

Abstract
States of growth hormone (GH) resistance, such those observed in Laron’s dwarf patients, are characterized by mutations in the GH receptor (GHR), decreased serum and tissue IGF-1 levels, impaired glucose tolerance, and impaired skeletal acquisition. IGF-1 replacement therapy in such patients increases growth velocity but does not normalize growth. Herein we combined the GH-resistant (GHR knockout, GHRKO) mouse model with mice expressing the hepatic Igf-1 transgene (HIT) to generate the GHRKO-HIT mouse model. In GHRKOHIT mice, serum IGF-1 levels were restored via transgenic expression of Igf-1 allowing us to study how endocrine IGF-1 affects growth, metabolic homeostasis, and skeletal integrity. We show that in a GH-resistant state, normalization of serum IGF-1 improved body adiposity and restored glucose tolerance but was insufficient to support normal skeletal growth, resulting in an osteopenic skeletal phenotype. The inability of serum IGF-1 to restore skeletal integrity in the total absence of GHR likely resulted from reduced skeletal Igf-1 gene expression, blunted GH-mediated effects on the skeleton that are independent of serum or tissue IGF-1, and from poor delivery of IGF-1 to the tissues. These findings are consistent with clinical data showing that IGF-I replacement therapy in patients with Laron’s syndrome does not achieve full skeletal growth.
Keywords: IGF-1, growth hormone receptor, bone, micro-computed tomography, betaislet, glucose tolerance
Introduction
The insulin-like growth factor (IGF)-1 and its receptor IGF-1R play a major role in tissue homeostasis during embryogenesis, development and pre-pubertal growth (1). IGF-1 acts both in endocrine and autocrine/paracrine fashions (2). The endocrine/circulating IGF-1, secreted by the liver, is mainly regulated by GH, whereas the autocrine/paracrine (tissue) IGF-1 is regulated by tissue-specific factors (3). In the circulation, IGFs bind to IGF-binding proteins (IGFBPs), which regulate their interaction with IGF-1R (3, 4). More than 90% of circulating IGF-1 is complexed with IGFBP-3 and an acid labile subunit (ALS) (5). Binding to IGFBPs and the ALS stabilizes IGF-1 in serum and regulates its bioavailability (5).
In the past few years, several groups, including ours, have focused their efforts on understanding the biological roles of each mode of IGF-1 in the whole body and skeletal growth. Studies in mice with a liver-specific gene deletion of Igf-1 LID) ((6)) or a null mutation in Als (ALSKO) (7, 8), in which serum IGF-1 levels are dramatically decreased, revealed only minor changes in linear growth, suggesting that tissue IGF-1 plays a major role in the determination of body size. An elegant study by Stratikopoulos et al. showed that in Igf-1 null mice (devoid of tissue IGF-1), endocrine IGF-1 was partially restored (~50% of normal) by re-expression of the Igf-1 gene in the liver (9). Characterization of those mice revealed partial restoration of body and skeletal size. Using a different strategy, we generated the KO-HIT mouse, where the sole source of endocrine IGF-1 production in the Igf-1 null background was a rat Igf-1 transgene expressed in the liver (HIT) (10). In this model, serum IGF-1 levels were 2-fold greater than controls, and, interestingly, body size and skeletal properties were restored starting at 8 weeks of age (11). Nonetheless, during early development (before 8 weeks), significant reductions in both body and skeletal size were evident in the KO-HIT mice, again suggesting that tissue IGF-1 plays a crucial role in determining body and skeletal size. However, increases in serum IGF-1 could permit postpubertal “catch up” growth. Importantly, however, overexpression of the Igf-2 transgene under the control of a PEPCK promoter in the liver of Igf-1 null mice resulted in increased circulating levels of IGF-2 but failed to restore growth (12). These findings suggest that despite the structural/functional similarities and despite the fact that their effects are mediated by the same IGF-1 receptor, IGF-1 and IGF-2 have distinct functions, and only treatment with the former is capable of restoring postnatal growth in a murine model system.
In the current study, we aimed to characterize the contribution of GH to skeletal growth and integrity. To that end, we generated the GHRKO-HIT mouse model in which Ghr was ablated in all tissues and the sole source of IGF-1 was the previously described hepatic Igf-1 transgene (HIT) (10). Our new data suggest that GH actions in extrahepatic tissues are vital for the establishment of body size and skeletal integrity.
Results
Generation and validation of the GHRKO-HIT mouse model
In our previous studies, we showed that the hepatic Igf-1 transgene (HIT) driven by the transthyretin (TTR) promoter was able to restore postnatal growth in the Igf-1 null mice (KO-HIT mouse model), such that their body size was indistinguishable from controls (10). Interestingly, however, despite the 2-fold increase in serum IGF-1 levels observed in the KO-HIT mice, GH levels remained normal (10). Thus, we could not rule out direct effects of GH on body size that are independent of tissue IGF-1. We therefore developed a new mouse model in which the hepatic Igf-1 transgene (HIT) driven by the transthyretin (TTR) promoter (Figure 1A) was expressed in Ghr null background (GHRKO-HIT). This was achieved via the crossing strategy depicted in Figure 1B.
Figure 1. Generation and validation of the GHRKO-HIT mouse model.
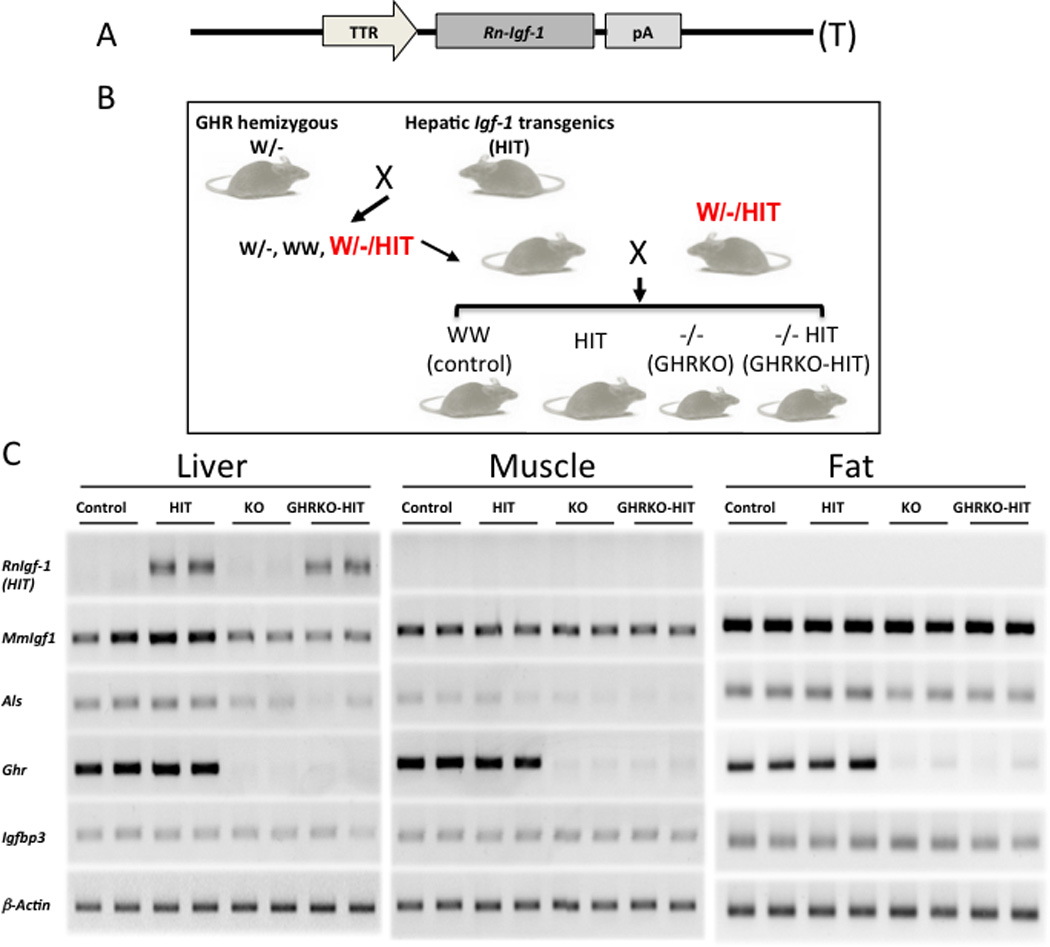
(A) Schematic diagram of the rat Igf-1 R-IGF-1) transgenic construct used to drive Igf-1 expression specifically in the liver under the TTR promoter. (B) Schematic diagram of the breeding strategy used to generate control GHRKO, GHRKO-HIT and HIT mice. GHR hemizygous mice (W/-) were crossed with HIT transgenic mice to generate W/-/HIT mice. Crossing of the W/-/HIT offspring gave rise to the desired genotypes. (C) Analyses of gene expression in liver, muscle and fat tissues dissected from control GHRKO, GHRKO-HIT and HIT mice. RT-PCR was used to detect the expression levels of RnIgf-1 rat Igf-1 transgene), MmIgf-1 mouse Igf-1), Als acid labile subunit), Ghr growth hormone receptor), and Igfbp-3 Igf-binding protein-3).
Gene expression studies in liver, muscle and fat showed that the rat Igf-1 transgene was expressed specifically in the livers of HIT and GHRKO-HIT mice but absent in all other tissues (Figure 1C). As expected, expression of the Ghr gene was detected in the liver, muscle and fat of all groups except the GHRKO and GHRKO-HIT mice. Likewise, expression of the Als gene, a surrogate marker of GH action, was decreased in the liver, muscle and fat of GHRKO and GHRKO-HIT mice. Expression of the endogenous mouse Igf-1 MmIgf-1) gene was detected in liver, muscle and fat, but, as expected, reduced levels were found in tissues of GHRKO and GHRKO-HIT mice (Figure 1C).
GHR action is essential for the establishment of body size
Postnatal growth of control (WT), HIT, GHRKO and GHRKO-HIT mice was assessed by body weight and body length in both males (Figure 2A, C) and females (Figure 2B, D) on two genetic backgrounds: C57Bl6/J (Figure 2A, B) and FVB/N (Figure 2C, D). As expected, both male and female GHRKO mice showed impaired growth and were significantly smaller than control mice on both the FVB/N and C57Bl6/J genetic backgrounds. HIT mice, on the other hand, showed a significant increase in body weight on the FVB/N genetic background (Figure 2C, D), as previously described (10, 11). On the C57Bl6/J genetic background, the body weight of HIT mice did not differ from that of control mice during 16 weeks (Figure 2A, B). Surprisingly, postnatal growth of the GHRKO-HIT mice was retarded in both genders on both genetic backgrounds. GHRKOHIT mice exhibited reduced body weight (by 40%) through 16 weeks of postnatal growth. These striking results show that in the absence of tissue GHR activity, serum IGF-1 is insufficient to restore normal growth and development, implying that tissue GHR is the master regulator of body size.
Figure 2. GHR action is essential for the establishment of body size.
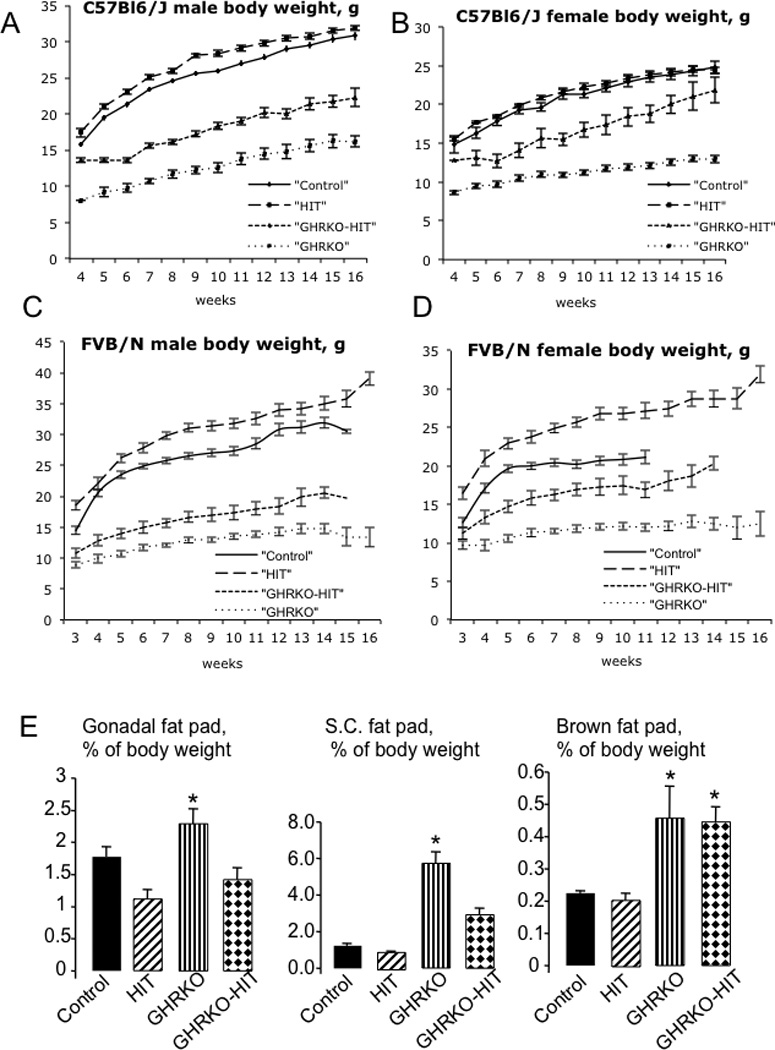
Body weights of male (A) and female (B) mice on the C57Bl6/J genetic background (n>20 mice per group genotype and age) and of male (C) and female (D) mice on the FVB/N genetic background (n>20 mice per group genotype and age) were followed from 3 to 16 weeks of age. Relative weights of gonadal-, subcutaneous- (S.C.) and brown-fat pads at 16 weeks of age in male mice (E) (n>20 mice per group genotype and age). Female mice showed similar results. Data are presented as the mean+/−SEM, and significance was determined at p<0.05. * significantly different from control.
Further characterization of these mouse models was performed on the C57Bl6/J genetic background. As with body weight, the body lengths of GHRKO mice were shorter than those of controls (Table 1), while HIT mice were similar to controls on the C57Bl6/J genetic background. Both male and female GHRKO-HIT mice, on the other had, were shorter than controls but longer than GHRKO mice. Interestingly, we also detected changes in body composition. Quadriceps weight was decreased in GHRKO mice, whereas normalization of serum IGF-1 levels in the GHRKO-HIT mice was associated with increased quadriceps weight (Table 1). Similarly, as previously described (13), relative liver weight was reduced in GHRKO mice, whereas expression of the rat Igf-1 transgene in the GHRKO-HIT mice normalized relative liver weight (Table 1). In accordance with previous studies (13), GHRKO mice exhibited increases in the weight of both gonadal and brown fat pads (Table 1Figure 2E). Normalization of serum IGF-1 levels (GHRKO-HIT) resulted in significant reductions in gonadal fat pad weight but did not influence the volume of brown adipose tissue. These data strongly suggest that GH regulates gonadal fat via liver-derived IGF-1.
Table 1.
Body weight, body length and organ weights of control, HIT, GHRKO and GHRKO-HIT male mice at 16 weeks of age (C57Bl6/j background).
| Control (n=12) | HIT (n=13) | GHRKO (n=10) | GHRKO-HIT (n=7) |
|
|---|---|---|---|---|
| Body weight, g | 29.26+/−1.08 bcd | 31.99+/−0.36abcd | 15.85+/−0.77 abd | 20.43+/−1.06 abc |
| Body length, cm | 10.24+/−0.04 cd | 10.38+/−0.07 cd | 7.66+/−0.06 abd | 8.48+/−0.11 abc |
| Liver, g (% body weight) |
1.31+/−0.06 (4.34+/−0.16) c |
1.48+/−0.05 (4.61+/−0.15) c |
0.56+/−0.02 (3.58+/−0.10) |
0.86+/−0.04 (4.24+/−0.12) c |
| Muscle, g (% body weight) |
0.35+/−0.01 (1.21+/−0.05) cd |
0.39+/−0.01 (1.24+/−0.03) cd |
0.13+/−0.00 (0.87+/−0.05) ab |
0.2+/−0.01 (0.98+/−0.05) ab |
| Gonadal fat, g (% body weight) |
0.49+/−0.05 (1.77+/−0.15) b |
0.35+/−0.04 (1.11+/−0.14) ac |
0.37+/−0.05 (2.29+/−0.22) bd |
0.30+/−0.05 (1.42+/−0.18) c |
| Brown fat, g (% body weight) |
0.07+/−0.00 (0.22+/−0.00) cd |
0.07+/−0.00 (0.20+/−0.02) cd |
0.07+/−0.02 (0.45+/−0.10) ab |
0.10+/−0.01 (0.44+/−0.04) ab |
| Kidney, g (% body weight) |
0.38+/−0.01 (1.29+/−0.03) bcd |
0.46+/−0.01 (1.46+/−0.04) acd |
0.13+/−0.00 (0.83+/−0.03) abd |
0.24+/−0.00 (1.20+/−0.03) abc |
| Spleen, g (% body weight) |
0.09+/−0.00 (0.32+/−0.03) bcd |
0.16+/−0.01 (0.50+/−0.04) acd |
0.03+/−0.00 (0.17+/−0.01) abd |
0.08+/−0.00 (0.38+/−0.02) abc |
| Heart, g (% body weight) |
0.15+/−0.01 (0.50+/−0.03) cd |
0.17+/−0.00 (0.51+/−0.02) cd |
0.07+/−0.00 (0.42+/−0.02) ab |
0.08+/−0.00 (0.40+/−0.03) ab |
| Lung, g (% body weight) |
0.16+/−0.00 (0.57+/−0.02) |
0.18+/−0.00 (0.54+/−0.01) |
0.09+/−0.00 (0.54+/−0.01) |
0.11+/−0.00 (0.55+/−0.05) |
significantly different from controls
significantly different from HIT
significantly different from GHRKO
significantly different from GHRKO-HIT
GHR regulates the endocrine pool of IGF-1
As previously described (13), serum IGF-1 levels were significantly decreased in GHRKO mice compared with controls (14.57+/−2.48 ng/ml vs. 292+/−13 ng/ml, respectively). In contrast, expression of the hepatic Igf-1 transgene (HIT) under the TTR promoter led to a 2-fold increase in serum IGF-1 levels (648+/−49 ng/ml vs. 292+/−13 ng/ml in controls), consistent with our previous observations (10, 11) (Figure 3A). However, despite harboring the same rat HIT transgene, serum IGF-1 levels in GHRKO-HIT mice were not elevated but were instead comparable to those of control mice (320+/−1 ng/ml vs. 292+/−13 ng/ml, respectively).
Figure 3. GHR regulates the endocrine pool of IGF-1.
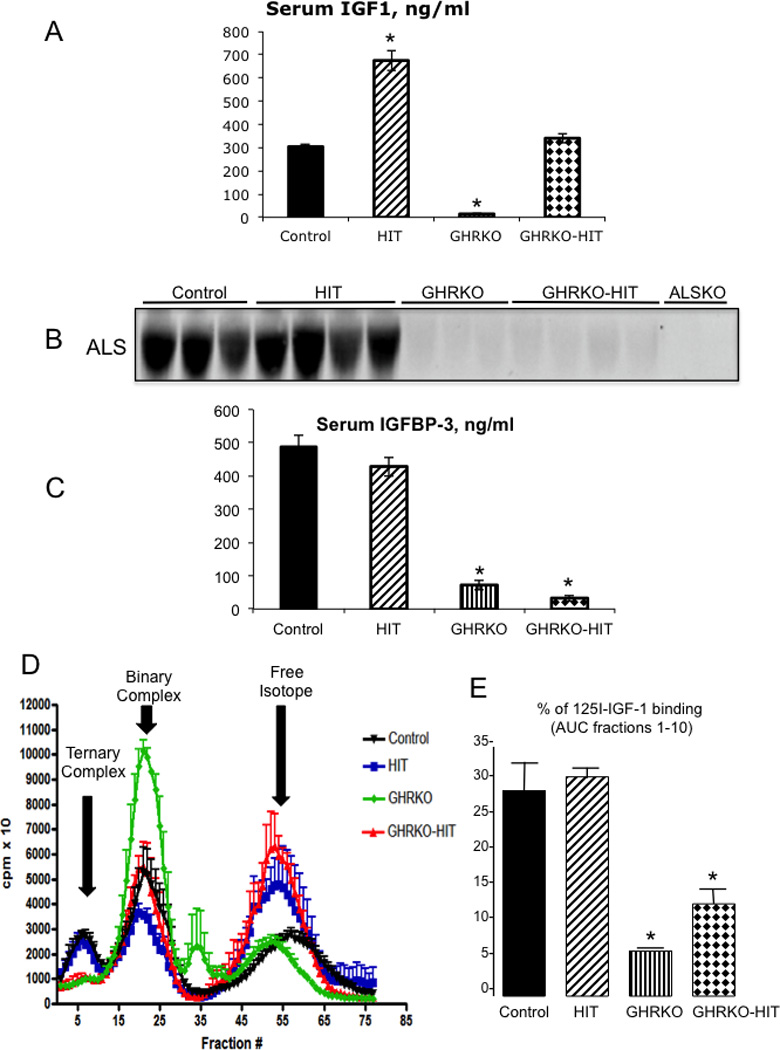
(A) Serum IGF-1 levels (n=10 mice per group genotype) and (B) ALS immunoreactivity in sera from control GHRKO, GHRKO-HIT and HIT mice (n=5 mice per group genotype). Serum from ALS null (ALSKO) mice was used as a negative control. (C) Plasma IGFBP-3 levels (n=10 mice per group genotype). (D) Formation of IGF complexes in sera from control GHRKO, GHRKO-HIT and HIT mice. Eluted fractions 1–10 represent ternary complexes of IGF-1/IGFBP-3/ALS, fractions 20–30 IGF represent binary complexes of IGF-1/IGFBP, and fractions 40–50 represent the unbound IGF-1 isotope (n=5 mice per group genotype). (E) Area under the curve of fractions 1–10, representing IGF-1 ternary complexes. Data are presented as the mean+/−SEM, and significance was determined at p<0.05. * significantly different from control.
To understand why serum IGF-1 levels were not elevated in the GHRKO-HIT mice, we measured serum levels of the IGFBPs and ALS, which stabilize IGF-1 in serum. Because hepatic Als gene expression was reduced in the GHRKO-HIT mice (Figure 1C), we speculated that it would show correspondingly reduced levels in the circulation. Indeed, as determined by a Western immunoblot assay, ALS was undetectable in the serum of GHRKO and GHRKO-HIT mice (Figure 3B). Surprisingly, this pattern correlated with reduced serum levels of IGFBP-3 in both GHRKO and GHRKO-HIT mice (Figure 3C), despite unchanged levels of hepatic Igfbp-3 gene expression (Figure 1C). Together, these results suggest that the absence of elevated serum IGF-1 levels in GHRKO-HIT mice can be attributed to decreased IGF-1 complex stability and that GHR regulates the pool of IGF-1 in serum by virtue of its control of als gene expression. It is important to note that both HIT and GHRKO-HIT mice harbor multiple copies of the rat Igf-1 transgene, which is constitutively expressed under the TTR promoter and thus is not regulated by GH. This may explain why there is a constant hepatic production and secretion of IGF-1 to the circulation reaching control levels.
Consequently, we examined the ability of serum from GHRKO, GHRKO-HIT, and HIT mice to support the formation of ternary complexes with 125I-IGF-1. This assay provides only qualitative information about the presence of the three serum components required for IGF-1 ternary complex formation, namely IGF-1, a binding protein and ALS. Importantly, the percent binding observed in the different fractions does not reflect the absolute concentration of any of the components. As presented in Figure 3D and E ternary complex formation (fractions 0–10) was significantly reduced in sera from GHRKO and GHRKOHIT mice, suggesting significant reductions in the levels of IGFBPs and ALS, as previously demonstrated (Figure 3B, C). However, IGF binary complexes (fractions 20–30) were present in all groups, including GHRKO, GHRKO-HIT, HIT and controls. These data suggest that in both GHRKO and the GHRKO-HIT mice, most of the serum IGF-1 is found in binary complexes with IGFBPs (other than IGFBP-3) or as a free peptide.
Normalization of serum IGF-1 levels is insufficient to restore skeletal growth in the complete absence of tissue GHR
Skeletal characterization of GHRKO-HIT mice on the C57Bl6/J genetic background was performed on femurs dissected at 16 weeks of age. As previously described (13), GHRKO mice showed significant decreases in all bone structural traits compared with control mice (Tables 23). These included reductions in femoral length (by 30%), total cross-sectional area (Tt.Ar; by 53%), cortical area (Ct.Ar; by 60%), cortical thickness (Ct.Th; by 40%) and polar moment of inertia (J0; by 80%). Cross-sectional areas of GHRKO femora were also smaller than controls when normalized to bone length i.e., robustness (Tt.Ar/Le) (Table 2), indicating that total inactivation of GHR action produced a slender bone phenotype.
Table 2.
Cortical bone analyses performed by mCT at the femoral midshaft of control, HIT, GHRKO and GHRKO-HIT mice at 16 weeks of age (C57Bl6/J background). Data are presented as the mean+/−SD, p<0.05.
| Cortical bone | Control (n=14) |
GHRKO (n=9) |
GHRKO-HIT (n=11) |
HIT (n=9) |
|---|---|---|---|---|
| Male | ||||
| Femur length (Le), mm | 15.12+/−0.26 | 10.44+/−0.67abd | 12.01+/−0.25abc | 15.25+/−0.25cd |
| Tt.Ar, mm2 | 1.81+/−0.20 | 1.13+/−0.38abd | 1.24+/−0.12abc | 1.86+/−0.22cd |
| Ct.Ar, mm2 | 0.86+/−0.05 | 0.39+/−0.09abd | 0.55+/−0.09abc | 0.86+/−0.14cd |
| Ma.Ar, mm2 | 0.91+/−0.2 | 0.73+/−0.28abd | 0.68+/−0.15ab | 1.01+/−0.12cd |
| Ct.Th, mm | 0.16+/−0.01 | 0.10+/−0.02abd | 0.14+/−0.01abc | 0.15+/−0.02cd |
| RCA | 0.48+/−0.05 | 0.36+/−0.06abd | 0.44+/−0.08c | 0.45+/−0.04d |
| J0, mm4 | 0.44+/−0.05 | 0.11+/−0.04abd | 0.18+/−0.05abc | 0.46+/−0.13cd |
| Robustness (Tt.Ar/Le) | 0.11+/−0.01 | 0.08+/−0.01ab | 0.09+/−0.00ab | 0.12+/−0.01cd |
| Female | ||||
|
Control (n=6) |
GHRKO (n=9) |
GHRKO-HIT (n=8) |
HIT (n=12) |
|
| Femur length (Le), mm | 15.03+/−0.36 | 11.29+/−0.73abd | 12.75+/−0.00abc | 15.10+/−0.17cd |
| Tt.Ar, mm2 | 1.61+/−0.14 | 0.91+/−0.22abd | 1.51+/−0.23abc | 1.71+/−0.26cd |
| Ct.Ar, mm2 | 0.74+/−0.06 | 0.37+/−0.10abd | 0.63+/−0.04abc | 0.79+/−0.03cd |
| Ma.Ar, mm2 | 0.94+/−0.21 | 0.53+/−0.15abd | 0.87+/−0.22bc | 0.91+/−0.24cd |
| Ct.Th, mm | 0.16+/−0.00 | 0.11+/−0.01abd | 0.16+/−0.00bc | 0.17+/−0.00cd |
| RCA | 0.45+/−0.04 | 0.41+/−0.06abd | 0.43+/−0.06bc | 0.47+/−0.06cd |
| J0, mm4 | 0.31+/−0.05 | 0.09+/−0.05abd | 0.20+/−0.03abc | 0.33+/−0.03cd |
| Robustness (Tt.Ar/Le) | 0.10+/−0.00 | 0.08+/−0.02abd | 0.09+/−0.00abc | 0.10+/−0.01cd |
significantly different from controls
significantly different from HIT
significantly different from GHRKO
significantly different from GHRKO-HIT
Table 3.
Trabecular bone parameters assayed by mCT at the distal femur of control, HIT, GHRKO and GHRKO-HIT mice at 16 weeks of age (C57Bl6/J background). Data are presented as the mean+/−SD, p<0.05.
| Trabecular bone |
Control (n=7) |
GHRKO (n=8) |
GHRKO-HIT (n=11) |
HIT (n=9) |
|---|---|---|---|---|
| Male | ||||
| BV/TV, % | 8.48+/−2.83 | 5.77+/−3.15 b | 5.58+/−2.30 ab | 10.62+/−2.90 acd |
| Tb.Th, µm | 48.51+/−3.53 | 43.55+/−10.29 | 50.16+/−6.40 | 47.87+/−3.29 |
| Tb.N, 1/mm | 1.7×10−3+/−0.4×10−3 | 1.2×10−3+/−0.4×10−3 b | 1.1×10−3+/−0.4×10−3 ab | 2.2×10−3+/−0.5×10−3 cd |
| Tb.Sp, µm | 234.13+/−21.30 | 261.56+/−43.10b | 282.46+/−48.85ab | 201.68+/−31.31acd |
| BMD, g/cc | 0.20+/−0.04 | 0.11+/−0.02abd | 0.15+/−0.03acd | 0.21+/−0.04cd |
| Female | ||||
|
Control (n=6) |
GHRKO (n=8) |
GHRKO-HIT (n=8) |
HIT (n=12) |
|
| BV/TV, % | 3.01+/−1.2 | 2.03+/−1.28d | 5.43+/−1.59acd | 2.77+/−0.71 d |
| Tb.Th, µm | 45.04+/−4.55 | 39.64+/−6.64 | 51.73+/−5.33 | 44.63+/−3.66 |
| Tb.N, 1/mm | 0.6×10−3+/−0.2×10−3 | 0.5×10−3+/−0.2×10−3 | 1.0×10−3+/−0.2×10−3 | 0.6×10−3+/−0.1×10−3 |
| Tb.Sp, µm | 301.93+/−31.16 | 345.61+/−39.80bd | 262.96+/−28.64cd | 298.25+/−20.98cd |
| BMD, g/cc | 0.10+/−0.02 | 0.08+/−0.02abd | 0.14+/−0.00acd | 0.11+/−0.01c |
significantly different from controls
significantly different from HIT
significantly different from GHRKO
significantly different from GHRKO-HIT
Our previous characterization of the HIT mouse model was performed in mice on the FVB/N genetic background (10, 11) and revealed significant increases in body size. However, on the C57Bl6/J genetic background, HIT mice do not exhibit increases in body size, and as a result, their skeletal phenotype at 16 weeks is similar to that of controls. It is conceivable that this discrepancy stems from genetic background differences.
Interestingly, normalization of serum IGF-1 levels failed to restore postnatal skeletal growth in GHRKO-HIT mice. GHRKO-HIT mice exhibited significantly shorter femora (20% decrease), reduced Tt.Ar (35% decrease) and cortical area (Ct.Ar; 30% decrease) and more slender, less robust bones (reduced Tt.Ar/Le) at 16 weeks of age compared with controls. Nonetheless, compared with GHRKO mice, we found that Tt.Ar, Ct.Ar, and Ct.Th were increased in GHRKO-HIT mice, whereas marrow area (Ma.Ar) did not differ significantly between GHRKO and GHRKO-HIT mice. This indicates that normalization of serum IGF-1 levels in GHRKO-HIT mice increased bone tissue area (Ct.Ar) and enhanced periosteal bone apposition.
Unlike the significant differences observed in the cortical shell of GHRKO vs. GHRKO-HIT mice, the trabecular bone phenotype at the distal femur was not different between the 2 groups (GHRKO and GHRKO-HIT) in male mice. However, it should be noted that both GHRKO and GHRKO-HIT mice show reductions in trabecular bone volume compared with control and HIT mice. In female mice however, we found an increase in BV/TV% and in BMD in the GHRKO-HIT mice as compared to the GHRKO mice (Table 3). The cellular or molecular mechanisms responsible for that increase are unclear and require further investigation.
To better understand the cellular mechanisms that lead to the GHRKO-HIT phenotype, we measured serum levels of osteocalcin, a bone formation marker. We found no significant differences between controls (53+/−17 ng/ml), GHRKO (49+/−18 ng/ml), GHRKO-HIT (49+/−15 ng/ml), or HIT (54+/−9 ng/ml) mice. We thus explored whether the kinetics of bone apposition differed between the groups using dynamic histomorphometry (Table 4, Figure 4). We found that the mineralized surface/bone surface (MS/BS) decreased in GHRKO mice at both the endosteal and periosteal surfaces as compared to controls. In contrast, GHRKO-HIT mice showed an increase in %MS/BS on both surfaces when compared to GHRKO mice. Calculations of mineral apposition rates (MAR) and bone formation rates (BFR) on both the endosteal and periosteal surfaces revealed large variability within the groups and thus did not reach significance. Nonetheless, we found that the MAR and BFR in both endosteal and periosteal surfaces decreased in GHRKO mice and reached control rates in GHRKO-HIT mice. HIT mice, however, tended to increase MAR and BFR at the endosteal surface as compared to control mice, while MAR and BFR at the periosteal surface were similar to controls. These data are in accordance with our previous findings (11), suggesting that increases in serum IGF-1 levels are associated with increased MAR and BFR. Overall our dynamic histomorphometry findings are in accordance with the morphology data obtained by mCT (Table 3).
Table 4.
Histomorphometry data from mid-diaphyseal cortical bone of female mice (on C57Bl/6 background) at 16 weeks of age.
| Surface | Parameter | Control | GHRKO | GHRKO-HIT | HIT |
|---|---|---|---|---|---|
| Endosteal | MS/BS, % | 45.34+/−7.13 | 39.88+/−30.49 | 65.95+/−19.79 | 71.14+/−10.89 |
| MAR, um/day | 0.59+/−0.56 | 0.30+/−0.40 | 0.75+/−0.04 | 0.97+/−0.34 | |
| BFR, um/day*100 | 29.96+/−29.18 | 21.83+/−34.01 | 49.69+/−15.38 | 67.14+/−26.40 | |
| Periosteal | MS/BS, % | 33.96+/−16.42 | 1.93+/−3.31 | 23.85+/−15.13 | 28.53+/−15.44 |
| MAR, um/day | 0.76+/−0.45 | 0.00+/−0.00 | 0.68+/−0.61 | 0.53+/−0.61 | |
| BFR, um/day*100 | 29.66+/−21.48 | 0.00+/−0.00 | 22.54+/−21.68 | 19.82+/−24.25 |
Values are the mean ± S.D., p<0.05. Calcein labels were injected twice in intervals of 10–14 days. In GHRKO and the GHRKO-HIT mice the intervals between injections were 17–23 days.
Figure 4. Serum IGF-1 does not restore the skeletal defects of GHRKO mice.
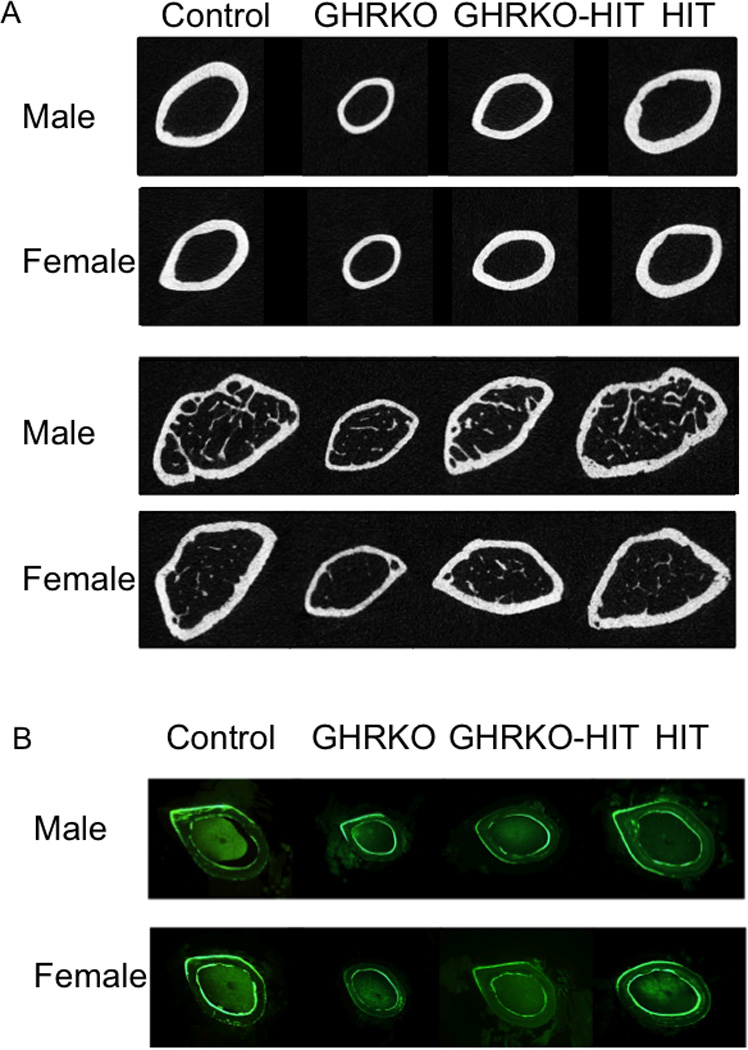
(A) mCT images taken at the femoral midshaft (cortical bone) or at the distal femur (trabecular bone) of control GHRKO, GHRKO-HIT and HIT mice. (B) Histological plastic sections (50 µm) of cortical bone taken at the femoral midshaft.
The impaired skeletal phenotype in the GHRKO mice attributed to global reductions in IGF-1 (in both serum and tissues) as well as to IGF-1-independent effects of GH on the skeleton (14). Thus, we assessed the levels of skeletal Igf-1 gene expression in femoral cortices of control, GHRKO, GHRKO-HIT and HIT mice at 16 weeks of age. Skeletal Igf-1 gene expression significantly reduced in GHRKO mice and was not restored in GHRKO-HIT mice (Figure 5A). As expected, Ghr gene expression was undetectable in both GHRKO and GHRKO-HIT mice (Figure 5B). Overall, these data suggest that one of the reason for the impaired skeletal growth in the GHRKO-HIT mice is likely the low levels of skeletal Igf-1 production that were not restored with expression of the hepatic Igf-1 transgene.
Figure 5. GHRKO and GHRKO-HIT mice show significant reductions in skeletal Igf-1 gene expression.
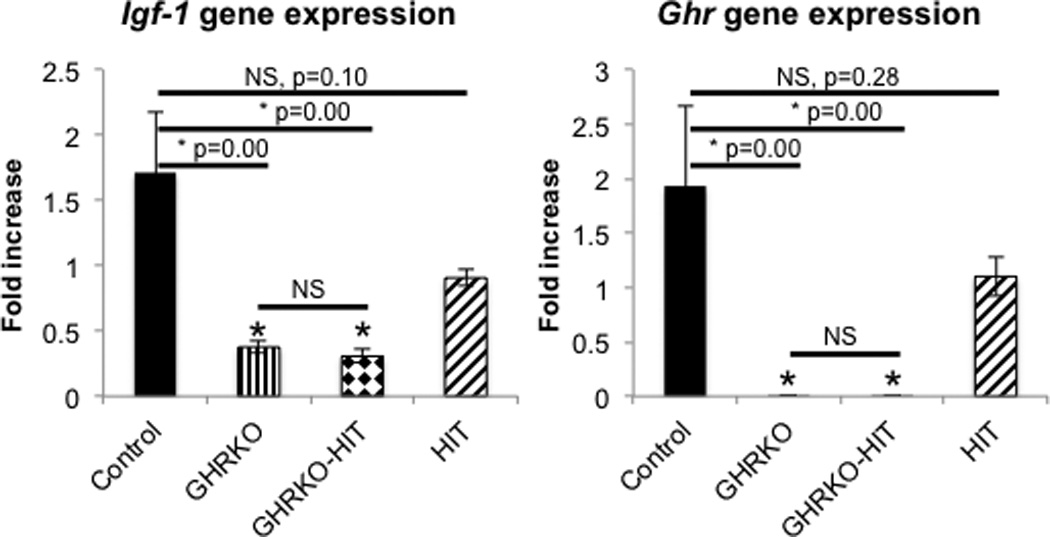
Femurs were dissected from 16 week-old mice. Marrow from the mid-diaphysis area flushed and RNA extracted from the cortical shells. (A) Igf-1 gene expression (assessed by real-time PCR) significantly decreased in GHRKO and GHRKO-HIT mice (n=5 mice per group genotype). (B) Ghr gene expression (assessed by real-time PCR) was undetectable in cortical bone RNA extracts from GHRKO and GHRKO-HIT mice. Data presented as mean+/−SEM of n=5 mice per group genotype and significance was determined at p<0.05, NS - not significant, * significantly different from control.
Normalization of serum IGF-1 levels in the complete absence of GHR is sufficient to restore glucose tolerance in GHRKO mice
GH affects multiple tissues, and its role in carbohydrate and lipid metabolism has been extensively investigated. GHRKO mice exhibit an enhanced insulin sensitivity reflected by decreases in serum levels of basal glucose and insulin (13). However, when challenged with a bolus of glucose, GHRKO mice exhibit impaired glucose clearance due to reduced β-cell mass and consequent reductions in insulin secretion (15). To address whether normalization of serum IGF-1 levels in GHRKO mice improved glucose tolerance, we measured insulin levels in serum and performed a glucose tolerance test (GTT). As expected, glucose tolerance was impaired in GHRKO mice and correlated with reduced β-cell mass (Figure 6). However, normalization of serum IGF-1 in GHRKO-HIT mice was associated with normalized GTT. This could be partially explained by increased β-cell mass compared with GHRKO mice (Figure 6).
Figure 6. Serum IGF-1 restores β-cell mass and glucose tolerance in GHRKO mice.
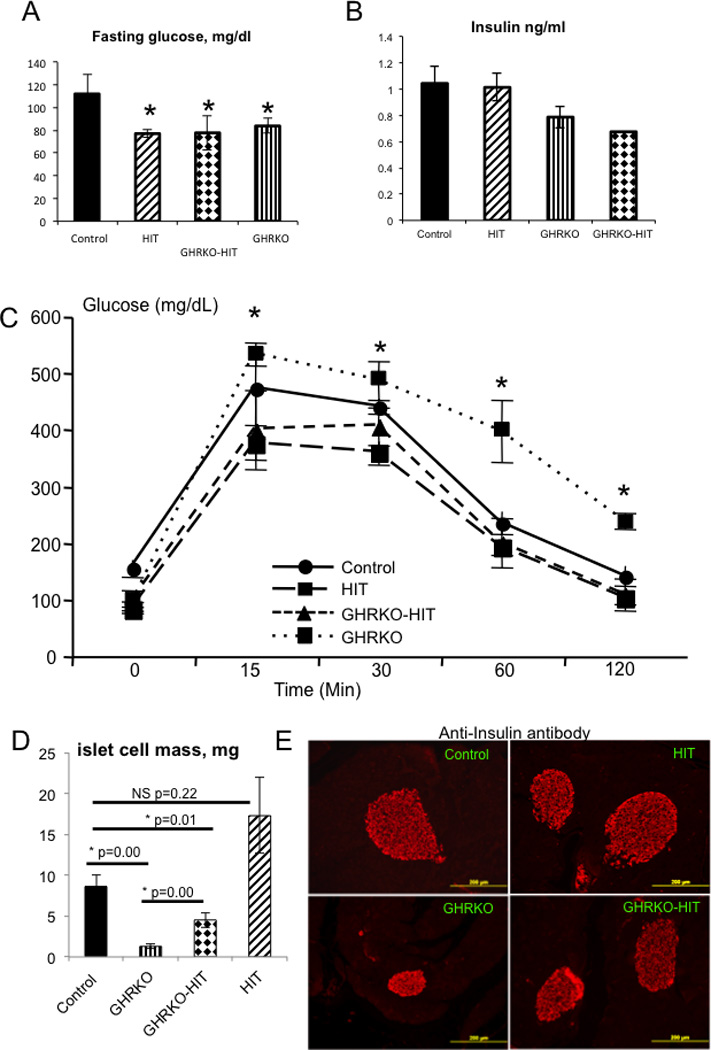
Fasting glucose levels (A) and serum insulin levels (B) were measured at 16 weeks of age. A glucose tolerance test (C) was performed in adult male mice at 14–16 weeks of age. β-cell mass (D) was assessed in pancreatic sections immunostained with an anti-insulin antibody. β-cell mass was calculated as the β-cell relative area multiplied by the pancreatic weights of 6 mice per group. Representative images of pancreatic β-islets immunostained with an anti-insulin antibody. Data are presented as the mean+/−SEM, and significance was determined at p<0.05. NS - not significant, * significantly different from control.
Discussion
In this study, we present a mouse model of GH resistance that, despite normal levels of serum IGF-1, shows significant growth retardation. We show that in a GH resistance state, normalization of serum IGF-1 levels improved body adiposity and restored glucose tolerance but was insufficient to support normal skeletal growth, resulting in an osteopenic bone phenotype. We show that skeletal Igf-1 gene expression significantly reduced in states of GH resistance and that serum IGF-1, although found in normal levels (GHRKO-HIT mice), is not bound to IGFBP-3 and ALS.
The GHR null (GHRKO) mice were generated over a decade ago and have proven to be a useful and suitable mammalian model for studying human Laron syndrome (16). The GHR defects that cause Laron syndrome are heterogeneous and include gene deletions and point mutations (17–19), but all are characterized by receptor inactivation and inhibition of downstream signaling pathways. This results in the failure to generate IGF-1 in response to endogenous or exogenous GH, which consequently leads to short stature, delayed bone age and bone maturation. Laron’s patients are resistant to GH treatment. However, cells isolated from these patients respond to insulin and IGF-1 (20), suggesting that the insulin and IGF-1 receptors are intact and active. Based on these in vitro data, several clinical studies verified the effects of recombinant IGF-1 treatment on skeletal growth in Laron dwarfs. Most studies (21–31) indicate a rapid increase in linear growth and height velocity during the first year of treatment, along with decreases in body adiposity. However, IGF-1 treatment did not normalize overall growth. Our study design resembles IGF-1 replacement therapy in Laron’s dwarfs in that serum IGF-1 levels were restored via transgenic expression. As with Laron’s dwarfs, we show here that skeletal GHR activity is vital for the establishment of normal growth.
GH is the primary regulator of ALS transcription in the liver (32). Deficiencies in GHR action in the liver lead to almost complete ablation of ALS expression and result in significant reductions in the formation of the IGF-1 ternary complex (ALS/IGFBP3/IGF-1), which is obligatory for effective IGF-1 delivery to tissues. In our previous study we have shown that serum IGF-1 levels decreased significantly in the ALSKO/BP3KO double knockout mice (5). In these mice the two endogenous copies of the Igf-1 gene are expressed under IGF-1 regulated promoter and reductions in serum IGF-1 levels result likely from rapid degradation of the peptide in serum. In the GHRKO-HIT mouse model presented herein, serum IGF-1 levels were restored to normal levels. However, in the GHRKO-HIT mouse model, the Igf-1 transgene presents in multiple copies and is constantly expressed under the TTR promoter, which is not regulated by GH. The normal level of IGF-1 in serum of GHRKO-HIT mice are likely the result of a constant high output of the peptide to circulation and possibly increases in the levels of IGFBPs (other than IGFBP3) and binary complex formation.
Our data suggest that serum IGF-1 in the GHRKO-HIT mice, which likely delivered to target tissues via binary complexes with IGFBPs, increase β-cell mass of GHRKO-HIT mice, leading to normal glucose tolerance. These data are also in agreement with a previous study (33) showing that ubiquitous restoration (via the metallothionein I promoter) of Igf-1 gene expression in GHRKO mice restored their β-cell mass and glucose tolerance. Here, we show that normalization of endocrine IGF-1 is in fact sufficient to restore glucose tolerance in a state of GHR resistance. Additionally, we show that normalization of IGF-1 levels was associated with reduced subcutaneous and gonadal fat pad weights, which may indirectly lead to enhanced insulin sensitivity.
Our previous studies with the LID (6) and Als null (ALSKO) mice (7), where serum IGF-1 levels were dramatically decreased, demonstrated that the GH/IGF-1 axis not only regulates linear bone growth but also controls the development of the diaphysis and thus has major effects on bone strength. Likewise, LID mice have more slender bones with reduced stiffness and reduced bending strength due to significant decreases in femoral total area (Tt.Ar) and cortical area (Ct.Ar) beginning at 8 weeks of age (8). The reductions in serum IGF-1 levels affected the cortical shell and prevented periosteal bone apposition during growth. The ALSKO mice show reductions in body weight and body length throughout growth (5) as well as slender bones with reduced Tt.Ar, Ct.Ar, and marrow area (Ma.Ar). The data from the LID and ALSKO models suggested that serum IGF-1 levels regulate periosteal bone apposition during growth. However, in LID mice, the reductions in serum IGF-1 levels led to increases in GH secretion, which could have direct compensatory effects on bone. Indeed, in a related study (34), we demonstrated that inhibition of GH action in LID mice resulted in significant reductions in Ct.Ar, indicating that antagonizing GH action caused deep reductions in the amount of cortical bone formed, leading to slender, mechanically inferior bones. To better understand the interplay between GH and IGF-1 in the skeleton, we previously generated transgenic mice with hepatocyte-specific expression of rat Igf-1 HIT). These mice show elevations in serum IGF-1 levels that lead to increases in body weight and femoral length, Tt.Ar, Ct.Ar, cortical thickness (Ct.Th), and Jo 11). When HIT mice were crossed to the IGF-1 null mice (IGF-1KO-HIT), serum IGF-1 levels increased ~2-fold, and skeletal growth normalized. IGF-1KO-HIT mice exhibited normal bone length, and in adults (16 weeks), the femoral cross-sectional area reached control levels (11). However, it should be noted that in the IGF-1KO-HIT mice, serum GH levels were normal, and the GHR pathway in bone was intact. Further, although skeletal igf-1 gene expression was ablated (due to the lack of the Igf-1 gene), circulating IGF-1 was bound to IGFBP-3 and ALS and was properly delivered to tissues. In sharp contrast, when Ghr gene was ablated, the same HIT transgene (GHRKO-HIT) was unable to restore growth. GHRKO-HIT mice displayed retarded linear growth and reduced transversal bone growth as indicated by smaller Tt.Ar values. In the GHRKO-HIT mice (as in the IGF-1KO-HIT) (i)-skeletal Igf-1 gene expression was blunted, (ii)-the GHR pathway was ablated (due to the lack of the Ghr gene), and (iii)-serum IGF-1 was not delivered bound in ternary or binary complexes with IGFBP-3 and ALS to tissues. Taken together the GHRKO-HIT and IGF-1KO-HIT mouse models, we suggest that IGF-1-independent actions of GH on the skeleton are vital for establishment of skeletal size and integrity and cannot be fully compensated for by increasing serum IGF-1 levels. Our findings are consistent with clinical studies reporting that Laron’s dwarf patients treated with recombinant IGF-1 do not reach normal height. They do, however, show improvement in height and growth velocity in response to treatment. Thus, in states of GHR resistance, strategies to increase skeletal expression of Igf-1 and improvement of IGF-1 delivery to tissues should be carefully considered to achieve better anabolic effects on the skeleton.
Methods
Animals
HIT mice (on the FVB/N) were generated as described previously (10). To establish a HIT mouse line on a C57Bl6/J background we have crossed the HIT-FVB/N mice 8 generations to wild-type C57Bl6/J mice. HIT mice were crossed to GHRKO mice (on the FVB/N or C57Bl6/J background) as described in Figure 1B. HIT and GHRKO-HIT mice were homozygous for the Igf-1 transgene. Mice were housed 4 per cage in a clean mouse facility, fed standard mouse chow (Purina Laboratory Chow 5001; Purina Mills) and water ad libitum, and kept on a 12 hour light:dark cycle. Animal care and maintenance were provided through NYU’s AAALAC Accredited Animal Facility. All procedures were approved by the Animal Care and Use Committee of NYU.
Serum hormones
Mice were bled between 7–9 AM through the mandibular vein before sacrifice. Serum samples were collected in a fed state at 16 weeks of age. Serum IGF-1 was measured by RIA (American Laboratory Products Company, Inc., Salem, NH, USA). Serum GH and IGFBP-3 levels were determined using commercial ELISA kits (Millipore, Temecula, CA, USA).
For measurement of ALS, sera were diluted 1:4 with saline and subsequently separated on a 4–12% acrylamide gel and transferred to nitrocellulose. Membranes were blocked with LICOR block solution (Lincoln, NE, USA) and incubated overnight at 4°C with a goat anti-ALS antibody (R&D Systems, Inc., Minneapolis, MN USA). Following washing, the membrane was incubated with IRDye800CW-donkey anti-goat antibody (LI-COR, Lincoln, NE, USA). Bound antibodies were detected by the LI-COR Odyssey infrared imaging system (LI-COR).
Formation of IGF complexes was performed in accordance to the protocol adopted from Baxter et al.(35). Serum samples (100 µl) were incubated overnight at 22°C with I125-IGF-1 (at a final concentration of 10 ng/ml) and then cross-linked with disuccinimidyl suberate as reported previously (36). Complexes were separated using HiPrep 16/60 Sephacryl S-200HR columns and 1-ml fractions were collected and counted.
Glucose tolerance test
Glucose (2 g/kg) was injected intraperitoneally to overnight-fasted mice. Glucose levels were measured in the tail vein at the indicated time points using an automatic Glucometer Elite (Bayer, Elkhart, IN).
MicroComputed Tomography
Cortical bone morphology at the mid-femoral diaphysis and trabecular bone volume fraction and microarchitecture in the excised distal femoral metaphysis were assessed as previously described (37) and according to JBMR guidelines (38). Femora were reconstructed at a 6 µm voxel resolution. For trabecular bone regions, we assessed the bone volume fraction (BV/TV, %), trabecular thickness (Tb.Th, µm), trabecular number (Tb.N), and trabecular spacing (Tb.Sp, µm). For cortical bone at the femoral midshaft, we measured the average total cross-sectional area inside the periosteal envelope (Tt.Ar, mm2), the cortical bone and medullary area within this same envelope (Ct.Ar, mm2 and Ma.Ar mm2 respectively), the relative cortical area (RCA; Ct.Ar/Tt.Ar, %), the average cortical thickness (Ct.Th, µm) and the polar moment of inertia (Jo). All regions of analysis were standardized according to anatomical landmarks.
Histomorphometry
Sixteen-week-old animals were injected twice with calcein (15 mg/Kg) with >10 days between the two injections (in the case of GHRKO and GHRKO-HIT mice the interval between the two injections was 17–23 days). Femora were fixed in 10% neutral buffered formalin, embedded in polymethylmethacrylate (PMMA) and sectioned (80 µm thickness) at the mid-diaphysis using a low-speed diamond-coated wafering saw (Leica, Bannockburn, IL, USA). Sections were adhered to either glass or acrylic slides using a nonfluorescing mounting medium. Final section thicknesses after polishing were between 30 and 40 µm. All measurements were performed using an OsteoMeasure system (Osteometrics, Atlanta, GA, USA) in accordance with standard protocols. Sections were imaged using a digital camera attached to a visible light/fluorescence microscope (Zeiss Axioplan2, Zeiss AxioVision, Thornwood, NY).
Histology
Tissues were fixed in 10% buffered formalin phosphate (Fisher Scientific) and embedded in paraffin. Immunohistochemistry with rabbit anti-human insulin (Cell Signaling #C27C9) was performed in 5-um sections of paraffin-embedded pancreata. β-cell mass was calculated from 3–5 pancreatic sections per mouse stained with the anti-insulin antibody. Relative β cell area was calculated using NIH Image J software, and β-cell mass was calculated as the β-cell relative area multiplied by the pancreatic weights of 6 mice per group.
Gene expression studies
Total RNA was extracted from tissues using TRIzol (Invitrogen, Carlsbad, CA). RNA samples (1 µg) were reverse-transcribed using oligo-(dT) primers (Invitrogen), and quantitative real-time PCR was performed following the manufacturer’s instructions using the QuantiTect™ SYBR® green PCR kit (Qiagen, Valencia, CA, USA) on an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA). Transcript levels were assayed 3 times in each sample, and the fold-change ratios between experimental and control samples were calculated relative to β-actin, GAPDH or 18S.
Statistical analysis
All differences in mean serum hormone levels, organ weights, and growth among the different groups were assessed by analyses of variance (ANOVA). Values are presented as the mean ± S.E., and P < 0.05 was considered statistically significant. All micro-CT measurements are presented as the mean ± S.D. One-way ANOVAs were used to test for differences among groups (Statview software version 5.0, SAS Institute Inc.). If the ANOVA revealed significant effects, the means were compared by Fisher’s test, considering P<0.05 as significant.
Acknowledgments
Shoshana Yakar is the principle investigator on the study and as such designed and oversaw all the experiments, data collection, and data analyses. Dr. Yakar also wrote the manuscript. Dr. Yingjie Wu and Mrs. Hui Sun collected the data and performed the metabolic studies. Mrs. Jelena Basta-Pljakic and Dr. Luis Cardoso performed the micro-CT analyses. Dr. Oran D Kennedy performed the histomorphometry analyses. Dr. Hector Jasper, Dr. Horacio Domené, Mrs. Liliana Karabatas, and Mrs. Clara Guida performed the serum IGF-1 complex-formation assay. Dr. Mitchell B Schaffler and Dr. Clifford J Rosen helped in data interpretation and manuscript preparation.
The authors would like to thank Sonali Lahoti, Ban Zhaojiang, and Brian Sunwoo (David B. Kriser Dental Center, Department of Basic Science and Craniofacial Biology, New York University College of Dentistry, New York, NY) for technical assistance.
Funding:
Financial support was received from funding agencies in the United States (NIH/NIAMS AR054919 & AR055141 to SY, AR45433 to CJR, AG034198 and MRI0723027 to LC and AR041210 and AR057139 to MBS) and Argentina (CONICET PIP 11420090100045 to HJ).
Footnotes
All authors concur with the submission. The material submitted for publication has not been previously reported and is not under consideration for publication elsewhere. None of the authors have conflicting financial interests.
References
- 1.Yakar S, Adamo ML. Insulin-like growth factor 1 physiology: lessons from mouse models. Endocrinol Metab Clin North Am. 2012;41:231–247. doi: 10.1016/j.ecl.2012.04.008. v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler AA, Yakar S, LeRoith D. Insulin-like growth factor-I: compartmentalization within the somatotropic axis? News Physiol Sci. 2002;17:82–85. doi: 10.1152/nips.01351.2001. [DOI] [PubMed] [Google Scholar]
- 3.Le Roith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocrine reviews. 2001;22:53–74. doi: 10.1210/edrv.22.1.0419. [DOI] [PubMed] [Google Scholar]
- 4.Yakar S, Kim H, Zhao H, Toyoshima Y, Pennisi P, Gavrilova O, Leroith D. The growth hormone-insulin like growth factor axis revisited: lessons from IGF-1 and IGF-1 receptor gene targeting. Pediatric nephrology (Berlin, Germany) 2005;20:251–254. doi: 10.1007/s00467-004-1613-y. [DOI] [PubMed] [Google Scholar]
- 5.Yakar S, Rosen CJ, Bouxsein ML, Sun H, Mejia W, Kawashima Y, Wu Y, Emerton K, Williams V, Jepsen K, Schaffler MB, Majeska RJ, Gavrilova O, Gutierrez M, Hwang D, Pennisi P, Frystyk J, Boisclair Y, Pintar J, Jasper H, Domene H, Cohen P, Clemmons D, LeRoith D. Serum complexes of insulin-like growth factor-1 modulate skeletal integrity and carbohydrate metabolism. Faseb J. 2009;23:709–719. doi: 10.1096/fj.08-118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yakar S, Canalis E, Sun H, Mejia W, Kawashima Y, Nasser P, Courtland HW, Williams V, Bouxsein M, Rosen C, Jepsen KJ. Serum IGF-1 determines skeletal strength by regulating subperiosteal expansion and trait interactions. J Bone Miner Res. 2009;24:1481–1492. doi: 10.1359/JBMR.090226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courtland HW, DeMambro V, Maynard J, Sun H, Elis S, Rosen C, Yakar S. Sex-specific regulation of body size and bone slenderness by the acid labile subunit. J Bone Miner Res. 2010;25:2059–2068. doi: 10.1002/jbmr.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D. Circulating levels of IGF-1 directly regulate bone growth and density. The Journal of clinical investigation. 2002;110:771–781. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19378–19383. doi: 10.1073/pnas.0809223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Sun H, Yakar S, LeRoith D. Elevated levels of insulin-like growth factor (IGF)-I in serum rescue the severe growth retardation of IGF-I null mice. Endocrinology. 2009;150:4395–4403. doi: 10.1210/en.2009-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elis S, Courtland HW, Wu Y, Rosen CJ, Sun H, Jepsen KJ, Majeska RJ, Yakar S. Elevated serum levels of IGF-1 are sufficient to establish normal body size and skeletal properties even in the absence of tissue IGF-1. J Bone Miner Res. 2010;25:1257–1266. doi: 10.1002/jbmr.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moerth C, Schneider MR, Renner-Mueller I, Blutke A, Elmlinger MW, Erben RG, Camacho-Hubner C, Hoeflich A, Wolf E. Postnatally elevated levels of insulin-like growth factor (IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except kidney weight. Endocrinology. 2007;148:441–451. doi: 10.1210/en.2006-0385. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Cataldo L, Coschigamo K, Wagner TE, Baumann G, Kopchick JJ. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proceedings of the National Academy of Sciences of the United States of America. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, Nauman E, Leary C, Halloran B. The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res. 2001;16:2320–2329. doi: 10.1359/jbmr.2001.16.12.2320. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Liu C, Sun H, Vijayakumar A, Giglou PR, Qiao R, Oppenheimer J, Yakar S, LeRoith D. Growth hormone receptor regulates beta cell hyperplasia and glucose-stimulated insulin secretion in obese mice. The Journal of clinical investigation. 2011;121:2422–2426. doi: 10.1172/JCI45027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaefer GB, Rosenbloom AL, Guevara-Aguirre J, Campbell EA, Ullrich F, Patil K, Frias JL. Facial morphometry of Ecuadorian patients with growth hormone receptor deficiency/Laron syndrome. J Med Genet. 1994;31:635–639. doi: 10.1136/jmg.31.8.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips JA., 3rd Molecular biology of growth hormone receptor dysfunction. Acta Paediatr Suppl. 1992;383:127–131. [PubMed] [Google Scholar]
- 18.Behncken SN, Rowlinson SW, Rowland JE, Conway-Campbell BL, Monks TA, Waters MJ. Aspartate 171 is the major primate-specific determinant of human growth hormone. Engineering porcine growth hormone to activate the human receptor. The Journal of biological chemistry. 1997;272:27077–27083. doi: 10.1074/jbc.272.43.27077. [DOI] [PubMed] [Google Scholar]
- 19.Souza SC, Frick GP, Wang X, Kopchick JJ, Lobo RB, Goodman HM. A single arginine residue determines species specificity of the human growth hormone receptor. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:959–963. doi: 10.1073/pnas.92.4.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geffner ME, Golde DW, Lippe BM, Kaplan SA, Bersch N, Li CH. Tissues of the Laron dwarf are sensitive to insulin-like growth factor I but not to growth hormone. The Journal of clinical endocrinology and metabolism. 1987;64:1042–1046. doi: 10.1210/jcem-64-5-1042. [DOI] [PubMed] [Google Scholar]
- 21.Laron Z, Anin S, Klipper-Aurbach Y, Klinger B. Effects of insulin-like growth factor on linear growth, head circumference, and body fat in patients with Laron-type dwarfism. Lancet. 1992;339:1258–1261. doi: 10.1016/0140-6736(92)91594-x. [DOI] [PubMed] [Google Scholar]
- 22.Backeljauw PF, Underwood LE. Prolonged treatment with recombinant insulin-like growth factor-I in children with growth hormone insensitivity syndrome--a clinical research center study. GHIS Collaborative Group. The Journal of clinical endocrinology and metabolism. 1996;81:3312–3317. doi: 10.1210/jcem.81.9.8784089. [DOI] [PubMed] [Google Scholar]
- 23.Backeljauw PF, Underwood LE. Therapy for 6.5–7.5 years with recombinant insulin-like growth factor I in children with growth hormone insensitivity syndrome: a clinical research center study. The Journal of clinical endocrinology and metabolism. 2001;86:1504–1510. doi: 10.1210/jcem.86.4.7381. [DOI] [PubMed] [Google Scholar]
- 24.Guevara-Aguirre J, Rosenbloom AL, Vasconez O, Martinez V, Gargosky SE, Allen L, Rosenfeld RG. Two-year treatment of growth hormone (GH) receptor deficiency with recombinant insulin-like growth factor I in 22 children: comparison of two dosage levels and to GH-treated GH deficiency. The Journal of clinical endocrinology and metabolism. 1997;82:629–633. doi: 10.1210/jcem.82.2.3743. [DOI] [PubMed] [Google Scholar]
- 25.Mauras N, Martinez V, Rini A, Guevara-Aguirre J. Recombinant human insulin-like growth factor I has significant anabolic effects in adults with growth hormone receptor deficiency: studies on protein, glucose, and lipid metabolism. The Journal of clinical endocrinology and metabolism. 2000;85:3036–3042. doi: 10.1210/jcem.85.9.6772. [DOI] [PubMed] [Google Scholar]
- 26.Tonella P, Fluck CE, Mullis PE. Insulin-like growth factor-I treatment in primary growth hormone insensitivity: effect of recombinant human IGF-I (rhIGF-I) and rhIGF-I/rhIGF-binding protein-3 complex. Horm Res Paediatr. 2010;73:140–147. doi: 10.1159/000277660. [DOI] [PubMed] [Google Scholar]
- 27.Guevara-Aguirre J, Vasconez O, Martinez V, Martinez AL, Rosenbloom AL, Diamond FB, Jr, Gargosky SE, Nonoshita L, Rosenfeld RG. A randomized, double blind, placebo-controlled trial on safety and efficacy of recombinant human insulin-like growth factor-I in children with growth hormone receptor deficiency. The Journal of clinical endocrinology and metabolism. 1995;80:1393–1398. doi: 10.1210/jcem.80.4.7536209. [DOI] [PubMed] [Google Scholar]
- 28.El Kholy M, Elsedfy HH. Effect of GnRH analogue on height potential in patients with severe growth hormone insensitivity syndrome treated with IGF-I. Journal of pediatric endocrinology – metabolism : JPEM. 2011;24:983–988. doi: 10.1515/jpem.2011.348. [DOI] [PubMed] [Google Scholar]
- 29.Klinger B, Laron Z. Three year IGF-I treatment of children with Laron syndrome. Journal of pediatric endocrinology – metabolism : JPEM. 1995;8:149–158. doi: 10.1515/jpem.1995.8.3.149. [DOI] [PubMed] [Google Scholar]
- 30.Laron Z, Klinger B, Eshet R, Kaneti H, Karasik A, Silbergeld A. Laron syndrome due to a post-receptor defect: response to IGF-I treatment. Isr J Med Sci. 1993;29:757–763. [PubMed] [Google Scholar]
- 31.Ranke MB, Savage MO, Chatelain PG, Preece MA, Rosenfeld RG, Blum WF, Wilton P. Insulin-like growth factor I improves height in growth hormone insensitivity: two years' results. Hormone research. 1995;44:253–264. doi: 10.1159/000184637. [DOI] [PubMed] [Google Scholar]
- 32.Ooi GT, Cohen FJ, Tseng LY, Rechler MM, Boisclair YR. Growth hormone stimulates transcription of the gene encoding the acid-labile subunit (ALS) of the circulating insulin-like growth factor-binding protein complex and ALS promoter activity in rat liver. Molecular endocrinology. 1997;11:997–1007. doi: 10.1210/mend.11.7.9942. [DOI] [PubMed] [Google Scholar]
- 33.Robertson K, Lu Y, De Jesus K, Li B, Su Q, Lund PK, Liu JL. A general and islet cell-enriched overexpression of IGF-I results in normal islet cell growth, hypoglycemia, and significant resistance to experimental diabetes. American journal of physiology. Endocrinology and metabolism. 2008;294:E928–E938. doi: 10.1152/ajpendo.00606.2007. [DOI] [PubMed] [Google Scholar]
- 34.Courtland HW, Sun H, Beth-On M, Wu Y, Elis S, Rosen CJ, Yakar S. Growth hormone mediates pubertal skeletal development independent of hepatic IGF-1 production. J Bone Miner Res. 2011;26:761–768. doi: 10.1002/jbmr.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baxter RC, Martin JL. Structure of the Mr 140,000 growth hormone-dependent insulin-like growth factor binding protein complex: determination by reconstitution and affinity-labeling. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:6898–6902. doi: 10.1073/pnas.86.18.6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Domene HM, Bengolea SV, Martinez AS, Ropelato MG, Pennisi P, Scaglia P, Heinrich JJ, Jasper HG. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. N Engl J Med. 2004;350:570–577. doi: 10.1056/NEJMoa013100. [DOI] [PubMed] [Google Scholar]
- 37.Elis S, Wu Y, Courtland HW, Cannata D, Sun H, Beth-On M, Liu C, Jasper H, Domene H, Karabatas L, Guida C, Basta-Pljakic J, Cardoso L, Rosen CJ, Frystyk J, Yakar S. Unbound (bioavailable) IGF1 enhances somatic growth. Dis Model Mech. 2011;4:649–658. doi: 10.1242/dmm.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]