

Abstract
Bypassing cellular senescence and becoming immortal is a prerequisite step in the tumorigenic transformation of a cell. It has long been known that loss of a key tumor suppressor gene, such as p53, is necessary, but not sufficient, for spontaneous cellular immortalization. Therefore, there must be additional mutations and/or epigenetic alterations required for immortalization to occur. Early work on these processes included somatic cell genetic studies to estimate the number of senescence genes, and microcell-mediated transfer of chromosomes into immortalized cells to identify putative senescence-inducing genetic loci. These principal studies laid the foundation for the field of senescence/immortalization, but were labor intensive and the results were somewhat limited. The advent of gene expression profiling and bioinformatics analysis greatly facilitated the identification of genes and pathways that regulate cellular senescence/immortalization. In this review, we present the findings of several gene expression profiling studies and supporting functional data, where available. We identified universal genes regulating senescence/immortalization and found that the key regulator genes represented six pathways: the cell cycle pRB/p53, cytoskeletal, interferon-related, insulin growth factor-related, MAP kinase and oxidative stress pathway. The identification of the genes and pathways regulating senescence/immortalization could provide novel molecular targets for the treatment and/or prevention of cancer.
Keywords: immortalization, senescence, cell cycle, interferon, cytoskeletal, IGF-related genes
Introduction
One of the critical steps in human carcinogenesis is cellular immortalization, a process in which cells must escape senescence and acquire an infinite lifespan. In the absence of immortalization although a cell might undergo malignant transformation it could not proliferate indefinitely. Frequently, the loss of tumor-suppressive genes contributes to cellular immortalization. One approach to understanding senescence and immortalization is comprehensive gene expression profiling of cells prior to and after their acquisition of an infinite lifespan. Although there are several studies that employ rodent cells as models for identifying candidate genes in replicative senescence (Benvenuti et al., 2002a, b), we have chosen not to include these studies in this review because of the differences between rodent and human cell immortalization. The differences between the mechanisms of human and mouse cell immortalization have been reviewed in Itahana et al. (2004).
A comparison of gene expression profiling studies is limited by the lack of overlap among the available genes on the various experimental platforms. Large profiling studies are only a tool for identifying potential genes that regulate a particular process, in the case of this review, immortalization and senescence. Researchers conducting these large-scale studies must be cognizant of such pitfalls including the potential lack of universality of the results, whether the gene expression change is primary or a consequence of other gene changes, and the potential for the identification of unrelated genes that coincidently respond to the same regulatory effectors (Chuaqui et al., 2002; Quackenbush, 2003). Our goal was not only to present data from several immortalization and senescence studies but also to determine whether there were gene(s) and/or pathway(s) that are regulated across cell models of senescence and immortalization. Furthermore, where available, we show supporting functional studies for genes identified as effectors of senescence and/or immortalization.
Replicative senescence
Normal somatic cells grown in culture cease to proliferate, senesce, after a finite number of divisions. This phenomenon, first described by Hayflick (1965), is referred to as replicative senescence or mortality stage 1 (M1) (Dimri et al., 1995; Coates, 2002; Cong et al., 2002). Apparent senescence can also be induced with chemicals, by the overexpression of tumor suppressor genes or with oncogenes, and is referred to as premature or induced senescence (Drayton and Peters, 2002). The validation for the involvement of a gene in cellular senescence is whether it is regulated during naturally occurring senescence of cells aging in culture. Senescent cells generally have a large, flattened morphology (Berube et al., 1998). These cells are growth arrested in G1 phase of the cell cycle, are incapable of synthesizing DNA and are unresponsive to stimulation with growth factors; yet interestingly, are still metabolically active (Berube et al., 1998; Lundberg et al., 2000). Senescent cells can be distinguished from pre-senescent, immortal, quiescent or terminally differentiated cells by histochemical detection of the biomarker β-galactosidase (SA β-gal) at pH 6 (Dimri et al., 1995). Other markers of cellular senescence include p16INK4a, p 21CIP1/WAF1, PAI-1, phosphorylated H2AX, activated CHK1 and CHK2 (Suzuki et al., 2001; d’Adda di Fagagna et al., 2003).
Cell types that are capable of entering replicative senescence include fibroblasts, epidermal keratinocytes, vascular smooth muscle cells, lens epithelial cells, glial cells, endothelial cells, melanocytes, T lymphocytes and adrenocortical cells (Cristofalo and Pignolo, 1993; Berube et al., 1998). Although there are similarities among the cell types in the senescent phenotype, the process by which a cell senesces is likely to have features that are cell-type specific.
Replicative senescence: fibroblast cells
In normal human fibroblast cells, telomeres become progressively shorter with each population doubling until they reach a critically short length (Cristofalo and Pignolo, 1993; Cong et al., 2002). The progressive shortening of telomeres in normal somatic cells is more prominent than in germ cells, and to a lesser extent stem cells, where the telomeres are maintained by telomerase (Cong et al., 2002). Critically, short telomeres may be one of the signals that can induce senescence or M1. Inactivation of p53, pRB/p16INK4a or another key proliferative checkpoint gene can extend the lifespan of a cell beyond M1. The telomeres of these checkpoint-defective cells continue to shorten and eventually the cells enter a stage called crisis, or M2. Only a rare cell in such populations is able to survive the widespread cell death characterized by M2. This rare cell escapes crisis and becomes immortalized through telomere stabilization either by telomerase activation or alternate lengthening of telomeres (Bischoff et al., 1990; Kim et al., 1994; Bodnar et al., 1998; Gollahon et al., 1998; Cong et al., 2002). Ectopic expression of the catalytic unit of telomerase, hTERT, can stabilize telomere length allowing cells to grow indefinitely (Bodnar et al., 1998).
Replicative senescence: epithelial cells
Epithelial cells have two stages of senescence. The first stage, termed M0, is a transient growth plateau that occurs after only a few cell divisions (Romanov et al., 2001; Yaswen and Stampfer, 2002; Stampfer and Yaswen, 2003; Zhang et al., 2003). Arrest of epithelial cells in M0 is associated with an increase in p16INK4a protein expression, but it is not a consequence of telomere shortening (Romanov et al., 2001; Yaswen and Stampfer, 2002; Stampfer and Yaswen, 2003; Zhang et al., 2003). In spite of there being no association of telomere shortening with M0-arrested epithelial cells, some consider this stage the most analogous to M1-senesced fibroblasts (Romanov et al., 2001). Following inactivation of p16INK4a, and in some cell types the inactivation of p53, epithelial cells are able to emerge from M0 and continue to proliferate for approximately another 20–70 population doublings before the second stage of growth arrest, termed M1 or agonoescence (Stampfer and Yaswen, 2003). Epithelial cell M1 growth arrest is associated with telomere shortening (Yaswen and Stampfer, 2002; Stampfer and Yaswen, 2003; Zhang et al., 2003).
Comparison of senescence in fibroblast and epithelial cells
There are several differences between M1 senescent fibroblasts, M0-arrested epithelial cells and M1-arrested epithelial cells (Romanov et al., 2001). M1-arrested epithelial cells do not express p16INK4a, whereas p16INK4a is expressed in M0 epithelial cells and M1 fibroblasts (Yaswen and Stampfer, 2002; Stampfer and Yaswen, 2003). There are a higher percentage of M1 epithelial cells stained for Annexin-V, a marker of apoptosis, than in M0-arrested epithelial cells or M1-arrested fibroblasts (Romanov et al., 2001). For more in depth information on senescence of fibroblast and epithelial cells, we refer readers to reviews by Cristofalo and Pignolo (1993) and Yaswen and Stampfer (2002), and to a study that compares senescence of isogenic sets of human mammary fibroblasts and epithelial cells (Romanov et al., 2001).
Correlation of senescence with age of a cell and with lifespan of an organism
It has been proposed that senescence reflects aging of cells, but this is controversial (Rubin, 2002). This theory is supported by evidence that shows a percentage of all cell cultures contain senescent cells and this percentage increases as the cell cultures age (Smith and Pereira-Smith, 1996; Campisi et al., 2001). For human fibroblasts, there is evidence from several studies of an inverse relationship between replicative potential and the age of the cell donor (Rohme, 1981; Hayflick, 1985; Smith and Pereira-Smith, 1996). There is also evidence suggesting that there is a correlation between the average life expectancy of a species and the number of doublings that cells from the organism undergo before senescing in culture (Rohme, 1981; Hayflick, 1985; Campisi et al., 2001). For example, cultured fibroblasts from a mouse, which has a maximal lifespan of 2 years, reach growth crisis after an average of nine population doublings, whereas fibroblasts from a human reach crisis after an average of 61 population doublings (Rohme, 1981). However, there are many who are skeptical of the correlation between lifespan of an organism and senescence. In part, this is because there are many exceptions to this correlation (Rubin, 2002).
Mechanisms by which cells escape senescence
Because cellular senescence is growth suppressive, it would seem that the accumulation of senescent cells during aging would result in a decrease in the incidence of cancer (Krtolica et al., 2001), yet we know that there is an increased incidence of cancer with age. This might be reconciled by the fact that there is also an accumulation of mutations with aging that can confer selective growth advantages to the cell and therefore mutations in key regulatory genes can result in bypassing senescence. Consequently, senescence is thought to be a tumor-suppressive mechanism. However, there is also evidence from a mouse model that suggests senescent fibroblasts can actually stimulate growth of immortalized and malignant epithelial cells (Krtolica et al., 2001). Tumors, whether naturally occurring or experimentally induced, have at the very least, an extended lifespan and are usually replicatively immortal.
Cancer cells that have escaped replicative senescence are immortal, capable of growing indefinitely. There are several mechanisms that contribute to a cell being able to escape senescence and become immortal, including genomic instability, telomere length stabilization, epigenetic gene silencing by selective promoter methylation, oxidative DNA damage, inactivation of cell cycle regulatory genes such as p16INK4a, p53, pRB or p21CIP1/WAF1, overexpression of a cellular oncogenic protein such as c-MYC or Bmi-1, or through expression of viral oncogenes (Berube et al., 1998; Bringold and Serrano, 2000; Lundberg et al., 2000; Neumeister et al., 2002; Itahana et al., 2003). Shortening of telomeres is another mechanism associated with replicative senescence. Telomeres are the tandem GT-rich repeats (TTAGGG) at the ends of chromosomes. Among their many roles telomeres act as ‘molecular clocks,’ determining the lifespan of cells (Cong et al., 2002; Harley, 2002). Telomeres become successively shorter, by 50–200 bp, with each round of replication. In most cases, maintenance of telomere length is required for immortalization. Telomere length is maintained through reactivation of telomerase or by the alternate lengthening of telomere pathway. Cellular senescence, apoptosis and genomic instability are the consequences resulting from dysfunctional telomeres (Campisi et al., 2001).
Methylation of CpG islands in gene promoters
There are several normal cellular processes that are regulated in part by epigenetic modification through DNA methylation, including developmental imprinting, X-chromosome inactivation and tissue-specific gene expression. In addition, aberrant promoter methylation has been found in growth-suppressive genes in human tumorigenesis (Baylin and Herman, 2000; Baylin et al., 2001; Esteller et al., 2001; Feinberg et al., 2002). CpG dinucleotides are typically methylated. In contrast CpG islands, which are CpG rich stretches of DNA ranging from 200 to 2000 bp in length in the regulatory regions of genes, are generally unmethylated. The transfer of a methyl group from S-adenosyl-L-methionine to the cytosines in CpG sites is catalysed by DNA-methyltransferases (DNMT) (Lopatina et al., 2002). There are three well-characterized DNMTs, DNMT1, DNMT3A and DNMT3B. DNMT1, the most abundant DNMT, is mainly responsible for maintenance methylation, whereas DNMT3a and DNMT3b are responsible for de novo methylation (Lopatina et al., 2002).
In human cancers, the silencing of tumor suppressor genes through aberrant DNA methylation of a CpG island(s) in the promoters in these genes is a common epigenetic change (Baylin and Herman, 2000). There are an assortment of pathways from which genes have been shown to be hypermethylated in cancer cells, including DNA repair, cell cycle control, invasion and metastasis. The tumor suppressor genes BRCA1, p16INK4a, p15INK4b, p14ARF, p73 and APC are among those that are silenced by hypermethylation, although the frequency of aberrant methylation is somewhat tumor-type specific (Esteller et al., 2001).
Aberrant hypermethylation of DNA can be reversed with chemical agents that inhibit DNMTs, which in effect ‘demethylate’ DNA. A commonly used inhibitor of DNMT is 5-aza-deoxycytidine (5-aza-dC), a cytosine analog. 5-aza-dC and related drugs work by substituting for cytosine during replication. DNMTs recognize and covalently bind 5-aza-dC in DNA. The covalently bound DNMT1 is unable to catalyse the transfer of methyl groups to the cytosine analog because the substituted nitrogen base cannot be methylated. Consequently, DNMT1 is depleted following several rounds of replication. This in turn results in DNA hypomethylation and the expression of genes that were silenced by methylation (Haaf, 1995; Kanai et al., 2001; Takebayashi et al., 2001).
Methylation of CpG Islands, a preneoplastic event
Vogt et al. (1998) found that when spontaneously immortalized fibroblasts with germline p53 mutations (from Li-Fraumeni patients (LFS)) were treated with 5-aza-dC, they growth arrested and senesced. They concluded that methylation of CpG islands may contribute to the spontaneous immortalization of LFS cells. Treatment with 5-aza-dC was found to restore the expression of genes such as p16INK4a and p21CIP1/WAF1 that were presumably silenced by aberrant DNA methylation (Vogt et al., 1998). We used 5-aza-dC as a tool to identify genes epigenetically silenced during immortalization. Our findings were the first to demonstrate pathway-specific epigenetic changes as a preneoplastic event (Kulaeva et al., 2003; Fridman et al., 2006).
Complementation groups of immortal cells
It is generally believed that many genes and pathways are involved in senescence. Losing the wild-type p53 allele in LFS human fibroblasts is necessary, but not sufficient, for spontaneous cellular immortalization as p53-deficient cells do senesce; however, the absence of p53 does extend their lifespan (Bischoff et al., 1990, 1991; Harvey et al., 1993; Tsutsui et al., 1997). The loss of p53 and the acquisition of the immortal phenotype can be accelerated by the treatment of p53+/− LFS fibroblasts with the chemical carcinogen aflatoxin B1 (Tsutsui et al., 1995). Because the loss of the wild-type p53 allele is insufficient to cause immortalization (Harvey et al., 1993), there must be additional mutations and/or epigenetic alterations necessary for immortalization to occur. Somatic cell genetics complementation studies were used to estimate the number of senescence genes. In somatic cell hybrids of mortal and immortal cells, the replicative senescence phenotype is dominant over the immortal phenotype (Smith and Pereira-Smith, 1996; Berube et al., 1998). Consequently, when two unrelated immortal cell lines with defects in different genes are fused together, the hybrid cells senesce and it can be concluded that they belong to different complementation groups (Smith and Pereira-Smith, 1996). To date four immortalization complementation groups have been identified to which 40 immortal human cell lines were assigned (Smith and Pereira-Smith, 1996; Berube et al., 1998). This suggests that there are at least four senescence genes or gene pathways that must be abrogated to achieve cellular immortalization (Smith and Pereira-Smith, 1996; Berube et al., 1998).
Chromosomes associated with a senescence-like phenotype
Microcell-mediated transfer of chromosomes into immortalized cells was used to identify putative senescence genes associated with the complementation groups (Smith and Pereira-Smith, 1996; Berube et al., 1998). Using this method, several chromosomes were identified as potentially encoding senescent genes, including chromosomes 1, 2, 3, 4, 6, 7, 10, 11, 16, 17, 18 and X (Smith and Pereira-Smith, 1996; Berube et al., 1998; Tominaga et al., 2002). Of these, only three chromosomes have been assigned to a complementation group: chromosome 4 to complementation group B, chromosome 1 to complementation group C, and chromosome 7 to complementation group D (Smith and Pereira-Smith, 1996). Complementation group A, thus far, does not have a chromosome associated with it, although some studies indicate that it may be chromosome 6 (Sandhu et al., 1994; Berube et al., 1998). A chromosome was assigned to a complementation group if it induced senescence in several cell lines within a complementation group, but did not induce senescence in cell lines from the other complementation groups (Smith and Pereira-Smith, 1996). Mapping known key senescence and immortalization regulatory genes to chromosomes does not reveal any gene clustering or pattern (MA Tainsky lab, unpublished data). However, we identified several clusters, defined as three or more genes within a distance of 2.5Mb, of coregulated genes on chromosome 1q that were commonly regulated in four independent immortal LFS cell lines (AL Fridman and MA Tainsky, unpublished data).
Senescence genes and pathways
Cellular senescence pathways are believed to have multiple layers of regulation with additional redundancy built into these layers (Smith and Pereira-Smith, 1996). On the basis of the complementation studies there are at least four senescence genes or pathways. There are, however, many more chromosomes that can induce senescence than there are senescence complementation groups. Furthermore, there are some immortal cell lines that have been assigned to multiple complementation groups (Duncan et al., 1993; Berry et al., 1994). This indicates that in any one immortal cell line there are probably multiple senescence genes/pathways that are abrogated (Sasaki et al., 1994; Vojta et al., 1996). Many of the functional studies, where a putative senescence gene is overexpressed in cells, indicate that although multiple genes/pathways may be abrogated in a particular cell line, as little as one gene/pathway is required for repair and subsequent reversion to senescence.
Two candidate regions on chromosome 1, 1q25 and 1q41–42 were identified as potentially encoding senescence genes (Berube et al., 1998). To date, the specific senescence gene on chromosome 1 has not been identified. On chromosome 6 the region 6q13–6q21 is believed to contain potential senescence genes as transfection of this region into ovarian tumor cells induced senescence (Morelli et al., 2000). The region containing the senescence gene was further narrowed down to fragile site FRA6F located at 6q21, but the gene in this region responsible for senescence has not yet been identified (Morelli et al., 2002). MORF4 is a senescence gene identified on chromosome 4 (Berube et al., 1998; Bertram et al., 1999) and complements cell lines in complementation group B.
Genes that have been shown to induce a senescence-like phenotype, or at the very least inhibit cell growth in tumor cells, include p14ARF (Dimri et al., 2000; Sekaric et al., 2007), E2F-1 (Dimri et al., 2000), IGFBP3 (Fridman et al., 2007), IGFBPrP1 (Wilson et al., 2002; Fridman et al., 2007), PAI-1 (Kortlever et al., 2006), MKK3 (Wang et al., 2002), MKK6 (Haq et al., 2002; Wang et al., 2002), Smurf2 (Zhang and Cohen, 2004) and HIC-5 (Shibanuma et al., 1997). Other genes that can induce a senescence-like phenotype are reviewed in Bringold and Serrano (2000), Lundberg et al. (2000) and Roninson (2003) and include p53, p63, p73, pRB, p16INK4a, p21CIP1/WAF1, p15INK4b, p57KIP2, RAF-1, E2 papillomavirus protein (inhibitor of E6 and E7), oncogenic ras and hTERT. Pathways known to regulate cellular senescence/immortalization, including the p16INK4a/pRB pathway, the p19ARF/p53/p21CIP1/WAF1 pathway, and the PTEN/p27KIP1 pathway are reviewed in Berube et al. (1998), Bringold and Serrano (2000), Lundberg et al. (2000) and Campisi (2001).
Genomic approaches to identify senescence/immortalization genes and pathways
There are a variety of model systems that have been used to identify and study senescence/immortalization genes and pathways. Normal cells grown in vitro senesce after a finite number of divisions (Hayflick, 1965) and such model systems are highly suitable for studying genes that are regulated during senescence. Cells that are derived from patients with Werner syndrome, a disease typified by premature aging and shortened lifespan, provide useful genetic variant systems for the study senescence as well as aging. Alternatively, senescence model systems have also employed immortalized cells that are chemically induced to senesce. A cellular senescence-like phenotype can be induced in immortalized cells that are treated with reagents such as 5-aza-dC, H2O2 or 5-bromodeoxyuridine (BrdU) (Chen et al., 1995; Vogt et al., 1998; Suzuki et al., 2001; Kulaeva et al., 2003; Fridman et al., 2006). Although not the focus of this review there are in vivo mouse models for studying senescence and aging (Sommer et al., 2006).
In vitro immortalization models have been developed using cells immortalized chemically, virally or with a biological agent. Mortal cells can be immortalized artificially by stabilizing the telomeres by overexpression of hTERT (Bodnar et al., 1998), through the addition of a chemical mutagen such as aflatoxin B1 (Tsutsui et al., 1995), or by transduction with viral oncogenes such as SV40 T-antigen, adenovirus E1a/E1B and HPV16-E6 or -E7 (Jha et al., 1998; Garbe et al., 1999; Schwarze et al., 2002; Boulet et al., 2007). These viral oncogenes act by inhibiting key genes such as the tumor suppressor gene proteins p53 and pRB, thus allowing the cell to bypass senescence and become immortal (Jha et al., 1998; Boulet et al., 2007). Alternatively, at a low but finite frequency spontaneous immortalization occurs in cells with a germline mutation such as p53+/− fibroblasts or epithelial cells derived from a patient with LFS (Bischoff et al., 1990; Rogan et al., 1995; Shay et al., 1995; Gollahon et al., 1998), or as in the case of APC+/− fibroblasts from a patient with familial adenomatous polyposis (Forsyth et al., 2004). Using fibroblasts from a patient with LFS, our lab was the first to spontaneously immortalize a human fibroblast cell line in vitro (Bischoff et al., 1990). As is typical for LFS patients, the fibroblasts had a germline p53 tumor suppressor gene mutation (Varley et al., 1997; Bachinski et al., 2005) that results in significant genomic instability. Consequently, low-passage precrisis LFS cells with genomic instability immortalize without intervention, thereby providing an ideal model of spontaneous cellular immortalization. The disadvantage of inducing immortalization with viral or chemical agents is that gene expression changes may be altered beyond those relevant to immortalization. However, because human cells have only been shown to spontaneously immortalize in culture using strains with certain pre-existing germline mutations (p53 and APC), all other human cell systems require pretreating cells with a chemical or by transducing them with a viral oncogenes to achieve cellular immortalization.
Immortalization is an essential step in the tumorigenic transformation of a cell, yet it can be reversed hence halting the tumorigenic transformation of a cell. Immortalization can be reversed and cellular senescence induced by reexpression of a key senescence gene that was abrogated as a consequence of mutation, infection with a viral oncogene or suppressed by epigenetic silencing. Furthermore, a cellular senescence-like phenotype can be induced in immortalized LFS cells as well as many tumor cell lines when they are treated with the DNMT inhibitor 5-aza-dC, further indicating that the immortal phenotype of cells is reversible (Vogt et al., 1998; Karpf and Jones, 2002; Kulaeva et al., 2003; Paz et al., 2003; Fridman et al., 2006). Therefore, understanding the process by which cells bypass senescence and become immortal could provide novel molecular targets for the treatment of preneoplastic lesions and/or prevention of cancer.
Senescence and immortalization model systems are essential for elucidating the genes and pathways involved in cellular senescence/immortalization. Recently model systems have been studied using functional genomics approaches, including differential proteomics, serial analysis of gene expression (SAGE) and microarrays, thus allowing researchers to identify relevant genes and pathways involved in senescence/immortalization. The findings of these studies, which are summarized in Supplemental Tables 1 and 2, are the focus of this review. The genomic approaches in combination with traditional methods including overexpression of putative senescence or immortalization genes has led to the discovery of six key distinct, yet interacting, senescence/immortalization pathways, including the cell cycle pathway, cytoskeletal genes, interferon (IFN) pathway, insulin growth factor (IGF)-related pathway genes, MAP kinase pathway and oxidative stress pathway (Figure 1).
Figure 1.
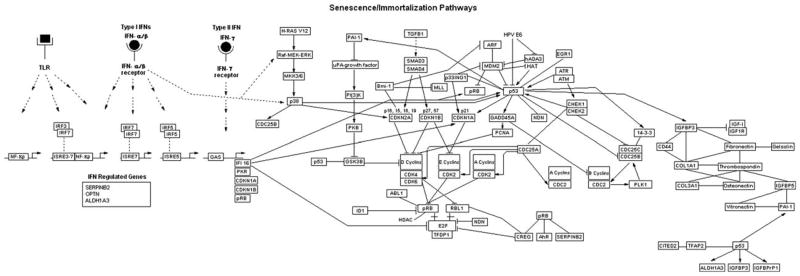
Senescence/immortalization pathways. This figure was drawn using GenMAPP version 2 (http://www.genmapp.org/; Salomonis et al. (2007)). Pathway and gene interactions were derived from literature referenced in this paper and the supplemental section of this paper, GenMAPP, KEGG Pathway (Kanehisa and Goto, 2000), NCBI Entrez Gene (http://www.ncbi.nlm.nih.gov/sites/entrez), InvivoGen (http://www.invivogen.com/docs/Insight200509.pdf), Schroder et al. (2004), and Takaoka and Yanai (2006).
SAGE was used to characterize genes differentially expressed during the senescence of prostate epithelial cells (Untergasser et al., 2002). Untergasser et al. (2002) found 157 mRNA (70 known genes) that were upregulated and 116 mRNA (65 known genes) that were repressed during senescence. Among these genes, 70 upregulated and 65 repressed genes were classified as functioning in the cell cycle, IFN-related, extracellular matrix or cytoskeletal pathways.
cDNA microarrays were used to identify genes in human prostate epithelial cells (HPECs) upregulated in senescence and repressed during the immortalization of the cells. Three genes, DOC1, BRAK and IGFBP3, were identified that were decreased in immortalized cells and upregulated in senescent cells (Schwarze et al., 2002). The identification of these genes is consistent with the involvement of the IFN, cytoskeletal, cell cycle and IGF pathway in immortalization; however, these genes did not overlap with those identified with expression changes in the study of prostate epithelial cells (Untergasser et al., 2002).
We used precrisis fibroblast LFS cell lines and spontaneously immortalized LFS fibroblast cell lines to investigate the nature of the genetic changes that might have given these cell lines the prerequisite growth advantage to become immortal (Kulaeva et al., 2003; Fridman et al., 2006). A cellular senescence-like phenotype can be induced in immortalized LFS cells that are treated with the DNMT inhibitor 5-aza-dC, thus reversing the immortal phenotype of these cells. On the basis of this observation, we hypothesized that genes epigenetically silenced by methylation are putative key regulatory cellular senescence genes. Consistent with previous studies, we found that the loss of checkpoint proteins such as p16INK4a, p 21CIP1/WAF1 and p53 led to immortalization-initiating events including genomic instability and telomere stabilization (Fridman et al., 2006). Using four independently derived immortal LFS cell lines, our analysis identified 149 upregulated genes and 187 downregulated genes after immortalization, 14 of which were epigenetically downregulated. Pathway analysis of the expression data with gene ontology bioinformatics software revealed a statistically significant contribution of IFN-related, cell cycle and cytoskeletal genes in the process of immortalization (Kulaeva et al., 2003; Fridman et al., 2006). In total, 12 of the 14 genes that are epigenetically regulated in all four immortal LFS cell lines have a known function and 8 of these genes could be categorized in one of the six senescence/immortalization pathways.
Cell Cycle
The cell cycle has long been known as one of the pathways that regulates senescence, in particular through pRB and p53. In our study of the spontaneously immortalized LFS cells, in addition to the genes known to be involved in the regulation of cellular senescence/immortalization (p16INK4a, p21CIP1/WAF1 and p53), we identified other cell cycle regulators among the 14 epigenetically regulated genes that were commonly regulated in all four immortal LFS cell lines. One of the 14 genes, HTATIP2, is categorized in the gene ontology cell cycle category. Two genes, IGFBPrP1 and ALDH1A3, are regulated by the cell cycle-regulating gene p53, and two genes, CREG and SERPINB2, have been shown to physically associate with the cell cycle regulator pRB. Our identification of these genes as epigenetically regulated in senescence/immortalization is supported by research from other laboratories. HTATIP2 is upregulated in senescent human mammary epithelial cells (HMECs) (Zhang et al., 2003). Overexpression of CREG in human teratocarcinoma NTERA-2 cells delays G1/S transition and inhibits growth (Di Bacco and Gill, 2003). SERPINB2 is one of only three genes that was upregulated in human HMEC during senescence on feeder layers, in HMEC during senescence on plastic and in senescent human fibroblasts (Zhang et al., 2003, 2004). Expression of SERPINB2 is also upregulated in the bladder tumor cell line T24 following 5-aza-dC treatment (Liang et al., 2002), as well as upregulated in senescent BJ fibroblast cells (a human dermal fibroblast cell line) (Linskens et al., 1995). In addition, expression of the p53-downregulated gene CDC25B is induced after immortalization and then following 5-aza-dC treatment its expression is repressed (MA Tainsky lab, unpublished data). CDC25B was also increased in immortalized HMEC, but not prestasis or postselection HMEC (Li et al., 2007).
The cell cycle protein inhibitor of growth 1 (p33ING1), located on chromosome 13q34, was identified as a senescence-inducing gene (Garkavtsev and Riabowol, 1997; Berube et al., 1998; Goeman et al., 2005). Expression of this tumor suppressor gene is increased in senescent human diploid fibroblasts, and when expression of it is blocked in pre-senescent cells the life span of the cells increases (Garkavtsev and Riabowol, 1997). Furthermore, ectopic expression of p33ING1 in IMR90 fibroblasts induces senescence (Goeman et al., 2005). There is a functional connection of p33ING1 with oncogenic ras (Goeman et al., 2005) and p14ARF (Gonzalez et al., 2006), which are both capable of inducing senescence. p33ING1 has also been shown to bind p53 (Garkavtsev et al., 1998; Leung et al., 2002). p33ING1 protein leads to the stabilization of p53 through disruption of the p53–MDM2 interaction (Leung et al., 2002). Additionally, the interaction of p33ING1 with p53 induces transcription of p21CIP1/WAF1 (Garkavtsev et al., 1998).
PAI-1, a downstream target of p53 (Kortlever et al., 2006), is among the genes that were overexpressed in senescent Werner syndrome fibroblasts (Murano et al., 1991; Lecka-Czernik et al., 1996). PAI-1 was also upregulated in senescent BJ fibroblasts (Shelton et al., 1999), senescent TIG-7 (a human embryonic lung fibroblast cell line) (Suzuki et al., 2001), in HeLa cells senesced with BrdU (Suzuki et al., 2001), and in senescent prostate epithelial cells (Untergasser et al., 2002). PAI-1 decreases in immortalized HMEC when compared with prestasis HMEC (Li et al., 2007). Functional studies of PAI-1 in mouse embryonic fibroblasts and human BJ fibroblasts show that PAI-1 is necessary and sufficient for senescence of these fibroblasts (Kortlever et al., 2006). PAI-1 and its connection to cytoskeletal genes are discussed below. The role of PAI-1 in senescence has been recently reviewed in Kortlever and Bernards (2006).
Cytoskeletal genes
Given the distinct morphological transition as cells transition from proliferation to senescence, it was no surprise that genes associated with the cytoskeleton have been found to be altered as cells senesce. Supporting evidence for the involvement of cytoskeletal genes in senescence comes from several laboratories that have shown an increase in expression of cytoskeletal genes, such as vimentin and fibronectin, in senescent human fibroblasts (Murano et al., 1991; Satoh et al., 1994; Kaneko et al., 1995). Among the genes that were overexpressed in senescent Werner syndrome fibroblasts, several are cytoskeletal related including fibronectin, PAI-1 and thrombospondin (Murano et al., 1991; Lecka-Czernik et al., 1996). Fibronectin was also upregulated in senescent human vascular endothelial cells (Shelton et al., 1999), senescent TIG-7 (Suzuki et al., 2001), HeLa cells senesced with BrdU (Suzuki et al., 2001), senescent prostate epithelial cells (Untergasser et al., 2002), senescent human oral keratinocytes (Kang et al., 2003) and senescent HMECs (Zhang et al., 2003). PAI-1 expression is induced in anchorage-dependent cells, but was not detectable in an anchorage-independent cell line (Lee et al., 2002) Fibronectin- and vitronectin-induced expression of PAI-1 (Lee et al., 2002). PAI-1 inhibits cleavage of focal adhesions, thus further connecting PAI-1 to senescence (Sorrell et al., 2006). DOC1 was identified by Schwarze et al. (2002) as upregulated in senescence and repressed during the immortalization of HPECs, and identified by Zhang et al. (2003) as upregulated in senescent fibroblasts. DOC1 is homologous to a mouse cytoskeleton-associated protein (Tandle et al., 2005). In our LFS study, there were a statistically significant number of cytoskeletal genes that were silenced during immortalization, including 2 of the 14 genes that were regulated across four LFS cell lines, HPS5 and MAP1LC3B. In their proteomic analysis of senescent rat fibroblasts, Benvenuti et al. (2002b) identified several cytoskeletal proteins upregulated during senescence.
IFN pathway
Probably the most unanticipated pathway found to be differentially regulated in senescence and immortalization was the IFN pathway. In our initial gene expression profiling on oligonucleotide microarrays where we compared gene expression profiles of precrisis with immortal LFS cells, and untreated with 5-aza-dC-treated LFS immortal cells, we demonstrated treatment of MDAH041 cells with 5-aza-dC induced senescence and resulted in the upregulation of 85 genes that appeared to be epigenetically silenced after immortalization (Kulaeva et al., 2003). Of these genes, there was a statistically significant portion that was linked with the IFN signaling pathway (39 out of 85) (Kulaeva et al., 2003). In an expanded study using four independent immortal LFS cell lines (Fridman et al., 2006), we confirmed our initial findings that the IFN pathway plays a significant role in immortalization. Furthermore, of the 14 genes that were epigenetically regulated in four LFS cell lines we found that 3 were regulated by IFN: ALDH1A3, OPTN and SERPINB2 (Fridman et al., 2006). Likewise, IFN-regulated genes were also found to be downregulated in tumorigenic benign prostatic hyperplasia cells (Shou et al., 2002), upregulated in senescent prostate epithelial cells (Untergasser et al., 2002) and upregulated in senescent human diploid fibroblasts (Yoon et al., 2004). In addition, IFN-regulated genes were sensitive to demethylation treatment in the bladder tumor cell line T24 following 5-aza-dC treatment (54% of upregulated genes) (Liang et al., 2002). BRAK, a chemokine that when over-expressed suppresses tumor growth (Ozawa et al., 2006), was identified as upregulated in senescence and repressed during the immortalization of HPEC (Schwarze et al., 2002). Treatment with IFN-γ reduced the tumorigenic potential of the benign prostatic hyperplasia cells (Shou et al., 2002). Expression of IFN-γ is increased in senescent BJ fibroblasts (Linskens et al., 1995). Furthermore, young and senescent cells respond differently to treatment with IFN-γ (Stratford et al., 2006), yet further proof that the IFN pathway plays a significant role in cellular senescence. We also find overexpression of either IRF-5 or IRF-7 is sufficient for inducing cellular senescence in LFS cells (Li et al., 2008). These data strongly corroborate the involvement of the IFN pathway in senescence/immortalization.
IGF pathway genes
The insulin-like growth factors, their receptors and their binding proteins have frequently been found to be altered in their expression during immortalization and complete carcinogenesis. IGFBP3 was overexpressed in senescent fibroblasts from a patient with Werner syndrome (Goldstein et al., 1991; Murano et al., 1991), in BrdU-senesced HeLa cells (Suzuki et al., 2001), in senescent TIG-7 fibroblasts (Suzuki et al., 2001), in senescent human oral keratinocytes (Kang et al., 2003), in senescent human diploid fibroblasts (Yoon et al., 2004) and in prestasis HMEC when compared with fully immortalized HMEC (Li et al., 2007). Consistent with these studies, IGFPB3 was identified by Schwarze et al. (2002) as upregulated in senescence and repressed during the immortalization of HPEC. We found that expression of IGFBP3 transcript and protein expression decreased during immortalization of LFS fibroblasts (Fridman et al., 2006), which further supported the involvement of IGFBP3 in this process. Functional studies have demonstrated that overexpression of this gene in immortalized LFS cell lines suppressed cell growth and inhibited colony formation (Fridman et al., 2007).
IGFBPrP1 was upregulated in senescent epithelial cells and senescent fibroblasts (Zhang et al., 2003), in HMECs (Swisshelm et al., 1995) and in HPECs (Lopez-Bermejo et al., 2000). Immortalization of LFS cells results in the repression of IGFBPrP1, and the epigenetic regulation of this gene was demonstrated by its upregulation by 5-aza-dC treatment (Fridman et al., 2006). IGFBPrP1 is upregulated in senescing normal fibroblasts and senescing precrisis LFS fibroblasts (Fridman et al., 2007). When IGFBPrP1 is expressed in LFS cells it inhibits both growth and colony formation (Fridman et al., 2007). Expression of IGFBPrP1 in MCF-7 cells induces a senescence-like state (Swisshelm et al., 1995; Wilson et al., 2002).
Other IGFBP genes, including IGFBP2, IGFBP4, IGFBP5, IGFBP6 and IGFBPrP2, have also been shown to be regulated during senescence/immortalization. IGFPB2 was upregulated in senescent cells, including BJ fibroblasts (Shelton et al., 1999), human retinal pigment epithelial cells (Shelton et al., 1999) and HMECs (Zhang et al., 2003). IGFBP6 mRNA expression increased in the senescent colon carcinoma cell line HCT116 (Chang et al., 2002). IGFBPrP2 expression increased in senescent HPECs (Lopez-Bermejo et al., 2000). IGFBP4 and IGFBP5 decrease during immortalization of LFS cells (Fridman et al., 2006, 2007). BrdU-induced senescence of HeLa cells results in the upregulation of IGFBP4 (Suzuki et al., 2001). IGFBP5 is upregulated in senescent cells, including IMR90 (Linskens et al., 1995), BJ fibroblasts (Linskens et al., 1995; Shelton et al., 1999), human retinal pigment epithelial cells (Shelton et al., 1999), human vascular endothelial cells (Shelton et al., 1999), TIG-7 fibroblasts (Suzuki et al., 2001) and HDF isolated from foreskin (Yoon et al., 2004). IGFBP5 was shown to decrease in E7-immortalized HPEC (Schwarze et al., 2002) and in fully immortalized HMEC when compared with prestasis HMEC (Li et al., 2007). Interestingly, the senescence cell cycle gene PAI-1 has been shown to bind IGFPB5 and partially protects it from proteolysis (Nam et al., 1997). However, there is some evidence that IGFBP5 may be inhibiting PAI-1 (Sorrell et al., 2006). Significantly, premature senescence was induced when IGFBP5 was overexpressed in human umbilical vein endothelial cells (Seok Kim et al., 2007). Induction of senescence by IGFBP5 in the human umbilical vein endothelial cells was p53 dependent (Seok Kim et al., 2007). The expression data and the functional studies of genes in the IGFBP family all support these genes as at the very least involved and potentially as key regulators of senescence/immortalization.
MAP kinase pathway
The upregulation of the MAP kinase pathway in cells with normal checkpoint control often results in growth arrest, apoptosis and/or senescence The decrease in MKK3 expression during immortalization of LFS fibroblasts (Fridman et al., 2006) is supported by functional studies of this pathway. Activation of p38HOG by constitutively active MKK3 or MKK6 was found to induce a senescence-like phenotype in BJ fibroblasts (Wang et al., 2002) and U2OS, a human osteogenic sarcoma cell line (Haq et al., 2002). Experimental evidence from Wang et al. (2002) shows oncogenic ras, which was previously shown to induce senescence-like growth arrest in human fibroblasts (Serrano et al., 1997), induces activation of p38HOG via the MEK-ERK pathway. Induction of premature senescence through the oncogenic ras-Raf/MEK/ERK-p38HOG pathway results in an increase in p53 and p16INK4a protein expression. Although Wang et al., found activation of p38HOG was sufficient to cause an increase in p53 and p16INK4a protein expression in BJ fibroblasts, Haq et al. (2002) did not find p53 protein levels increased following activation of p38HOG, but there was an increase in p21CIP1/WAF1 protein expression in U2OS cells; U2OS cells do not express p16INK4a. That there are multiple known senescence genes that can be activated by p38HOG indicates that even within a senescence pathway there is redundancy.
Oxidative stress pathway
Oxidative DNA damage has long been known as a contributing factor in the senescence of human diploid fibroblasts grown in culture (Chen et al., 1995). An increase in reactive oxygen species (ROS) has been demonstrated in cellular senescence (Chen et al., 1995) with senescing and aged cells having a higher level of ROS than normal cells (Hagen et al., 1997). Treatment of human fibroblasts with H2O2 induces a replicative senescence-like phenotype (Chen et al., 1998). In normal diploid cells, ras oncogenes (Lee et al., 1999) and p21CIP/WAF1 (Macip et al., 2002) can induce senescence with increased intracellular ROS. Treatment of cells with H2O2 (Chen et al., 1998) or the induction of hyperoxia (von Zglinicki et al., 1995; Chen et al., 1998) induces senescence through telomere shortening through a mechanism relying on proper cell cycle control (Chen and Ames, 1994). H2O2-treated cells have elevated levels of p53 and p21CIP/WAF1, and hypophosphorylated pRB relative to untreated cells (Chen et al., 1998). Interestingly however, oxidative stress pathway genes were also found to be increased in hTERT and spontaneously immortalized primary breast tumors (Dairkee et al., 2007). Because ROS is involved in senescence and p53 can enhance ROS levels, the ROS/oxidative stress pathway likely plays a role in senescence. p53-regulated genes such as PIG3 and FDXR genes are involved in the response to ROS (Polyak et al., 1997). In addition, ROS alone can induce p21CIP/WAF1 expression even in the absence of p53, but to a lesser extent (Russo et al., 1995). Treatment of three p53-deficient human fibroblast strains with five different antioxidants failed to inhibit their progression toward immortalization using intermediate markers such as anchorage-independent growth or cytogenetic abnormalities, although one antioxidant, oltipraz, was significantly effective in transiently delaying a shift to hyperdiploidy in all three cell strains (Kraniak et al., 2006). Therefore, ROS inhibition alone is not sufficient to block the induction of senescence. p53 provides a critical switch in cell fate between entering apoptosis or reversible cell cycle arrest events such as G1, G0 or senescence. It is therefore no surprise that cells deficient in one or both copies of p53 are prone to spontaneous immortalization (Bischoff et al., 1990), but resistance to ROS-induced growth arrest is not sufficient to escape senescence.
Common senescence/immortalization pathways
Shelton et al. (1999) using cDNA microarrays studied replicative senescence in three cell types: dermal fibroblasts, retinal pigment epithelial cells and vascular endothelial cells. They came to the conclusion that although these cell types have similar senescence phenotypes, the genes and pathways involved vary among them. In our analysis of the spontaneously immortalized cell lines, we were able to identify common pathways that were dysregulated during immortalization despite somatic cell hybrid studies showing the LFS cell lines used were in different complementation groups. Somatic cell hybrids were made between MDAH041 (tel+, p 53−/−, N-ras-transformed) or MDAH087 (tel-,p53−/−, N-ras-transformed) and HT1080 (tel+, p 53wt, N-ras) (Gollahon et al., 1998), a fibrosarcoma cell line that had been assigned to senescence complementation groupA (Pereira-Smith and Smith, 1988). Both the MDAH041–HT1080 and the MDAH087–HT1080 hybrids were telomerase positive (Gollahon et al., 1998). The MDAH041–HT1080 hybrids senesced rapidly whereas the MDAH087–HT1080 grew indefinitely (Gollahon et al., 1998). It can be concluded that MDAH041 and HT1080 are in different complementation groups. MDAH087 is probably in the same complementation group as HT1080, complementation group A. That we were able to identify both genes and pathways that were dysregulated during immortalization that were in common to cell lines in different complementation groups suggests that it is likely there are certain senescence/immortalization pathways that are consistently involved in this process. In our review of senescence and immortalization studies, we find that across cell types and model systems consistently genes in the cell cycle pathway, cytoskeletal genes, IFN pathway, IGF pathway genes, MAP kinase pathway and oxidative stress pathway were identified as key regulators of senescence/immortalization.
Conclusion
Carcinogenesis is widely accepted to be a multistep process resulting from the accrual of mutations in tumor suppressor genes and oncogenes that confers growth advantages and/or genomic instability to the cell. One of the critical steps in this process is immortalization. Senescence is a mechanism by which cells can suppress unregulated growth by arresting cell proliferation. In contrast, cancer cells have the ability to grow indefinitely because they bypassed replicative senescence and become immortal. In the absence of immortalization, a cell is unable to undergo malignant transformation. Therefore, identification of the genes and pathways regulating the process by which cells bypass senescence and become immortal could provide novel early molecular targets for the treatment and/or prevention of cancer.
In this review, we have presented what we believe are the critical pathways in cellular senescence and immortalization based on information gathered from several gene expression profiling studies and some of these genes have functionally implicated certain cellular processes in senescence and immortalization (Figure 1 and Supplemental Table 1). There are a variety of cellular factors and mechanisms involved in bypassing senescence leading to the cellular immortalization including telomere length, genomic instability and epigenetic gene silencing due to gene methylation. The loss of growth-suppressive genes, and the ensuing cellular immortalization, can result from multiple mechanisms including chromosomal recombination, numerical changes in chromosomes, point mutations and epigenetic silencing. These changes have been considered cell origin dependent as well as dependent on their cell culture conditions (Zhang et al., 2004). However, there is considerable overlap of the genes and pathways that regulate senescence/immortalization, even across cell types, indicating at least some commonality among these regulatory pathways (Supplemental Tables 1 and 2).
In our analysis, there are several common genes regulating these processes and many of these genes that were identified as regulators of senescence/immortalization fall into six main pathways: the cell cycle pRB/p53 pathway, cytoskeletal genes, IFN pathway, IGF pathway genes, MAP kinase pathway and oxidative stress pathway. In reality, these pathways are so interwoven that it is difficult to describe them as individually involved in the transition from senescence to immortalization (Figure 1).
Our conclusion is that a comprehensive, well-controlled gene-profiling study is necessary to fully understand senescence/immortalization. Ideally, this mega study would be performed using different cell systems on a single experimental gene-profiling platform. It would necessarily require functional data on the most common senescence/immortalization regulatory genes identified to produce a universal picture of the genes and pathways that regulate senescence/immortalization. A study of this proportion may be difficult to fund, thus we propose the next step should be to extend the work in this review and compile data from gene expression profiling experiments and functional data of senescence/immortalization regulatory genes into a large database. An in-depth analysis of this database could potentially help define and identify novel senescence/immortalization genes.
Supplementary Material
Acknowledgments
This study was supported by the Barbara and Fred Erb Endowed Chair in Cancer Genetics (to MAT), and by funds from the Barbara Ann Karmanos Cancer Institute, the State of Michigan Life Sciences Corridor, Wayne State University and Michigan Center for Genomic Technologies Applied Genomics Technology Center (MEDC 085P10009816), and the Genomics and Biostatistics Cores of the Barbara Ann Karmanos Cancer Institute (P30CA022453). We thank the numerous members of the Tainsky lab who over the past 20 years have contributed to the studies of the immortalization of Li-Fraumeni cells, including Sun Yim, Farideh Bischoff, Tian-Ai Wu, Eliayhu Kraus, Sylvia Dryden, Olga Kulaeva, Lin Tang, Rita Rosati, Janice Kraniak and Qunfang Li. We acknowledge Louise C Strong whose insight into human cancer genetics led to the establishment of cell lines from Li-Fraumeni patients. We thank Dr Janice Kraniak for critical reading of the manuscript.
References
- Bachinski LL, Olufemi SE, Zhou X, Wu CC, Yip L, Shete S, et al. Genetic mapping of a third Li-Fraumeni syndrome predisposition locus to human chromosome 1q23. Cancer Res. 2005;65:427–431. [PubMed] [Google Scholar]
- Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet. 2001;10:687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- Benvenuti S, Cramer R, Bruce J, Waterfield MD, Jat PS. Identification of novel candidates for replicative senescence by functional proteomics. Oncogene. 2002a;21:4403–4413. doi: 10.1038/sj.onc.1205525. [DOI] [PubMed] [Google Scholar]
- Benvenuti S, Cramer R, Quinn CC, Bruce J, Zvelebil M, Corless S, et al. Differential proteome analysis of replicative senescence in rat embryo fibroblasts. Mol Cell Proteomics. 2002b;1:280–292. doi: 10.1074/mcp.m100028-mcp200. [DOI] [PubMed] [Google Scholar]
- Berry IJ, Burns JE, Parkinson EK. Assignment of two human epidermal squamous cell carcinomas cell lines to more than one complementation group for the immortal phenotype. Mol Carcinog. 1994;9:134–142. doi: 10.1002/mc.2940090305. [DOI] [PubMed] [Google Scholar]
- Bertram MJ, Berube NG, Hang-Swanson X, Ran Q, Leung JK, Bryce S, et al. Identification of a gene that reverses the immortal phenotype of a subset of cells and is a member of a novel family of transcription factor-like genes. Mol Cell Biol. 1999;19:1479–1485. doi: 10.1128/mcb.19.2.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berube NG, Smith JR, Pereira-Smith OM. The genetics of cellular senescence. Am J Hum Genet. 1998;62:1015–1019. doi: 10.1086/301848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff FZ, Strong LC, Yim SO, Pratt DR, Siciliano MJ, Giovanella BC, et al. Tumorigenic transformation of spontaneously immortalized fibroblasts from patients with a familial cancer syndrome. Oncogene. 1991;6:183–186. [PubMed] [Google Scholar]
- Bischoff FZ, Yim SO, Pathak S, Grant G, Siciliano MJ, Giovanella BC, et al. Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: aneuploidy and immortalization. Cancer Res. 1990;50:7979–7984. [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Boulet G, Horvath C, Broeck DV, Sahebali S, Bogers J. Human papillomavirus: E6 and E7 oncogenes. Int J Biochem Cell Biol. 2007;39:2006–2011. doi: 10.1016/j.biocel.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35:317–329. doi: 10.1016/s0531-5565(00)00083-8. [DOI] [PubMed] [Google Scholar]
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11:S27–S31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- Campisi J, Kim SH, Lim CS, Rubio M. Cellular senescence, cancer and aging: the telomere connection. Exp Gerontol. 2001;36:1619–1637. doi: 10.1016/s0531-5565(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA. 2002;99:389–394. doi: 10.1073/pnas.012602599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Ames BN. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci USA. 1994;91:4130–4134. doi: 10.1073/pnas.91.10.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci USA. 1995;92:4337–4341. doi: 10.1073/pnas.92.10.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QM, Bartholomew JC, Campisi J, Acosta M, Reagan JD, Ames BN. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: p53 and Rb control G1 arrest but not cell replication. Biochem J. 1998;332(Part 1):43–50. doi: 10.1042/bj3320043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuaqui RF, Bonner RF, Best CJ, Gillespie JW, Flaig MJ, Hewitt SM, et al. Post-analysis follow-up and validation of microarray experiments. Nat Genet. 2002;32(Suppl):509–514. doi: 10.1038/ng1034. [DOI] [PubMed] [Google Scholar]
- Coates PJ. Markers of senescence? J Pathol. 2002;196:371–373. doi: 10.1002/path.1073. [DOI] [PubMed] [Google Scholar]
- Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66:407–425. doi: 10.1128/MMBR.66.3.407-425.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofalo VJ, Pignolo RJ. Replicative senescence of human fibroblast-like cells in culture. Physiol Rev. 1993;73:617–638. doi: 10.1152/physrev.1993.73.3.617. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Dairkee SH, Nicolau M, Sayeed A, Champion S, Ji Y, Moore DH, et al. Oxidative stress pathways highlighted in tumor cell immortalization: association with breast cancer outcome. Oncogene. 2007;26:6269–6279. doi: 10.1038/sj.onc.1210452. [DOI] [PubMed] [Google Scholar]
- Di Bacco A, Gill G. The secreted glycoprotein CREG inhibits cell growth dependent on the mannose-6-phosphate/insulin-like growth factor II receptor. Oncogene. 2003;22:5436–5445. doi: 10.1038/sj.onc.1206670. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Itahana K, Acosta M, Campisi J. Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol. 2000;20:273–285. doi: 10.1128/mcb.20.1.273-285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayton S, Peters G. Immortalisation and transformation revisited. Curr Opin Genet Dev. 2002;12:98–104. doi: 10.1016/s0959-437x(01)00271-4. [DOI] [PubMed] [Google Scholar]
- Duncan EL, Whitaker NJ, Moy EL, Reddel RR. Assignment of SV40-immortalized cells to more than one complementation group for immortalization. Exp Cell Res. 1993;205:337–344. doi: 10.1006/excr.1993.1095. [DOI] [PubMed] [Google Scholar]
- Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- Feinberg AP, Cui H, Ohlsson R. DNA methylation and genomic imprinting: insights from cancer into epigenetic mechanisms. Semin Cancer Biol. 2002;12:389–398. doi: 10.1016/s1044-579x(02)00059-7. [DOI] [PubMed] [Google Scholar]
- Forsyth NR, Morales CP, Damle S, Boman B, Wright WE, Kopelovich L, et al. Spontaneous immortalization of clinically normal colon-derived fibroblasts from a familial adenomatous polyposis patient. Neoplasia. 2004;6:258–265. doi: 10.1593/neo.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman AL, Rosati R, Li Q, Tainsky MA. Epigenetic and functional analysis of IGFBP3 and IGFBPrP1 in cellular immortalization. Biochem Biophys Res Commun. 2007;357:785–791. doi: 10.1016/j.bbrc.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Fridman AL, Tang L, Kulaeva OI, Ye B, Li Q, Nahhas F, et al. Expression profiling identifies three pathways altered in cellular immortalization: interferon, cell cycle, and cytoskeleton. J Gerontol A Biol Sci Med Sci. 2006;61:879–889. doi: 10.1093/gerona/61.9.879. [DOI] [PubMed] [Google Scholar]
- Garbe J, Wong M, Wigington D, Yaswen P, Stampfer MR. Viral oncogenes accelerate conversion to immortality of cultured conditionally immortal human mammary epithelial cells. Oncogene. 1999;18:2169–2180. doi: 10.1038/sj.onc.1202523. [DOI] [PubMed] [Google Scholar]
- Garkavtsev I, Grigorian IA, Ossovskaya VS, Chernov MV, Chumakov PM, Gudkov AV. The candidate tumour suppressor p33ING1 cooperates with p53 in cell growth control. Nature. 1998;391:295–298. doi: 10.1038/34675. [DOI] [PubMed] [Google Scholar]
- Garkavtsev I, Riabowol K. Extension of the replicative life span of human diploid fibroblasts by inhibition of the p33ING1 candidate tumor suppressor. Mol Cell Biol. 1997;17:2014–2019. doi: 10.1128/mcb.17.4.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeman F, Thormeyer D, Abad M, Serrano M, Schmidt O, Palmero I, et al. Growth inhibition by the tumor suppressor p33ING1 in immortalized and primary cells: involvement of two silencing domains and effect of Ras. Mol Cell Biol. 2005;25:422–431. doi: 10.1128/MCB.25.1.422-431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S, Moerman EJ, Jones RA, Baxter RC. Insulin-like growth factor binding protein 3 accumulates to high levels in culture medium of senescent and quiescent human fibroblasts. Proc Natl Acad Sci USA. 1991;88:9680–9684. doi: 10.1073/pnas.88.21.9680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollahon LS, Kraus E, Wu TA, Yim SO, Strong LC, Shay JW, et al. Telomerase activity during spontaneous immortalization of Li-Fraumeni syndrome skin fibroblasts. Oncogene. 1998;17:709–717. doi: 10.1038/sj.onc.1201987. [DOI] [PubMed] [Google Scholar]
- Gonzalez L, Freije JM, Cal S, Lopez-Otin C, Serrano M, Palmero I. A functional link between the tumour suppressors ARF and p33ING1. Oncogene. 2006;25:5173–5179. doi: 10.1038/sj.onc.1209526. [DOI] [PubMed] [Google Scholar]
- Haaf T. The effects of 5-azacytidine and 5-azadeoxycytidine on chromosome structure and function: implications for methylation-associated cellular processes. Pharmacol Ther. 1995;65:19–46. doi: 10.1016/0163-7258(94)00053-6. [DOI] [PubMed] [Google Scholar]
- Hagen TM, Yowe DL, Bartholomew JC, Wehr CM, Do KL, Park JY, et al. Mitochondrial decay in hepatocytes from old rats: membrane potential declines, heterogeneity and oxidants increase. Proc Natl Acad Sci USA. 1997;94:3064–3069. doi: 10.1073/pnas.94.7.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Brenton JD, Takahashi M, Finan D, Finkielsztein A, Damaraju S, et al. Constitutive p38HOG mitogen-activated protein kinase activation induces permanent cell cycle arrest and senescence. Cancer Res. 2002;62:5076–5082. [PubMed] [Google Scholar]
- Harley CB. Telomerase is not an oncogene. Oncogene. 2002;21:494–502. doi: 10.1038/sj.onc.1205076. [DOI] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, et al. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 1993;8:2457–2467. [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The cell biology of aging. Clin Geriatr Med. 1985;1:15–27. [PubMed] [Google Scholar]
- Itahana K, Campisi J, Dimri GP. Mechanisms of cellular senescence in human and mouse cells. Biogerontology. 2004;5:1–10. doi: 10.1023/b:bgen.0000017682.96395.10. [DOI] [PubMed] [Google Scholar]
- Itahana K, Zou Y, Itahana Y, Martinez JL, Beausejour C, Jacobs JJ, et al. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003;23:389–401. doi: 10.1128/MCB.23.1.389-401.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha KK, Banga S, Palejwala V, Ozer HL. SV40-mediated immortalization. Exp Cell Res. 1998;245:1–7. doi: 10.1006/excr.1998.4272. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Ushijima S, Saito Y, Nakanishi Y, Sakamoto M, Hirohashi S. mRNA expression of genes altered by 5-azacytidine treatment in cancer cell lines is associated with clinicopathological parameters of human cancers. J Cancer Res Clin Oncol. 2001;127:697–706. doi: 10.1007/s004320100284. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Satoh Y, Ikemura K, Konishi T, Ohji T, Karasaki Y, et al. Alterations of expression of the cytoskeleton after immortalization of human fibroblasts. Cell Struct Funct. 1995;20:107–115. doi: 10.1247/csf.20.107. [DOI] [PubMed] [Google Scholar]
- Kang MK, Kameta A, Shin KH, Baluda MA, Kim HR, Park NH. Senescence-associated genes in normal human oral keratinocytes. Exp Cell Res. 2003;287:272–281. doi: 10.1016/s0014-4827(03)00061-2. [DOI] [PubMed] [Google Scholar]
- Karpf AR, Jones DA. Reactivating the expression of methylation silenced genes in human cancer. Oncogene. 2002;21:5496–5503. doi: 10.1038/sj.onc.1205602. [DOI] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- Kortlever RM, Bernards R. Senescence, wound healing and cancer: the PAI-1 connection. Cell Cycle. 2006;5:2697–2703. doi: 10.4161/cc.5.23.3510. [DOI] [PubMed] [Google Scholar]
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraniak JM, Abrams J, Nowak JE, Tainsky MA. Antioxidant agents transiently inhibit aneuploidy progression in Li-Fraumeni cell strains. Mol Carcinog. 2006;45:141–156. doi: 10.1002/mc.20145. [DOI] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva OI, Draghici S, Tang L, Kraniak JM, Land SJ, Tainsky MA. Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene. 2003;22:4118–4127. doi: 10.1038/sj.onc.1206594. [DOI] [PubMed] [Google Scholar]
- Lecka-Czernik B, Moerman EJ, Jones RA, Goldstein S. Identification of gene sequences overexpressed in senescent and Werner syndrome human fibroblasts. Exp Gerontol. 1996;31:159–174. doi: 10.1016/0531-5565(95)02014-4. [DOI] [PubMed] [Google Scholar]
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem. 1999;274:7936–7940. doi: 10.1074/jbc.274.12.7936. [DOI] [PubMed] [Google Scholar]
- Lee CC, Shyu KG, Lin S, Wang BW, Liu YC, Chang H. Cell adhesion regulates the plasminogen activator inhibitor-1 gene expression in anchorage-dependent cells. Biochem Biophys Res Commun. 2002;291:185–190. doi: 10.1006/bbrc.2002.6415. [DOI] [PubMed] [Google Scholar]
- Leung KM, Po LS, Tsang FC, Siu WY, Lau A, Ho HT, et al. The candidate tumor suppressor ING1b can stabilize p53 by disrupting the regulation of p53 by MDM2. Cancer Res. 2002;62:4890–4893. [PubMed] [Google Scholar]
- Li Q, Tang L, Roberts PC, Kraniak JM, Fridman AL, Kulaeva OI, et al. Interferon regulatory factors IRF5 and IRF7 inhibit growth and induce senescence in immortal Li-Fraumeni fibroblasts. Mol Cancer Res. 2008;6:770–784. doi: 10.1158/1541-7786.MCR-07-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Pan J, Li JL, Lee JH, Tunkey C, Saraf K, et al. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol Cancer. 2007;6:7. doi: 10.1186/1476-4598-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Gonzales FA, Jones PA, Orntoft TF, Thykjaer T. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Res. 2002;62:961–966. [PubMed] [Google Scholar]
- Linskens MH, Feng J, Andrews WH, Enlow BE, Saati SM, Tonkin LA, et al. Cataloging altered gene expression in young and senescent cells using enhanced differential display. Nucleic Acids Res. 1995;23:3244–3251. doi: 10.1093/nar/23.16.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatina N, Haskell JF, Andrews LG, Poole JC, Saldanha S, Tollefsbol T. Differential maintenance and de novo methylating activity by three DNA methyltransferases in aging and immortalized fibroblasts. J Cell Biochem. 2002;84:324–334. doi: 10.1002/jcb.10015. [DOI] [PubMed] [Google Scholar]
- Lopez-Bermejo A, Buckway CK, Devi GR, Hwa V, Plymate SR, Oh Y, et al. Characterization of insulin-like growth factor-binding protein-related proteins (IGFBP-rPs) 1, 2, and 3 in human prostate epithelial cells: potential roles for IGFBP-rP1 and 2 in senescence of the prostatic epithelium. Endocrinology. 2000;141:4072–4080. doi: 10.1210/endo.141.11.7783. [DOI] [PubMed] [Google Scholar]
- Lundberg AS, Hahn WC, Gupta P, Weinberg RA. Genes involved in senescence and immortalization. Curr Opin Cell Biol. 2000;12:705–709. doi: 10.1016/s0955-0674(00)00155-1. [DOI] [PubMed] [Google Scholar]
- Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli C, Karayianni E, Magnanini C, Mungall AJ, Thorland E, Negrini M, et al. Cloning and characterization of the common fragile site FRA6F harboring a replicative senescence gene and frequently deleted in human tumors. Oncogene. 2002;21:7266–7276. doi: 10.1038/sj.onc.1205573. [DOI] [PubMed] [Google Scholar]
- Morelli C, Magnanini C, Mungall AJ, Negrini M, Barbanti-Brodano G. Cloning and characterization of two overlapping genes in a subregion at 6q21 involved in replicative senescence and schizophrenia. Gene. 2000;252:217–225. doi: 10.1016/s0378-1119(00)00231-6. [DOI] [PubMed] [Google Scholar]
- Murano S, Thweatt R, Shmookler Reis RJ, Jones RA, Moerman EJ, Goldstein S. Diverse gene sequences are overexpressed in Werner syndrome fibroblasts undergoing premature replicative senescence. Mol Cell Biol. 1991;11:3905–3914. doi: 10.1128/mcb.11.8.3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam TJ, Busby W, Jr, Clemmons DR. Insulin-like growth factor binding protein-5 binds to plasminogen activator inhibitor-I. Endocrinology. 1997;138:2972–2978. doi: 10.1210/endo.138.7.5230. [DOI] [PubMed] [Google Scholar]
- Neumeister P, Albanese C, Balent B, Greally J, Pestell RG. Senescence and epigenetic dysregulation in cancer. Int J Biochem Cell Biol. 2002;34:1475–1490. doi: 10.1016/s1357-2725(02)00079-1. [DOI] [PubMed] [Google Scholar]
- Ozawa S, Kato Y, Komori R, Maehata Y, Kubota E, Hata R. BRAK/CXCL14 expression suppresses tumor growth in vivo in human oral carcinoma cells. Biochem Biophys Res Commun. 2006;348:406–412. doi: 10.1016/j.bbrc.2006.07.070. [DOI] [PubMed] [Google Scholar]
- Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, et al. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res. 2003;63:1114–1121. [PubMed] [Google Scholar]
- Pereira-Smith OM, Smith JR. Genetic analysis of indefinite division in human cells: identification of four complementation groups. Proc Natl Acad Sci USA. 1988;85:6042–6046. doi: 10.1073/pnas.85.16.6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- Quackenbush J. Genomics. Microarrays—guilt by association. Science. 2003;302:240–241. doi: 10.1126/science.1090887. [DOI] [PubMed] [Google Scholar]
- Rogan EM, Bryan TM, Hukku B, Maclean K, Chang AC, Moy EL, et al. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol Cell Biol. 1995;15:4745–4753. doi: 10.1128/mcb.15.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohme D. Evidence for a relationship between longevity of mammalian species and life spans of normal fibroblasts in vitro and erythrocytes in vivo. Proc Natl Acad Sci USA. 1981;78:5009–5013. doi: 10.1073/pnas.78.8.5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov SR, Kozakiewicz BK, Holst CR, Stampfer MR, Haupt LM, Tlsty TD. Normal human mammary epithelial cells spontaneously escape senescence and acquire genomic changes. Nature. 2001;409:633–637. doi: 10.1038/35054579. [DOI] [PubMed] [Google Scholar]
- Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–2715. [PubMed] [Google Scholar]
- Rubin H. The disparity between human cell senescence in vitro and lifelong replication in vivo. Nat Biotechnol. 2002;20:675–681. doi: 10.1038/nbt0702-675. [DOI] [PubMed] [Google Scholar]
- Russo T, Zambrano N, Esposito F, Ammendola R, Cimino F, Fiscella M, et al. A p53-independent pathway for activation of WAF1/CIP1 expression following oxidative stress. J Biol Chem. 1995;270:29386–29391. doi: 10.1074/jbc.270.49.29386. [DOI] [PubMed] [Google Scholar]
- Salomonis N, Hanspers K, Zambon AC, Vranizan K, Lawlor SC, Dahlquist KD, et al. GenMAPP 2: new features and resources for pathway analysis. BMC Bioinformatics. 2007;8:217. doi: 10.1186/1471-2105-8-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu AK, Hubbard K, Kaur GP, Jha KK, Ozer HL, Athwal RS. Senescence of immortal human fibroblasts by the introduction of normal human chromosome 6. Proc Natl Acad Sci USA. 1994;91:5498–5502. doi: 10.1073/pnas.91.12.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Honda T, Yamada H, Wake N, Barrett JC, Oshimura M. Evidence for multiple pathways to cellular senescence. Cancer Res. 1994;54:6090–6093. [PubMed] [Google Scholar]
- Satoh Y, Kashimura M, Kaneko S, Karasaki Y, Higashi K, Gotoh S. Cloning of cDNAs with possible association with senescence and immortalization of human cells. Mutat Res. 1994;316:25–36. doi: 10.1016/0921-8734(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J Biol Chem. 2002;277:14877–14883. doi: 10.1074/jbc.M200373200. [DOI] [PubMed] [Google Scholar]
- Sekaric P, Shamanin VA, Luo J, Androphy EJ. hAda3 regulates p14ARF-induced p53 acetylation and senescence. Oncogene. 2007;26:6261–6268. doi: 10.1038/sj.onc.1210462. [DOI] [PubMed] [Google Scholar]
- Seok Kim K, Bae Seu Y, Baek SH, Jin Kim M, Jun Kim K, Hye Kim J, et al. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol Biol Cell. 2007;18:4543–4552. doi: 10.1091/mbc.E07-03-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shay JW, Tomlinson G, Piatyszek MA, Gollahon LS. Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome. Mol Cell Biol. 1995;15:425–432. doi: 10.1128/mcb.15.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol. 1999;9:939–945. doi: 10.1016/s0960-9822(99)80420-5. [DOI] [PubMed] [Google Scholar]
- Shibanuma M, Mochizuki E, Maniwa R, Mashimo J, Nishiya N, Imai S, et al. Induction of senescence-like phenotypes by forced expression of hic-5, which encodes a novel LIM motif protein, in immortalized human fibroblasts. Mol Cell Biol. 1997;17:1224–1235. doi: 10.1128/mcb.17.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou J, Soriano R, Hayward SW, Cunha GR, Williams PM, Gao WQ. Expression profiling of a human cell line model of prostatic cancer reveals a direct involvement of interferon signaling in prostate tumor progression. Proc Natl Acad Sci USA. 2002;99:2830–2835. doi: 10.1073/pnas.052705299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science. 1996;273:63–67. doi: 10.1126/science.273.5271.63. [DOI] [PubMed] [Google Scholar]
- Sommer M, Poliak N, Upadhyay S, Ratovitski E, Nelkin BD, Donehower LA, et al. DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle. 2006;5:2005–2011. doi: 10.4161/cc.5.17.3194. [DOI] [PubMed] [Google Scholar]
- Sorrell AM, Shand JH, Tonner E, Gamberoni M, Accorsi PA, Beattie J, et al. Insulin-like growth factor-binding protein-5 activates plasminogen by interaction with tissue plasminogen activator, independently of its ability to bind to plasminogen activator inhibitor-1, insulin-like growth factor-I, or heparin. J Biol Chem. 2006;281:10883–10889. doi: 10.1074/jbc.M508505200. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, Yaswen P. Human epithelial cell immortalization as a step in carcinogenesis. Cancer Lett. 2003;194:199–208. doi: 10.1016/s0304-3835(02)00707-3. [DOI] [PubMed] [Google Scholar]
- Stratford FL, Chondrogianni N, Trougakos IP, Gonos ES, Rivett AJ. Proteasome response to interferon-gamma is altered in senescent human fibroblasts. FEBS Lett. 2006;580:3989–3994. doi: 10.1016/j.febslet.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Minagawa S, Michishita E, Ogino H, Fujii M, Mitsui Y, et al. Induction of senescence-associated genes by 5-bromodeoxyuridine in HeLa cells. Exp Gerontol. 2001;36:465–474. doi: 10.1016/s0531-5565(00)00223-0. [DOI] [PubMed] [Google Scholar]
- Swisshelm K, Ryan K, Tsuchiya K, Sager R. Enhanced expression of an insulin growth factor-like binding protein (mac25) in senescent human mammary epithelial cells and induced expression with retinoic acid. Proc Natl Acad Sci USA. 1995;92:4472–4476. doi: 10.1073/pnas.92.10.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- Takebayashi S, Nakao M, Fujita N, Sado T, Tanaka M, Taguchi H, et al. 5-Aza-2′-deoxycytidine induces histone hyperacetylation of mouse centromeric heterochromatin by a mechanism independent of DNA demethylation. Biochem Biophys Res Commun. 2001;288:921–926. doi: 10.1006/bbrc.2001.5863. [DOI] [PubMed] [Google Scholar]
- Tandle AT, Mazzanti C, Alexander HR, Roberts DD, Libutti SK. Endothelial monocyte activating polypeptide-II induced gene expression changes in endothelial cells. Cytokine. 2005;30:347–358. doi: 10.1016/j.cyto.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Tominaga K, Olgun A, Smith JR, Pereira-Smith OM. Genetics of cellular senescence. Mech Ageing Dev. 2002;123:927–936. doi: 10.1016/s0047-6374(02)00030-1. [DOI] [PubMed] [Google Scholar]
- Tsutsui T, Fujino T, Kodama S, Tainsky MA, Boyd J, Barrett JC. Aflatoxin B1-induced immortalization of cultured skin fibroblasts from a patient with Li-Fraumeni syndrome. Carcinogenesis. 1995;16:25–34. doi: 10.1093/carcin/16.1.25. [DOI] [PubMed] [Google Scholar]
- Tsutsui T, Tanaka Y, Matsudo Y, Hasegawa K, Fujino T, Kodama S, et al. Extended lifespan and immortalization of human fibroblasts induced by X-ray irradiation. Mol Carcinog. 1997;18:7–18. doi: 10.1002/(sici)1098-2744(199701)18:1<7::aid-mc2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Untergasser G, Koch HB, Menssen A, Hermeking H. Characterization of epithelial senescence by serial analysis of gene expression: identification of genes potentially involved in prostate cancer. Cancer Res. 2002;62:6255–6262. [PubMed] [Google Scholar]
- Varley JM, McGown G, Thorncroft M, Santibanez-Koref MF, Kelsey AM, Tricker KJ, et al. Germ-line mutations of TP53 in Li-Fraumeni families: an extended study of 39 families. Cancer Res. 1997;57:3245–3252. [PubMed] [Google Scholar]
- Vogt M, Haggblom C, Yeargin J, Christiansen-Weber T, Haas M. Independent induction of senescence by p16INK4a and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ. 1998;9:139–146. [PubMed] [Google Scholar]
- Vojta PJ, Futreal PA, Annab LA, Kato H, Pereira-Smith OM, Barrett JC. Evidence for two senescence loci on human chromosome 1. Genes Chromosomes Cancer. 1996;16:55–63. doi: 10.1002/(SICI)1098-2264(199605)16:1<55::AID-GCC8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Saretzki G, Docke W, Lotze C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: a model for senescence? Exp Cell Res. 1995;220:186–193. doi: 10.1006/excr.1995.1305. [DOI] [PubMed] [Google Scholar]
- Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22:3389–3403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HM, Birnbaum RS, Poot M, Quinn LS, Swisshelm K. Insulin-like growth factor binding protein-related protein 1 inhibits proliferation of MCF-7 breast cancer cells via a senescence-like mechanism. Cell Growth Differ. 2002;13:205–213. [PubMed] [Google Scholar]
- Yaswen P, Stampfer MR. Molecular changes accompanying senescence and immortalization of cultured human mammary epithelial cells. Int J Biochem Cell Biol. 2002;34:1382–1394. doi: 10.1016/s1357-2725(02)00047-x. [DOI] [PubMed] [Google Scholar]
- Yoon IK, Kim HK, Kim YK, Song IH, Kim W, Kim S, et al. Exploration of replicative senescence-associated genes in human dermal fibroblasts by cDNA microarray technology. Exp Gerontol. 2004;39:1369–1378. doi: 10.1016/j.exger.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cohen SN. Smurf2 up-regulation activates telomere-dependent senescence. Genes Dev. 2004;18:3028–3040. doi: 10.1101/gad.1253004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Herbert BS, Pan KH, Shay JW, Cohen SN. Disparate effects of telomere attrition on gene expression during replicative senescence of human mammary epithelial cells cultured under different conditions. Oncogene. 2004;23:6193–6198. doi: 10.1038/sj.onc.1207834. [DOI] [PubMed] [Google Scholar]
- Zhang H, Pan KH, Cohen SN. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc Natl Acad Sci USA. 2003;100:3251–3256. doi: 10.1073/pnas.2627983100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.