRLR-L with PRLR-S results in suppression of PRL-triggered transient effects seen in male and female TG neurons expressing PRLR-S alone. Altogether, PRL-induced transient enhancement was observed only in TG neurons from male and female mice expressing PRLR-S alone (Fig. 7A-7C), but not co-expressing PRLR-S and PRLR-L at an approximately equal ratio (Fig. 7D).
FIGURE 7.

A, modulation of ICAP (50 nm) by PRL (0.1 μg/ml) in TG neurons from male WT mice and in TG neurons from male PRLR KO mice transfected with PRLR-S or PRLR-L isoforms. Treatment with PRL was 5 min in all experiments. CAP was applied for ≈40 s. Statistical analysis was performed using an unpaired t test (**, p < 0.01; n = 8–14; error bars, S.E.). Veh, vehicle. B, representative ICAP traces in TG neurons from male PRLR KO mice transfected with PRLR-S or PRLR-L isoforms. Pretreatments with vehicle or PRL are indicated. C, modulation of ICAP (50 nm) by PRL (0.1 μg/ml) in TG neurons from female WT mice and in TG neurons from female PRLR KO mice transfected with PRLR-S or PRLR-L isoforms. CAP was applied for ≈40 s. Statistical analysis was performed using an unpaired t test (**, p < 0.01; ***, p < 0.001; n = 8–12). D, modulation of ICAP (50 nm) by PRL (0.1 μg/ml) in TG neurons from male or female PRLR KO mice (indicated below x axis) co-transfected (1:1 molar ratio) with both PRLR-S and PRLR-L isoforms. CAP was applied for ≈40 s. Statistical analysis was performed using unpaired t test. NS, nonsignificant; n = 7–10).
Because PRLR-L and PRLR-S differ in intracellular domains (1), we examined a possible role for the PRLR-L intracellular domain in inhibition of PRLR-S function. PRLR-L mutant constructs were generated by deletion of intracellular portions (Fig. 8A). PRLR-L (≈100 kDa), PRLR-S (≈40 kDa) as well as PRLR-L deletion mutants (marked Del1, Del2, and Del3; ≈35, 58, and 80 kDa, respectively) that were effectively expressed in CHO cells (Fig. 8B). PRLR-L, PRLR-L-Del3, and PRLR-L-Del2, but not PRLR-L-Del1 co-expressed with rat STAT5b in CHO cells can constitutively activate tyrosine phosphorylation of STAT5b (Fig. 8C and Ref. 1). This STAT5b phosphorylation is substantially enhanced upon treatment (30 min) of CHO cells with PRL (0.1 μg/ml; Fig. 8C). Further, co-expression of PRLR-L and PRLR-S reduced constitutive and PRL-evoked phosphorylation of STAT5b (Fig. 8C). Evaluation of functional interaction between PRLR-L mutants and PRLR-S showed that deletions of PRLR-L C-terminal domains in amino acids 540–592 (Del3; Fig. 8A) and 430–592 (Del2; Fig. 8A) did not significantly affect suppression of PRLR-S by PRLR-L (Fig. 8D). In contrast, additional removal of the 293–430-amino acid C-terminal portion of PRLR-L ablated inhibitory property of PRLR-L on transient effects of PRLR-S (Fig. 8D). Overall, it appears that the 293–430 portion of the PRLR-L C-terminal domain containing Box-2 and sequences required for phosphorylation of STAT5b (Fig. 8, A–C) is important for inhibitory action of PRLR-L on PRLR-S (Fig. 8D).
FIGURE 8.
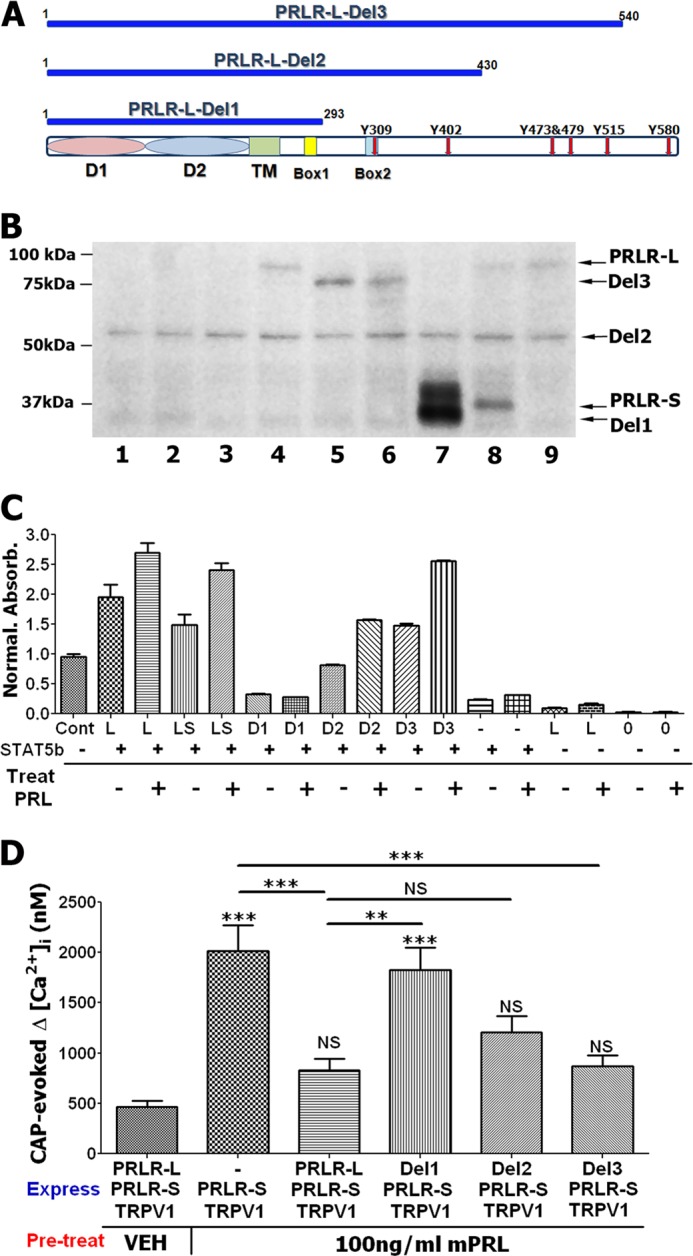
Effect of mutant PRLR-L on PRLR-S function. A, schematic representations of PRLR-L mutants marked as PRLR-L-Del1 (Del1), PRLR-L-Del2 (Del2), and PRLR-L-Del3 (Del3). Extracellular domains (D1 and D2), TM (transmembrane domain), Box1 and Box2, and tyrosine phosphorylation sites (Y red arrows) are indicated. B, expression of rat PRLR-L, PRLR-L mutants, PRLR-S, and STAT5b in CHO cells. PRLR-L, PRLR-L mutant, and PRLR-S bands are noted. Equal amounts of protein extracts (≈4 μg) were loaded. Lanes: 1, no transfection; 2, GFP; 3, STAT5b; 4, PRLR-L+STAT5b; 5, Del3; 6, Del2+Del3 (1:1 ratio); 7, Del1; 8, PRLR-L+ PRLR-S (1:1 ratio); and 9, PRLR-L. Anti-PRLR antibody is U5 (1:1000 dilution). C, ELISA for pSTAT5b in protein extracts isolated from PRLR-STAT5b co-transfected CHO cells. Transfected CHO cells were pretreated (30 min) with vehicle (−) or PRL (0.1 μg/ml) (+). Transfection abbreviations: Cont, a positive control supplied with pSTAT5 ELISA kit; STAT5b co-transfection CHO cells are marked with (+); L, PRLR-L; LS, PRLR-L+ PRLR-S (1:1 ratio); D1, PRLR-L-Del1; D2, PRLR-L-Del2; D3, PRLR-L-Del3; and 0, no transfection. Number of trials is 2–3. D, effects of PRLR-L and PRLR-L mutants (Del1, Del2, and Del3) on PRLR-S-mediated sensitization of CAP (50 nm)-evoked intracellular Ca2+ accumulation by PRL (0.1 μg/ml). Co-transfection ratios for PRLR-S with PRLR-L and PRLR-L mutants are 1:1. PRL pretreatments were for 5 min. CAP application was 1 min. Statistic is one-way ANOVA with Bonferroni's post hoc test (**, p < 0.01; ***, p < 0.001; NS, nonsignificant; n = 35–50; error bars, S.E.).
Expression of PRLR-L and PRLR-S in TG Sensory Neurons/Nerves from Rats and Humans
Differential expression patterns for PRLR isoforms in certain regions of brain and in non-neuronal cells are well documented (16, 23, 47). A possible differential expression in sensory neurons appears important because cell signaling pathways via PRLR-L and PRLR-S isoforms are different (Figs. 6–8 and Ref. 1). Furthermore, a co-expression of PRLR isoforms could affect the function of each (Figs. 7D and 8 and Refs. 44, 46). Even so, the precise expression patterns of PRLR isoforms in various types of neurons are still not clear.
Here, we investigated expression patterns for PRLR-L and PRLR-S in TG neurons from female and male rats and human female nerves. The main technical difficulty for this study is that rat isoform-specific PRLR antibodies are not available, thus mandating the use of in situ hybridization for rat studies. In addition, specific probes for PRLR-S produced a weak in situ hybridization signal hampering interpretation for cell distributions of PRLR-S. Therefore, to evaluate the expression of PRLR isoforms in rat TG neurons, we have employed antibodies recognizing all PRLR forms and in situ hybridization with PRLR-L-specific probes. Subtraction of cell numbers for PRLR-L-positive neurons from overall PRLR expression allows an estimate of the percentage of TG neurons expressing PRLR-S. In situ hybridization with the PRLR-L-specific antisense, but not sense probes showed that PRLR-L is expressed in only 4.6% (n = 31 of 672) female and 3.8% (n = 24 of 624) male rat TG neurons (Fig. 9, A–C). In contrast, IHC with the 1A2B1 antibody, which recognizes both PRLR-L and PRLR-S isoforms, labeled 59.3% female rat TG neurons (n = 308 from 522; Fig. 9D). Approximately 50–60% of PRLR-positive neurons also expressed TRPV1 (Fig. 9D). Expression of PRLR is also prominent in satellite glial cells (Fig. 9E).
FIGURE 9.

Expression of PRLR isoforms in female rat TG. A, in situ hybridization of rat female TG section with PRLR-L-specific antisense digoxigenin cRNA probe. B, in situ hybridization of rat female TG section with control PRLR-L sense digoxigenin cRNA probe. C, percentage of rat female TG neurons expressing both PRLR isoforms and only PRLR-L form. D, IHC showing expression of PRLR (green) and TRPV1 (red) in rat female TG neurons. PRLR antibody is 1A2B1 clone. E, high magnification image of rat female TG neurons expressing TRPV1 (red) in neurons and PRLR (green) in satellite cells. Nuclei are revealed by TO-PRO. PRLR antibody is U5 clone.
Recently, isoform-specific antibodies for human PRLR have been developed and characterized (23). These antibodies do not label rat and mouse PRLR isoforms. Therefore, we have utilized these to evaluate PRLR isoform expression in female human dental pulps containing sensory nerves originating from TG. IHC of longitudinally sectioned female dental pulps show that both PRLR isoforms (long identified as PRLR-LF and short as PRLR-SF1a) are present in nerve fibers (Fig. 10, A–D). The axoplasm of many dental nerve fibers expresses NFH (seen as green in Fig. 10, A and C), and the myelin of myelinated nerve fibers can be stained with MBP. Therefore, we used NFH and MBP antibodies in combination with PRLR isoform-specific antibodies to critically evaluate expression in nerve fiber axoplasm with the use of transverse nerve sections from the female human dental pulp. Fig. 10E (left panel) illustrates that PRLR-LF is predominantly co-expressed with MBP (green), but not in NFH-identified nerve fiber axoplasm (blue). Conversely, PRLR-SF1a is co-expressed with both MBP (green) and NFH (blue) in a subset of female human dental pulp nerves (Fig 10E, right panel). Similar patterns were observed for antibodies against PRLR-SF1b (data not shown). In summary, PRLR-L is expressed mainly in satellite glial cells of female rat TG, with expression in <4% neurons and nerves. In human female dental pulp nerves, PRLR-L isoform is expressed predominantly in myelin sheath wrapping nerves. The predominant PRLR isoform of TG neurons and nerves in female rats and humans is the PRLR-S isoform that is also present in the myelin sheath.
FIGURE 10.

Expression of PRLR isoforms in female human dental pulp. A and B, IHC shows expression of PRLR-L (red) in a human female dental pulp. Nerves are revealed by marker NFH (green), and nuclei are stained by TO-PRO (blue). C and D, IHC shows expression of PRLR-S (red) in a human female dental pulp. NFH (green) is nerve fiber marker, and nuclei are stained by TO-PRO (blue). E, IHC shows expression of PRLR-L (red; left panel) and PRLR-S (red; right panels) in cross-sections of human female dental nerve fibers. Nerve fiber marker is NFH (blue), and myelin sheath is identified with MBP (green).
DISCUSSION
PRL can modulate the function of CNS and PNS neurons, and it plays a critical role in diverse bodily functions such as maternal behavior (48), appetite (49), sexual receptivity (50), and hypersensitivity to painful stimuli (2–4), where the action of PRL on neurons can be either acute/transient or long term. Transient effects of PRL have been characterized in many neuronal types including gonadotropin-releasing hormone neurons, tuberoinfundibular dopamine neurons, the anteroventral-periventricular nucleus, and dorsal root ganglion and TG sensory neurons (2, 7, 17, 51, 52). Even though it is well known that long term effects of PRL are mainly mediated via the JAK/STAT pathways controlling gene expression modulation (15, 43), the molecular mechanisms underlying the transient effect of PRL in neurons are not known. Thus, a major focus of the present study was to identify the mechanisms responsible for the acute actions of PRL in TG sensory neurons.
In this study, we show that PRL evokes transient effects in sensory neurons via activation of PKCϵ or PI3K pathways. PRLR belongs to the class 1 cytokine receptor family, and this finding is consistent with the ability of other members of this family to independently activate both PKC and PI3K pathways (12). Class 1 cytokine receptors could activate PKC and PI3K pathways downstream from each other (53). However, it is not clear whether these kinases are activated independently in TG sensory neurons. Activation of these cellular signaling pathways by PRL in non-neuronal cells has been demonstrated previously and includes PRL activation of PKC in immune cells (54), PKC and PI3K in prostate cells (55), PI3K in decidual cells (56), and PKC and PI3K in breast tumor and lymphoma cell lines (57, 58). Involvement of PI3K in the acute action of PRL on CNS neurons has recently been reported (17). Our previous studies have also shown that PRL is able to rapidly (within 15 min) phosphorylate the TRPV1 channel in TG neurons (2, 7).
Acute effects of hormones, such as PRL, and neurotransmitters include a regulation of the activity of many ligand-gated and voltage-gated channels, and this regulation has critical implications for nervous system function. For example, PRL can transiently modulate Ca2+-dependent K+ channels (BK channels) and TRP-like channels in tuberoinfundibular dopamine neurons (17) where this modulation could enhance the activity in tuberoinfundibular dopamine neurons leading to dopamine release, eventually affecting lactation, sexual libido, fertility, and body weight (17). PRL also acutely sensitizes the TRP channel in dorsal root ganglion and TG neurons (2, 7). This sensitization can result from the elevated local PRL levels seen in tissues after inflammation or surgical procedures, thus contributing to the development of thermal hyperalgesia (2–4). Our findings suggest that the increased PRL levels seen in a variety of painful pathological conditions could rapidly regulate neuronal activities via activation of PKC and PI3K pathways and where this regulation contributes to nociceptor sensitization.
The PRLR gene is alternatively spliced to generate isoforms, which differ in the length and amino acid sequences at their cytoplasmic tails, whereas the extracellular PRL-binding domain is identical for all PRLR isoforms (13, 14). Two main isoforms of PRLR in human and rats are short and long (1, 15, 59). The PRL-induced JAK/STAT pathway is triggered by activation of PRLR-L, but not PRLR-S isoforms, containing both required domain box-1 and box-2 (Fig. 8, A and C, and Refs. 1, 43). The roles of PRLR isoforms in mediating the transient effects of PRL in neurons were not clear, so we addressed this issue, and our results demonstrate that the transient effects of PRL in TG sensory neurons of male and female rats can be exclusively mediated by PRLR-S (Figs. 6 and 7). Interestingly, the action of PRLR-S can be suppressed by co-expression with PRLR-L (Figs. 7D and 8D). PRLR-L and PRLR-S can heterodimerize when heterologously expressed and in breast cancer cells (44, 45). Heterodimerization between PRLR-S and PRLR-L can also lead to a suppression of PRLR-L-induced gene transcription in non-neuronal cells (44, 46). It has previously been shown that two intramolecular disulfide (S-S) bonds within the extracellular subdomain 1 (D1; see Fig. 8A) of PRLR-S contributes to the inhibition of PRLR-L functions (46). Our data imply that intracellular domain(s) of PRLR-L located within the 293–430 amino acid region are involved in inhibition of PRLR-S transient effects (Fig. 8D). This region also contain box-2 (Fig. 8A), which is critical for tyrosine phosphorylation of STAT5 (Fig. 8, B and C, and Ref. 1). It is not clear how the PRLR-L suppresses expression of PRLR-S or whether PRLR-L/PRLR-S forms nonfunctional heterodimers. However, our data show that PRLR-L does not eliminate expression of PRLR-S (lanes 8 and 9 in Fig. 8B; see also Fig. 8C).
The functional interaction between PRLR-L and PRLR-S suggests that the relative expression levels of these isoforms in neurons and other cells could have critically important effects (46). Our data indicate that PRLR-L is expressed predominantly in glial cells of TG in female rats and in women dental pulpal tissues (Figs. 9 and 10). In contrast, PRLR-S is present in TG neurons/nerves as well as in glial cells in female rat TG and female human dental pulps (Figs. 9 and 10). Such a differential expression of PRLR-L and PRLR-S isoforms has been reported in different regions of the brain (16, 47, 60). Further, relative levels of PRLR isoforms could change during pathological conditions, and in this respect, our findings and the findings of others on the functional interaction between PRLR isoforms appear physiologically relevant.
It is well documented that the nature and magnitude of PRL responses are regulated by estrogen (7, 61) and during lactation in rodents (16) and to a lesser extent in humans (53). Thus, in the male, compared with the ovariectomized with estradiol replacement (OVX-E) rat, PRL expression was less in the hypothalamus and lacking in the corpus striatum (61). This estrogen regulation of PRL responses may be an important contributor to sex-dependent pain. For example, transient effects of PRL on the TRPV1 channel are weak or absent in TG neurons from male and female OVX rats, yet present or restored in neurons from estrus female or OVX-E rats, respectively (Fig. 7 and Refs. 2, 7). Nevertheless, overexpression of PRLR-S in male sensory neurons restores transient PRL effects on TRPV1 channel (Fig. 7, A and B). This observation implies that PRLR-S expression is higher in TG neurons of females than males, but this point requires further investigation. It is noteworthy that quantitative methods (such as RT-PCR) that could be employed to evaluate the relative expression of PRLR isoforms are not suitable for this study because both PRLR isoforms are expressed in non-neuronal cells in TG. Thus, the characterization of the PRLR isoform expression with isoform-specific antibodies is crucial to understand that difference in signaling and effect in neuronal versus non-neuronal cells.
PRL contributes to hyperalgesia/pain in sex-dependent fashion (2–4). Because local trauma and inflammation lead to an increase in peripheral levels of PRL in female and to a lesser extent in male rats, it could be suggested that transient modulation of TRP channels by PRL in the periphery is one of underlying mechanisms for PRL-induced thermal (i.e. heat and cold) hyperalgesia (2–4). TRP channels are also involved in hypersensitivity to thermal and mechanical stimuli at presynaptic levels in the dorsal horn of the spinal cord and certain brain stem regions (62, 63). In this respect, PRLR isoforms may also contribute to the transient presynaptic regulation of TRPs (3). Further, expression patterns of PRLR isoforms could be altered during pathological pain conditions, such as systemic inflammation, autoimmune diseases, stress, and trauma. Taking this into account, PRL could produce hypersensitivity to thermal and mechanical stimuli (possibly in a sex-dependent manner) by more efficiently up-regulating the nociceptive transmission between central terminals of nociceptors and dorsal horn spinal cord neurons via different mechanisms and PRLR isoforms. Collectively, this study reports the molecular mechanism underlying transient effects of PRL in sensory neurons of female and male rats. Our findings also suggest that the possible interaction between PRLR-L and PRLR-S and cell signaling differs between transient and long term effects of PRL in neuronal and non-neuronal cells.
Acknowledgments
We thank Dr. Kenneth Hargreaves, Dr. Shivani Ruparel, Dustin Green, Jei Li, Mei Li, and Lola Miranda for help in accomplishing these studies.
This work was supported, in whole or in part, by National Institutes of Health Grant DE017696 (to A. N. A.) and by the Intramural Research Program of the NCI/National Institutes of Health (to E. G.).
- PRL
- prolactin
- ANOVA
- analysis of variance
- BIS
- bisindolylmaleimide I
- CAP
- capsaicin
- CHO
- Chinese hamster ovary
- ICAP
- capsaicin-gated current
- iCGRP
- immunoreactive calcitonin gene-related peptide
- IHC
- immunohistochemistry
- MBP
- myelin basic protein
- NFH
- neurofilament heavy
- OVX
- ovariectomized
- OVX-E
- ovariectomized with estradiol replacement
- PMA
- phorbol 12-myristate 13-acetate
- PRLR-L
- prolactin receptor long form
- PRLR-S
- prolactin receptor short form
- TG
- trigeminal ganglia
- TRP
- transient receptor potential
- TRPV1
- transient receptor potential type V1.
REFERENCES
- 1. Bole-Feysot C., Goffin V., Edery M., Binart N., Kelly P. A. (1998) Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr. Rev. 19, 225–268 [DOI] [PubMed] [Google Scholar]
- 2. Patil M. J., Ruparel S. B., Henry M. A., Akopian A. N. (2013) Prolactin regulates TRPV1, TRPA1, and TRPM8 in sensory neurons in sex-dependent manner: contribution of prolactin receptor to inflammatory pain. Am. J. Physiol. Endocrinol. Metab., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patil M. J., Green D. P., Henry M. A., Akopian A. N. (2013) Sex-dependent roles of prolactin and prolactin receptor in postoperative pain and hyperalgesia in mice. Neuroscience 253, C132–C141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scotland P. E., Patil M., Belugin S., Henry M. A., Goffin V., Hargreaves K. M., Akopian A. N. (2011) Endogenous prolactin generated during peripheral inflammation contributes to thermal hyperalgesia. Eur. J. Neurosci. 34, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yardeni I. Z., Shavit Y., Bessler H., Mayburd E., Grinevich G., Beilin B. (2007) Comparison of postoperative pain management techniques on endocrine response to surgery: a randomised controlled trial. Int. J. Surg. 5, 239–243 [DOI] [PubMed] [Google Scholar]
- 6. Reiner Z., Oresković M., Ribarić K. (1987) Endocrine responses to head and neck surgery in men. Acta Otolaryngol. 103, 665–668 [PubMed] [Google Scholar]
- 7. Diogenes A., Patwardhan A. M., Jeske N. A., Ruparel N. B., Goffin V., Akopian A. N., Hargreaves K. M. (2006) Prolactin modulates TRPV1 in female rat trigeminal sensory neurons. J. Neurosci. 26, 8126–8136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mena F., González-Hernández A., Navarro N., Castilla A., Morales T., Rojas-Piloni G., Martínez-Lorenzana G., Condés-Lara M. (2013) Prolactin fractions from lactating rats elicit effects upon sensory spinal cord cells of male rats. Neuroscience 248, C552–C561 [DOI] [PubMed] [Google Scholar]
- 9. Ochoa-Amaya J. E., Malucelli B. E., Cruz-Casallas P. E., Nasello A. G., Felicio L. F., Carvalho-Freitas M. I. (2011) Dual effects of hyperprolactinemia on carrageenan-induced inflammatory paw edema in rats. Neuroimmunomodulation 18, 245–253 [DOI] [PubMed] [Google Scholar]
- 10. Zheng J. (2013) Molecular mechanism of TRP channels. Compr. Physiol. 3, 221–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brederson J. D., Kym P. R., Szallasi A. (2013) Targeting TRP channels for pain relief. Eur. J. Pharmacol. 716, 61–76 [DOI] [PubMed] [Google Scholar]
- 12. Boutin J. M., Jolicoeur C., Okamura H., Gagnon J., Edery M., Shirota M., Banville D., Dusanter-Fourt I., Djiane J., Kelly P. A. (1988) Cloning and expression of the rat prolactin receptor, a member of the growth hormone/prolactin receptor gene family. Cell 53, 69–77 [DOI] [PubMed] [Google Scholar]
- 13. Kelly P. A., Djiane J., Postel-Vinay M. C., Edery M. (1991) The prolactin/growth hormone receptor family. Endocr. Rev. 12, 235–251 [DOI] [PubMed] [Google Scholar]
- 14. Clevenger C. V., Kline J. B. (2001) Prolactin receptor signal transduction. Lupus 10, 706–718 [DOI] [PubMed] [Google Scholar]
- 15. Goffin V., Kelly P. A. (1996) Prolactin and growth hormone receptors. Clin. Endocrinol. 45, 247–255 [DOI] [PubMed] [Google Scholar]
- 16. Pi X. J., Grattan D. R. (1999) Increased expression of both short and long forms of prolactin receptor mRNA in hypothalamic nuclei of lactating rats. J. Mol. Endocrinol. 23, 13–22 [DOI] [PubMed] [Google Scholar]
- 17. Lyons D. J., Hellysaz A., Broberger C. (2012) Prolactin regulates tuberoinfundibular dopamine neuron discharge pattern: novel feedback control mechanisms in the lactotrophic axis. J. Neurosci. 32, 8074–8083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ormandy C. J., Camus A., Barra J., Damotte D., Lucas B., Buteau H., Edery M., Brousse N., Babinet C., Binart N., Kelly P. A. (1997) Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev. 11, 167–178 [DOI] [PubMed] [Google Scholar]
- 19. Marcondes F. K., Bianchi F. J., Tanno A. P. (2002) Determination of the estrous cycle phases of rats: some helpful considerations. Braz. J. Biol. 62, 609–614 [DOI] [PubMed] [Google Scholar]
- 20. Patil M. J., Belugin S., Akopian A. N. (2011) Chronic alteration in phosphatidylinositol 4,5-bisphosphate levels regulates capsaicin and mustard oil responses. J. Neurosci. Res. 89, 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Akopian A. N., Ruparel N. B., Jeske N. A., Hargreaves K. M. (2007) Transient receptor potential TRPA1 channel desensitization in sensory neurons is agonist dependent and regulated by TRPV1-directed internalization. J. Physiol. 583, 175–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Henry M. A., Freking A. R., Johnson L. R., Levinson S. R. (2006) Increased sodium channel immunofluorescence at myelinated and demyelinated sites following an inflammatory and partial axotomy lesion of the rat infraorbital nerve. Pain 124, 222–233 [DOI] [PubMed] [Google Scholar]
- 23. Ginsburg E., Alexander S., Lieber S., Tarplin S., Jenkins L., Pang L., Heger C. D., Goldsmith P., Vonderhaar B. K. (2010) Characterization of ductal and lobular breast carcinomas using novel prolactin receptor isoform specific antibodies. BMC Cancer 10, 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cesare P., Dekker L. V., Sardini A., Parker P. J., McNaughton P. A. (1999) Specific involvement of PKC-ϵ in sensitization of the neuronal response to painful heat. Neuron 23, 617–624 [DOI] [PubMed] [Google Scholar]
- 25. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 26. Jeske N. A., Patwardhan A. M., Gamper N., Price T. J., Akopian A. N., Hargreaves K. M. (2006) Cannabinoid WIN 55,212-2 regulates TRPV1 phosphorylation in sensory neurons. J. Biol. Chem. 281, 32879–32890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patil M., Patwardhan A., Salas M. M., Hargreaves K. M., Akopian A. N. (2011) Cannabinoid receptor antagonists AM251 and AM630 activate TRPA1 in sensory neurons. Neuropharmacology 61, 778–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patwardhan A. M., Diogenes A., Berg K. A., Fehrenbacher J. C., Clarke W. P., Akopian A. N., Hargreaves K. M. (2006) PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the δ-opioid receptor. Pain 125, 114–124 [DOI] [PubMed] [Google Scholar]
- 29. Vellani V., Mapplebeck S., Moriondo A., Davis J. B., McNaughton P. A. (2001) Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J. Physiol. 534, 813–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhave G., Zhu W., Wang H., Brasier D. J., Oxford G. S., Gereau R. W., 4th (2002) cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35, 721–731 [DOI] [PubMed] [Google Scholar]
- 31. Lopshire J. C., Nicol G. D. (1998) The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. J. Neurosci. 18, 6081–6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhuang Z. Y., Xu H., Clapham D. E., Ji R. R. (2004) Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J. Neurosci. 24, 8300–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Camps M., Rückle T., Ji H., Ardissone V., Rintelen F., Shaw J., Ferrandi C., Chabert C., Gillieron C., Françon B., Martin T., Gretener D., Perrin D., Leroy D., Vitte P. A., Hirsch E., Wymann M. P., Cirillo R., Schwarz M. K., Rommel C. (2005) Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 11, 936–943 [DOI] [PubMed] [Google Scholar]
- 34. Fukazawa H., Uehara Y., Murakami Y., Mizuno S., Hamada M., Takeuchi T. (1994) Labeling of v-Src and BCR-ABL tyrosine kinases with [14C]herbimycin A and its use in the elucidation of the kinase inactivation mechanism. FEBS Lett. 340, 155–158 [DOI] [PubMed] [Google Scholar]
- 35. Martiny-Baron G., Kazanietz M. G., Mischak H., Blumberg P. M., Kochs G., Hug H., Marmé D., Schächtele C. (1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J. Biol. Chem. 268, 9194–9197 [PubMed] [Google Scholar]
- 36. Sobhia M. E., Grewal B. K., Ml S.P., Patel J., Kaur A., Haokip T., Kokkula A. (2013) Protein kinase C inhibitors: a patent review (2010–present). Expert Opin. Ther. Pat. 23, 1297–1315 [DOI] [PubMed] [Google Scholar]
- 37. Wu-Zhang A. X., Newton A. C. (2013) Protein kinase C pharmacology: refining the toolbox. Biochem. J. 452, 195–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brandman R., Disatnik M. H., Churchill E., Mochly-Rosen D. (2007) Peptides derived from the C2 domain of protein kinase C ϵ (ϵPKC) modulate ϵPKC activity and identify potential protein-protein interaction surfaces. J. Biol. Chem. 282, 4113–4123 [DOI] [PubMed] [Google Scholar]
- 39. Bright R., Raval A. P., Dembner J. M., Pérez-Pinzón M. A., Steinberg G. K., Yenari M. A., Mochly-Rosen D. (2004) Protein kinase Cδ mediates cerebral reperfusion injury in vivo. J. Neurosci. 24, 6880–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lim S., Choi J. W., Kim H. S., Kim Y. H., Yea K., Heo K., Kim J. H., Kim S. H., Song M., Kim J. I., Ryu S. H., Suh P. G. (2008) A myristoylated pseudosubstrate peptide of PKC-ζ induces degranulation in HMC-1 cells independently of PKC-ζ activity. Life Sci. 82, 733–740 [DOI] [PubMed] [Google Scholar]
- 41. Dai Y., Moriyama T., Higashi T., Togashi K., Kobayashi K., Yamanaka H., Tominaga M., Noguchi K. (2004) Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J. Neurosci. 24, 4293–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patwardhan A. M., Berg K. A., Akopain A. N., Jeske N. A., Gamper N., Clarke W. P., Hargreaves K. M. (2005) Bradykinin-induced functional competence and trafficking of the δ-opioid receptor in trigeminal nociceptors. J. Neurosci. 25, 8825–8832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goupille O., Daniel N., Bignon C., Jolivet G., Djiane J. (1997) Prolactin signal transduction to milk protein genes: carboxyl-terminal part of the prolactin receptor and its tyrosine phosphorylation are not obligatory for JAK2 and STAT5 activation. Mol. Cell. Endocrinol. 127, 155–169 [DOI] [PubMed] [Google Scholar]
- 44. Gadd S. L., Clevenger C. V. (2006) Ligand-independent dimerization of the human prolactin receptor isoforms: functional implications. Mol. Endocrinol. 20, 2734–2746 [DOI] [PubMed] [Google Scholar]
- 45. Qazi A. M., Tsai-Morris C. H., Dufau M. L. (2006) Ligand-independent homo- and heterodimerization of human prolactin receptor variants: inhibitory action of the short forms by heterodimerization. Mol. Endocrinol. 20, 1912–1923 [DOI] [PubMed] [Google Scholar]
- 46. Xie Y. L., Hassan S. A., Qazi A. M., Tsai-Morris C. H., Dufau M. L. (2009) Intramolecular disulfide bonds of the prolactin receptor short form are required for its inhibitory action on the function of the long form of the receptor. Mol. Cell. Biol. 29, 2546–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bakowska J. C., Morrell J. I. (2003) The distribution of mRNA for the short form of the prolactin receptor in the forebrain of the female rat. Brain Res. Mol. Brain Res. 116, 50–58 [DOI] [PubMed] [Google Scholar]
- 48. Bridges R. S., Numan M., Ronsheim P. M., Mann P. E., Lupini C. E. (1990) Central prolactin infusions stimulate maternal behavior in steroid-treated, nulliparous female rats. Proc. Natl. Acad. Sci. U.S.A. 87, 8003–8007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Noel M. B., Woodside B. (1993) Effects of systemic and central prolactin injections on food intake, weight gain, and estrous cyclicity in female rats. Physiol. Behav. 54, 151–154 [DOI] [PubMed] [Google Scholar]
- 50. Dudley C. A., Jamison T. S., Moss R. L. (1982) Inhibition of lordosis behavior in the female rat by intraventricular infusion of prolactin and by chronic hyperprolactinemia. Endocrinology 110, 677–679 [DOI] [PubMed] [Google Scholar]
- 51. Diogenes A., Akopian A. N., Hargreaves K. M. (2007) NGF up-regulates TRPA1: implications for orofacial pain. J. Dent. Res. 86, 550–555 [DOI] [PubMed] [Google Scholar]
- 52. Brown R. S., Piet R., Herbison A. E., Grattan D. R. (2012) Differential actions of prolactin on electrical activity and intracellular signal transduction in hypothalamic neurons. Endocrinology 153, 2375–2384 [DOI] [PubMed] [Google Scholar]
- 53. Ben-Jonathan N., LaPensee C. R., LaPensee E. W. (2008) What can we learn from rodents about prolactin in humans? Endocr. Rev. 29, 1–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kumar A., Singh S. M., Sodhi A. (1997) Effect of prolactin on nitric oxide and interleukin-1 production of murine peritoneal macrophages: role of Ca2+ and protein kinase C. Int. J. Immunopharmacol. 19, 129–133 [DOI] [PubMed] [Google Scholar]
- 55. Gorski E., Zou J., Costello L. C., Franklin R. B. (1999) Protein kinase C mediates prolactin regulation of mitochondrial aspartate aminotransferase gene expression in prostate cells. Mol. Urol. 3, 17–23 [PubMed] [Google Scholar]
- 56. Amaral M. E., Cunha D. A., Anhê G. F., Ueno M., Carneiro E. M., Velloso L. A., Bordin S., Boschero A. C. (2004) Participation of prolactin receptors and phosphatidylinositol 3-kinase and MAP kinase pathways in the increase in pancreatic islet mass and sensitivity to glucose during pregnancy. J. Endocrinol. 183, 469–476 [DOI] [PubMed] [Google Scholar]
- 57. Ahonen T. J., Härkönen P. L., Rui H., Nevalainen M. T. (2002) PRL signal transduction in the epithelial compartment of rat prostate maintained as long-term organ cultures in vitro. Endocrinology 143, 228–238 [DOI] [PubMed] [Google Scholar]
- 58. Gutzman J. H., Rugowski D. E., Schroeder M. D., Watters J. J., Schuler L. A. (2004) Multiple kinase cascades mediate prolactin signals to activating protein-1 in breast cancer cells. Mol. Endocrinol. 18, 3064–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis J. A., Linzer D. I. (1989) Expression of multiple forms of the prolactin receptor in mouse liver. Mol. Endocrinol. 3, 674–680 [DOI] [PubMed] [Google Scholar]
- 60. Pi X., Voogt J. L., Grattan D. R. (2002) Detection of prolactin receptor mRNA in the corpus striatum and substantia nigra of the rat. J. Neurosci. Res. 67, 551–558 [DOI] [PubMed] [Google Scholar]
- 61. Pi X. J., Grattan D. R. (1998) Distribution of prolactin receptor immunoreactivity in the brain of estrogen-treated, ovariectomized rats. J. Comp. Neurol. 394, 462–474 [DOI] [PubMed] [Google Scholar]
- 62. Gregus A. M., Doolen S., Dumlao D. S., Buczynski M. W., Takasusuki T., Fitzsimmons B. L., Hua X. Y., Taylor B. K., Dennis E. A., Yaksh T. L. (2012) Spinal 12-lipoxygenase-derived hepoxilin A3 contributes to inflammatory hyperalgesia via activation of TRPV1 and TRPA1 receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 6721–6726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Patwardhan A. M., Scotland P. E., Akopian A. N., Hargreaves K. M. (2009) Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc. Natl. Acad. Sci. U.S.A. 106, 18820–18824 [DOI] [PMC free article] [PubMed] [Google Scholar]