

Abstract
Rheumatoid arthritis (RA) is a complex, multi-system disease whose primary site of inflammatory tissue damage is the joint. The increasing evidences indicate that activated RA fibroblast-like synoviocytes (FLS) play a critical role in the development of pannus by migrating into cartilage and bone. Furthermore FLS and T cells can activate each other in vitro and in vivo, which is crucial for the progress of RA. Deoxycytidine kinase (DCK) has been linked to peripheral T cell homeostatic proliferation and survival, which is very important for RA. Yet, the function of DCK in FLS is still unknown. Here, we present a story that DCK could regulate the migration and invasion of FLS through AKT pathway in RA patients. Moreover, DCK seems to be the upstream of AKT and FAK, and AKT inhibitor exerted the similar effect on FLS motility. In summary, our study characterized the new role of DCK in human primary FLS cells, and figured out the possible pathway DCK involved in, and these findings might propose DCK as a novel target for controlling joint destruction of RA.
Keywords: Rheumatoid arthritis, deoxycytidine kinase, fibroblast-like synoviocyte, v-akt murine thymoma viral oncogene homolog 1, focal adhesion kinase
Introduction
Rheumatoid arthritis (RA) is a common chronic inflammatory disorder characterized by abnormal synovial hyperplasia and progressive destruction of cartilage and bone [1]. Several cell types, including T cells, macrophages, B cells, osteoclasts and chondrocytes, are involved in destructive processes of the RA joint [2-4]. However, increasing evidences indicate that activated RA fibroblast-like synoviocytes (FLS), which are present in great numbers in rheumatoid arthritis synovium, exhibit the characteristics of malignant cells and play a critical role in the development of pannus by migrating into cartilage and bone [5-10]. More importantly, FLS and T cells can activate each other in vitro and in vivo, which is crucial for the progress of RA. Similar to professional antigen-presenting cell (APC)-T cell interactions, FLS and T cells in co-culture have been shown to interact with each other in antigen-dependent systems [11-13].
Deoxycytidine kinase (DCK) is a rate-limiting enzyme in deoxyribonucleoside salvage, a metabolic pathway that recycles DNA degradation products [14,15]. DCK phosphorylates and therefore activates nucleoside analog prodrugs frequently used in cancer, autoimmunity, and viral infections. In contrast to its well established therapeutic relevance, the biological function of DCK remains unknown. DCK is highly expressed in the thymus and bone marrow, indicating a possible role in lymphopoiesis [16-18]. Toy et al. had established DCK knockout (KO) mice and found that DCK inactivation selectively and profoundly affected T and B cell development [19]. Lymphocyte numbers in DCK KO mice were 5 to 13-fold below normal values. Choi et al reported that a deficiency in DCK affected peripheral T cell homeostatic proliferation and survival [20].
T cell receptor (TCR) engagement of MHC/antigen triggers complex signaling cascades in T cells and participates in T cell-FLS interactions [21,22]. However, the role of DCK in the pathogenesis of RA has not been explored. The goal of this study was to investigate the role of DCK in regulating the migration and invasion of rheumatoid arthritis FLS. To this end, wound healing, transwell migration and invasion assays were performed to investigate the effects of DCK knockdown on the migration and invasion of FLS cells, and F-actin reorganization was detected by phalloidin staining. Furthermore, we also found that DCK silencing inhibited the phosphorylation of Akt and focal adhesion kinase (FAK), the activation of NF-КB and the AKT inhibitor exhibited similar effects on FLS like DCK silencing. In summary, DCK might play an important role in the regulation of migration, invasion, some MMPs expression and cytoskeletal reorganization in RA FLS through AKT and FAK pathways. These findings could propose DCK as a novel target for controlling joint destruction of RA.
Materials and methods
Isolation and culture of RA FLS cells
All synovium were obtained from 12 RA patients (2 men and 10 women) undergoing total joint replacement. Patients had a mean age of 54 years (range 30-71 years). RA diagnosis was based on the presence of at least four of the seven criteria developed by the American College of Rheumatology for RA [23]. The synovial tissue was isolated from the synovium, minced and incubated with 0.5 mg/ml collagenase (Sigma) for 2 h at 37 °C in DMEM (Hyclone), filtered through a nylon mesh, washed extensively with phosphate buffered saline (PBS), and cultured in DMEM supplemented with 20% FCS (endotoxin content < 0.006 ng/ml; Gibco), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified 5% CO2 atmosphere. Following overnight culture, non-adherent cells were removed and adherent cells were cultivated in DMEM supplemented with 20% FCS until confluence. FLS were identified as reported previously [24], and the cells were used for experiment at three to six passages.
RA FLS cells were pretreated for 24 h with the Akt specific inhibitor MK2206 (Selleckchem, USA) at various concentrations (240, 480 and 960 nM) for investigating the effect of inhibition of Akt activity on RA FLS migration and invasion.
The study protocol was in accordance with the standards of the responsible local committee or with the Helsinki Declaration and approved by the ethics committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University. All patients gave their informed written and signed consent to participate in the study and consent to publish.
shRNAs construction and virus infection
Target shRNA against human DCK gene (GenBank accession NM_00078) for RNAi were designed as follows: shRNA 1 (5′-GCAGCCTGCTATAAAGTTAAA-3′) and shRNA 2 (5′-CCTTGAATTGGATGGAATCAT-3′). Oligonucleotides were synthesized according to the sequences and annealed into double-strands (Life Technologies). DCK shRNAs were inserted into a lentivirus vector expressing tomato fluorescence when transfected into the cells. Empty vector served as negative control (NC). 293T cells were co-transfected with lentivirus vector and packaging plasmids (pHelper 1.0 including gag/pol and pHelper 2.0 including VSVG) using the Lipofectamine (Invitrogen). Infectious lentivirus vectors were harvested at 48 h and 72 h following transfection.
FLS Cells were divided into two groups: NC (infection with empty lentiviral vector) and DCK-shRNA (infection with DCK-shRNA lentiviral vector). FLS cells were seeded at a density of 1x105 cells/well in 6-well tissue culture plates. After 24 h of culture, the cells were infected with specific or negative control lentiviral vectors, at the multiplicity of infection (MOI) 5. To determine infection efficiency, cells expressing tomato fluorescence were observed using fluorescence microscopy 2009 (Olympus, Tokyo, Japan) at 72 h following infection. The cells were collected for analyses at 96 h after infection.
Real-time quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from FLS cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Reverse transcription was performed with Primerscript RT reagent kit (Takara). Real-time PCR was performed using SYBR Premix Ex Taq (Applied Takara) using the following primers: GAPDH, sense 5’-GTGAAGGTCGGAGTCAACG-3’ and antisense 5’-TGAGGTCAATGAAGGGGTC-3’; MMP-1, sense 5’-ATCCCTTCTACCCGGAAGTTG-3’ and antisense 5’-TCATCTCTGTCGGCAAATT-CG-3’; MMP-3, sense 5’-GCGCAAATCCCTCAGGAA-3’ and antisense 5’-CATCCACGCCTGAAGGAAGA-3’; tissue inhibitors of metalloproteinase (TIMP)-2, sense 5’-GAAGGAGCCCCATCAATCCT-3’ and antisense 5’-CTCCCATTTCTACAAGGCTCAGA-3’; DCK, sense 5’-CAGTTTAGTCCCACCTCTTACTTCCT-3’ and antisense 5’-AGCAGGCTGCCTAATTAATCCA-3’. Amplification conditions were as follows: initial denaturation at 95 °C for 15 s, 5 s at 95 °C and 30 s at 60 °C for 40 cycles. Data were collected and analyzed on an ABI PRISM 7900 sequence detection system (Applied Biosystems, USA). GAPDH gene was used as an endogenous control. The amount of gene expression was then calculated as the difference cycle threshold (ΔCT) between the CT value of the target gene and the GAPDH. ΔΔCT were difference between ΔCT values of test sample and the control. Relative expression of target genes was calculated as 2-ΔΔCT.
Western blot analyses
FLS cells were lysed with 100 μl lysis buffer (PH 6.8, 50 mM Tris, 2% SDS, 0.1% Bromophenol blue, 10% Glycerol, 100 mM DL-Dithiothreitol) supplemented with protease and phosphatase inhibitors (1 mM phenylmethyl sulfonylfluoride, 5 mM NaF, 1 mM Na3VO4, complete protease inhibitor cocktail) (Roche). Cell lysates were collected by scraping and centrifuged at 12,000 rpm for 15 min. Protein concentration was measured with a BCA protein Assay. 20 μg of total protein from each sample were electrophoresis on a 10% SDS-PAGE gel and transferred onto nitrocellulose membranes (Millipore). Membranes were blocked in 5% non-fat milk in TBS containing 0.1% Tween 20 (TBST) for 1 h at room temperature, and then incubated with specific primary antibodies DCK (Epitomics, USA), FAK, phospho-FAK (Cell Signaling Technologies, Danvers, MA, USA), phospho-Akt (Santa Cruz, CA, USA), Akt (Santa Cruz, CA, USA), I kappa B kinase α (IKKα) (Epitomics, USA), and GAPDH (Cell Signaling Technologies, Danvers, MA, USA) at 4 °C overnight. Following washing and incubating with HRP-conjugated anti-rabbit-IgG-HRP (Cell Signaling Technologies, Danvers, MA, USA), the membranes were detected using ECL regent (Millipore Immobilon, CAT: WBKLS0500).
Determination of MMP-1 and MMP-3
After infection with lentiviruses for 72 h, supernatants of FLS cells were harvested and the concentrations of MMP-1 and MMP-3 were determined using commercially available Bio-plex kits (R&D, CA, USA) according to the manufacturers’ instructions.
Cell migration, invasion and wound healing assay
FLS cells were seeded at a density of 4,000 cells/well in a 96-well plate. After 72 h of infection with the lentiviruses, the cells formed a fluent monolayer and were observed under a fluorescent scope. A linear scratch was formed using a 10 μl pipette tip at 120 h after infection. Wounded monolayers were washed with PBS to remove detached cells and debris. The ability of FLS cells to close the wounded space was used to assess their migration ability. The width of the wound was recorded at 0 h and 24 h following scratching (original magnification, x40).
Transwell migration assays were performed using a 24-well Boyden chamber (6.5 mm diameter, 8.0 μm; BD) according to the manufacturer’s instructions. In brief, FLS cells were infected with lentiviruses and cultured for 96 h. Cells were then harvested by trypsinization, washed and resuspended in serum-free media at a density of 2.5x104 cells/well. A 100 μL cell suspension was placed onto the upper and the lower chambers of the transwell were filled with 500 μL of media containing serum as an adhesive substrate. Cells were incubated at 37 °C under 5% CO2 for 24 h and then non-migrating cells on the upper side of the membrane were removed with a cotton swab. Migrating cells on the lower side of the membrane were fixed with 4% methanol for 20 min and stained with 0.1% crystal violet for 1 min. Photomicrographs of five random fields were obtained (original magnification, x100), and cells were counted to calculate the average number of cells that had migrated.
For the in vitro invasion assay, similar experiments were performed using inserts coated with a Matrigel basement membrane matrix. The Matrigel (Matrigel basement membrane matrix, Becton Dickinson, USA) was diluted in serum-free cold media and placed into upper chambers of a 24 well Transwell and incubated at 37 °C for 1 h. Cells were resuspended with serum-free media at a density of 5x104 cells/well and incubated for 48 h to evaluate cell migration. All experiments were performed in duplicate.
MTS assay
FLS cells infected with lentiviruses were seeded in a 96-well plate at a density of 4,000 cells/well and incubated at 37 °C in a humidified 5% CO2 atmosphere for 120 h. Cell proliferation was determined using a CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega, WI, USA) according to the manufacturer’s instructions. The spectrophotometric absorbance of each sample was measured at 590 nm using the TECAN spectra (Thermo, Austria). All cultures were assayed in triplicate.
Filamentous actin (F-actin) staining
FLS cells were seeded in a 24-well plate at a density of 2x104 cells/well. After 96 h of infection with the lentiviruses, cells were washed twice with phosphate buffered saline (PBS) and then fixed with 0.3 ml/well of 4% paraformaldehyde at room temperature for 10 min. Then cells were permeabilized with 0.1% Triton X-100 in PBS for 1 min at room temperature, and blocked in 2% bovine serum albumin (BSA) for 20 min. For the detection of cytoskeletal F-actin, the cells were incubated with FITC-conjugated phalloidin (SIGMA) in PBS (1:1000) for 20 min at room temperature. The coverslips were mounted on glass slides and examined under confocal fluorescence microscopy (Leica TCS SP5, German).
Statistical analyses
All data are expressed as mean ± SD. Unpaired t-test (two-tailed) was applied. Statistical analyses were performed using the Prism5 software (GraphPad). *p<0.05 was considered statistically significant.
Results
DCK regulated the migration and invasion of FLS cells
To examine the function of DCK on the migration and invasion of FLS cells, DCK was silenced by lentiviral shRNAs in primary FLS cells. After 72 h, there were more than 90% cells expressing tomato fluorescent protein, which confirmed the high infection efficiency. The efficacy of DCK knockdown at mRNA and protein levels in those FLS cells were then determined by qPCR and Western blot. The qPCR result showed that the mRNA level of DCK in shRNA-1 and shRNA-2 subgroups was remarkably reduced by 73% and 68%, respectively (Figure 1A). And the knockdown efficiency was also confirmed by Western blot, which showed that the protein level of DCK was dramatically decreased in shRNA1 and shRNA2 subgroups (Figure 1B). Then, FLS cells with or without DCK knockdown were subjected to wound-healing and transwell assay. The wound healing assay showed that FLS cells with DCK knockdown have weaker migration ability (Figure 1C, 1D). And the DCK knockdown also inhibited the migration in transwell migration assay comparing with the FLS without DCK knockdown (Figure 1E, 1F). The above results suggest that DCK knockdown weakened the ability of FLS cells to migrate. To evaluate the effect of DCK knockdown on the invasion of FLS cells, transwell invasion assay was performed. Compared with the control cells, DCK-shRNA (shRNA-1 and shRNA-2) infected cells showed a substantial reduction in invasive capacity (Figure 1G, 1H).
Figure 1.
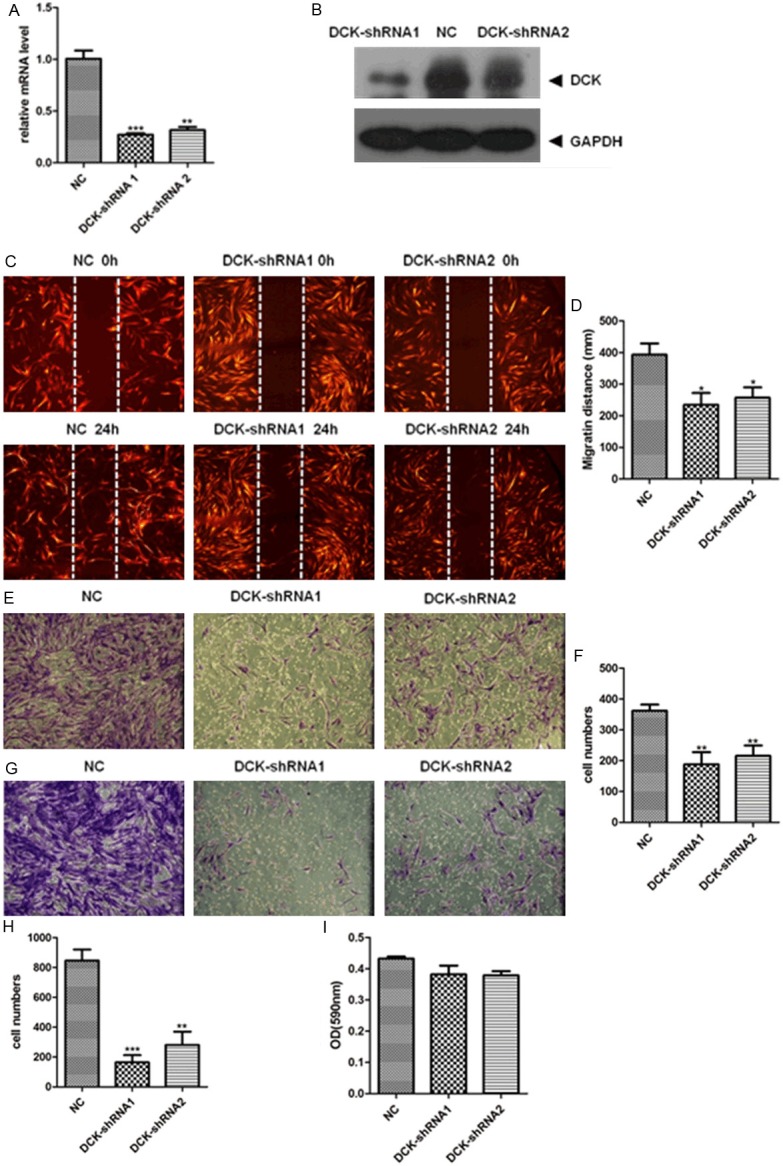
DCK knockdown suppressed migration and invasion of FLS cells. A: qPCR assessment of DCK mRNA levels after infection with DCK-shRNA compared to negative control (NC) cells. B: Western blot for DCK protein expression levels in FLS cells infected with DCK-shRNA or control shRNA. C and D: Wound healing assay showed decreased migration of FLS cells with DCK-shRNA lentivirus. The images were taken at 0 and 24 h after scratch applied (original magnification 40x). The migratory distances of the DCK-shRNA1 or -shRNA2 from 0 to 24 h were significantly decreased compared with that of NC group (p<0.05). E and F: FLS cells on transwell inserts were stained (original magnification 100x). The number of migratory cells across the polycarbonate membrane in the DCK-shRNA1 group and DCK-shRNA2 group were lower than that in NC group (p<0.01). G and H: Invasion of FLS cells was inhibited by DCK shRNA knockdown. Cell invasion assay was performed in Matrigel invasion chambers in 24-well culture plates. After 48 h of incubation, the Transwell inserts were stained with 0.1% viola crystallina. For quantification, the cells were counted under a microscope (x100). The number of migratory DCK-shRNA group cells were significantly decreased compared with control group cells (DCK-shRNA1, p<0.001; DCK-shRNA2, p<0.01). I: MTS assay for cell viability. Error bars represent the SD (standard errors) from three independent experiments. In whole figure, *, p<0.05; **, p<0.01 and ***, p<0.001.
In order to exclude the possibility that the inhibitory effect on cell migration and invasion by DCK knockdown was due to the cytotoxicity of shRNA knockdown, we performed an MTS assay to examine the cell viability. The result showed that the DCK knockdown did not affect the cell viability comparing with the NC group (Figure 1I). Thus, we concluded that DCK involved in primary FLS migration and invasion regulation.
DCK knockdown reduced MMP production in RA FLS
MMPs are necessary for cell migration and invasion. Since DCK could regulate the migration and invasion in FLS, we then examined the relationship between MMPs production and DCK expression level. The RA FLS cells were infected with DCK-shRNA lentivirus or empty virus for 96 h and then were subjected to qPCR and Bio-plex analysis. DCK-shRNA infected cells had much lower mRNA expression levels of MMP-1, MMP-3 and higher level of TIMP2 (MMPs inhibitor) comparing with cells infected with empty virus (NC) (Figure 2A-C). To further confirm the reduction of MMP1 and MMP3 caused by DCK knockdown, the MMP1 and MMP3 production levels in supernatant were also examined after DCK-shRNA or empty vector virus infection. The Bio-plex analyses showed that protein production of MMP-1 and MMP-3 was markedly decreased in the DCK-shRNA (shRNA-1 and shRNA-2) group (p<0.05, Figure 2D, 2E), which was consistent with the previous qPCR result. These findings suggest that DCK knockdown reduced the production of MMP-1 and MMP-3 in RA FLS.
Figure 2.
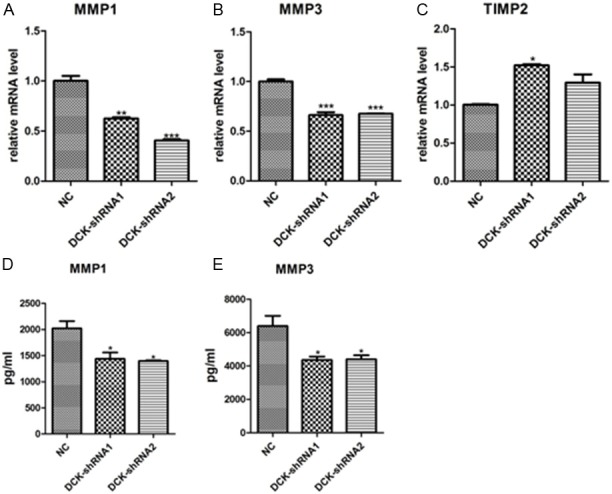
Effect of DCK knockdown on MMPs production in RA FLS. A-C: Total RNA from the DCK-shRNA cells and the control cells were used to analyze the mRNA expression levels of MMP1, MMP3, TIMP-2 by qPCR. D and E: Protein production of MMP-1 and MMP-3 were measured in FLS supernatants by Bio-plex. All the error bars represent the SD from three independent experiments, each measured in triplicate. *p<0.05, **p <0.01 and ***p<0.001 compared to the control.
DCK affected the cytoskeletal reorganization in RA FLS
The reorganization of cytoskeleton proteins (such as F-actin) has been linked with cell migration and invasion. To further confirm the effect of DCK knockdown on FLS migration and invasion, we assessed whether F-actin reorganization was affected in response to DCK knockdown. Being consistent with previous results, the F-actin stained with phalloidin has a dramatic decrease in the condensation of actin stress fiber and adhesion plaque formation in the DCK shRNA (shRNA-1 and shRNA-2) FLS (Figure 3). Our data suggested that DCK down-regulation inducing a reduction of FLS cell motility strongly correlated with the inhibition of F-actin reorganization.
Figure 3.
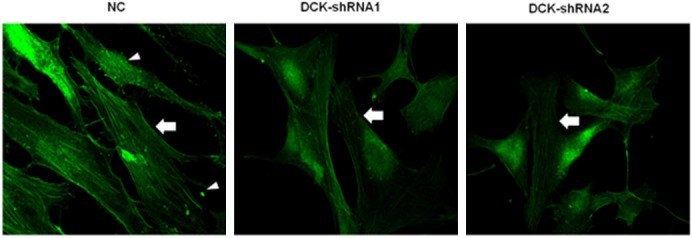
Effect of DCK knockdown on cytoskeletal filamentous actin (F-actin) alterations in RA FLS. Negative control (NC) cells form focal adhesion-anchored stress fibers (arrow indicating) and adhesion plaque (triangles indicating) as shown by phalloidin staining. The actin stress fiber and adhesion plaque formation were obviously affected in DCK-shRNA1 or -shRNA2 cells (arrow indicating).
DCK regulated AKT and FAK pathway in RA FLS
The previous results had linked DCK expression level to the motility of RA primary FLS cells, to further characterize the mechanism of DCK in the motility regulation, the signaling pathways by which DCK affected the migration and invasion of RA FLS cells were examined in these cells. AKT plays a pivotal role in promoting the motility of fibroblasts, and focal adhesion kinase (FAK) is a major target of AKT in promoting cell motility [25-27]. Therefore, we evaluated the effects of DCK knockdown on AKT and FAK phosphorylation (activation) in FLS cells. Comparing with NC cells, the phosphorylation of AKT and FAK was significantly decreased in FLS infected with DCK-shRNA1 or DCK-shRNA2, and the total protein levels of AKT and FAK were not changed (Figure 4). These results indicated that DCK participated in the regulation of phosphorylation of AKT and FAK, by which DCK might control the migration and invasion in FLS.
Figure 4.
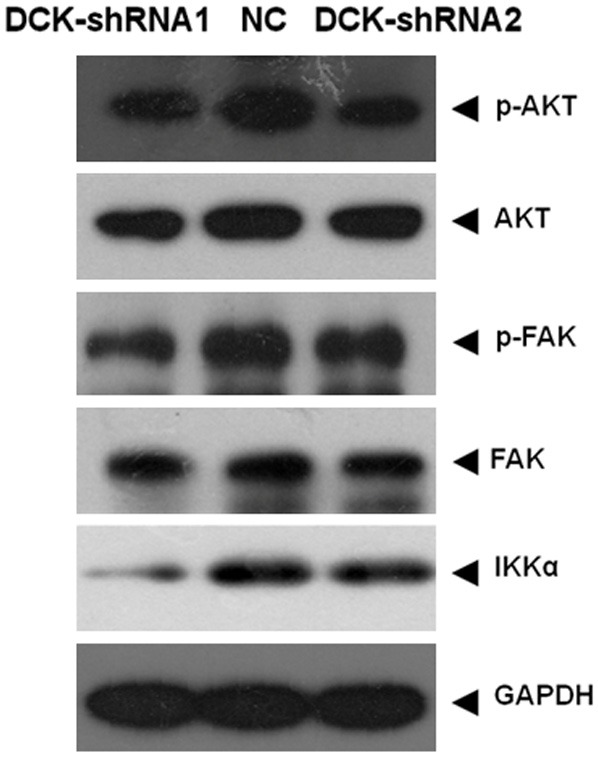
The activation of AKT and FAK were examined by western blot. The phosphorylation level of AKT and FAK decreased, yet total protein was not changed in DCK knockdown cells (DCK-shRNA1 and DCK-shRNA2), the IKKα expression level changing was also corresponding to AKT phosphorylation alteration. GAPDH was loaded as internal control.
AKT activation was necessary for the migration and invasion of FLS cells
We had proven that DCK could regulate the migration and invasion of FLS and DCK also involved in AKT pathway activation, yet it is still not known whether AKT pathway contributes the motility of FLS cells. To confirm whether AKT activation is required for DCK-mediated migration and invasion of RA FLS cells, we performed wound healing, transwell and invasion assays with FLS cells treated with the Akt inhibitor MK2206 at various concentrations (240, 480 and 960 nM). The MK2206 inhibition effect on AKT pathway were examined by western blot (Figure 5), the de-phosphorylation of AKT and FAK, degradation of IKKα [28-30] were similar with DCK knockdown in FLS cells. The migration and invasion of RA FLS cells were inhibited by MK2206 in a dose-dependent manner (Figure 6A-E). The cytotoxicity of MK2206 was also detected in FLS cells by MTS assay, the results showed even pretreatment with MK2206 at the highest concentration of 960 nM for 24 h, the FLS cell viability was not affected significantly (Figure 6F). These results suggested that AKT pathway activation is very important for FLS motility regulation, the similar pattern of AKT inhibitor and DCK knockdown on FLS migration and invasion indicated that DCK might involve in RLS motility through regulating AKT pathway.
Figure 5.
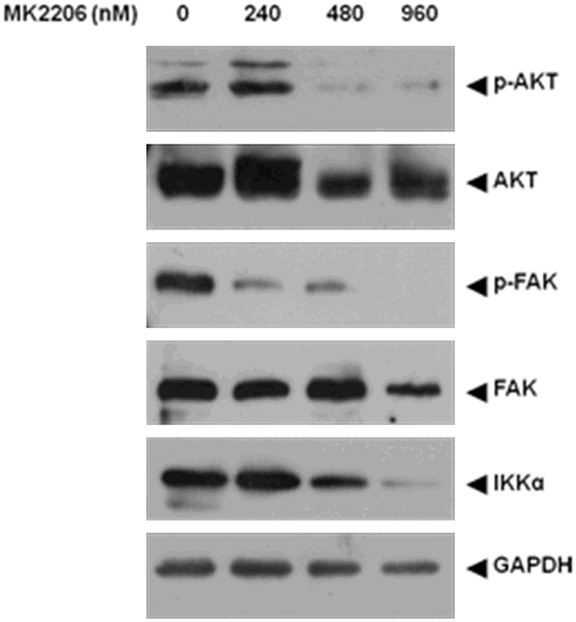
RA FLS cells were pretreated for 24 h with AKT specific inhibitor MK2206 at different concentrations (240, 480 and 960 nM). The phosphorylation protein levels of AKT, FAK, their respective total proteins and IKKα in FLS, were blotted using standard Western techniques. GAPDH was loading as a control.
Figure 6.
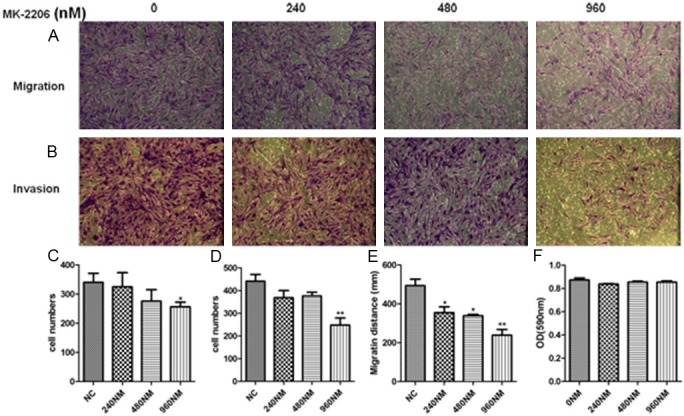
AKT activation was necessary for migration and invasion of RA FLS. RA FLS cells were pretreated for 24 h with AKT specific inhibitor MK2206 at indicated concentrations (240, 480 and 960 nM). A and C: Transwell migration assay. B and D: Transwell Invasion Assay. E: Wound healing assay. F: MTS assay. Error bars represent the SD from three independent experiments. *p<0.05 and **p<0.01 compared to the control.
Discussion
To our knowledge, this is the first report to show that DCK plays an important role in regulation of biological behaviors of FLS isolated from RA patients. To investigate the functional significance of DCK, we used shRNA to knock down DCK expression in RA FLS cells. Our data shows that DCK silencing had a strong inhibitory effect on the migration and invasion of FLS cells and this effect was not due to the toxic effects of viruses. Furthermore, DCK shRNA knockdown not only decreased the production of MMP-1 and MMP-3, but also prevented cytoskeletal reorganization of RA FLS cells. These results provide novel evidence that DCK plays a major role in the modulation of RA FLS migration and invasion.
DCK plays a well-known role in the nucleoside salvage pathway, which affects immune function, as revealed by the severe combined immunodeficiency caused by a genetic deficiency in adenosine deaminase [16,31-33]. DCK deficient mice possess reduced levels of circulating B and T lymphocytes [34]. However, direct function of DCK in FLS cells has not been studied yet. In the pathogenesis of RA, FLS movement is regarded as an important mechanism causing the invasion of cartilage and bone. Once they arrive at the bone, FLS can activate osteoclasts to enhance bone erosion and destruction [35,36]. Migration of FLS to cartilage and bone has been considered as a critical step in the aggravation of RA, and regulation of FLS migration and invasion may be a new therapeutic strategy for destructive progress of RA. Therefore, our findings that DCK may be a key regulator of RA FLS migration and invasion extend our understanding of the important role of DCK in RA, implying that DCK is a potential target for RA treatment.
Having established that RA FLS migration and invasion are regulated by DCK, we further investigated the signaling mechanisms involved in DCK-mediated migration and invasion. In RA FLS, DCK knockdown inhibited AKT and FAK phosphorylation. AKT has been regarded as a key regulator of biological processes including cell migration [37,38]. Enhanced AKT activity is known to be associated with accelerated tumor cell invasion [39-42]. FAK is present in focal adhesions and is essential for reorganization of the cytoskeleton and cell migration [43,44]. AKT promotes FAK-mediated cell migration through direct phosphorylation of FAK, while the inhibition of AKT activation suppressed phosphorylation of FAK in fibroblasts [27]. In the present study, we demonstrated that AKT inhibitor suppressed migration, invasion and phosphorylation of FAK in FLS from RA patients in a dose-dependent manner, and these inhibitory effects are not due to decreased cell viability, these findings were consistent with DCK knockdown effect on FLS cells. These results confirm that AKT played an essential role in cell motility and DCK regulated the motility of FLS cells through AKT pathway. Yet, how DCK involving in AKT signaling pathway is still unknown, which needs to be further addressed. And due to the limited amount of primary FLS cells, we could not confirm the over-expression effect of DCK on FLS cells motility, those will be carefully analyzed in our FLS cell model in vitro.
MMPs, mainly produced by FLS in RA, are proteases that participate in the remodeling of the extracellular matrix and play important roles in the progressive destruction of joints in RA [45,46]. Levels of MMP-1 and MMP-3 were higher in SF than in the systemic circulation, which were mainly produced by FLS [46,47]. MMP-3 has been proposed as an important indicator of radiological progression in early RA [48]. Similar to these findings, we demonstrated that DCK knockdown decreased the production of MMP-1 and MMP-3 in RA primary FLS cells. Moreover, mRNA expression levels of TIMP-2 were higher in DCK-shRNA cells than in control cells. In normal tissue, MMPs exist in balance with their inhibitors, primarily the tissue inhibitors of metalloproteinases (TIMPs) [44-46]. An imbalance of MMPs and TIMPs contributes to the cartilage destruction in RA. Abundant activation of NF-КB was detected in rheumatoid synovium [49], nuclear translocation of NF-КB in cultured fibroblast-like synoviocytes occurs rapidly through the activation of IKK signaling complexes and induces cytokine production and MMP expression. In this study, we found that the level of IKKα in RA FLS cells infected with DCK shRNA or treated with an Akt inhibitor were dramatically reduced, which might inactivate NF-KB and suppress MMPs expression and secretion. Therefore, DCK mediates the activation of AKT, which in turn activates NF-КB signaling through upregulation of IKKα in FLS, and leads to high expression of MMPs.
Taken together, we mapped out a new function of DCK in FLS cells from RA patients. And DCK knockdown reduced the motility of FLS, inactivated the AKT pathway. These findings could be repeated by applying AKT inhibitor on FLS, which suggested that DCK involves in migration and invasion in FLS of RA patients at least partially through AKT pathway. Our result indicates DCK might be a novel target for controlling joint destruction of RA.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (Grant No. 81001332), Shanghai Leading Academic Discipline Project (Grant No. T0203) and Shanghai Clinical Research Resource Sharing Platform Project (Grant No. SHDC12007205).
Disclosure of conflict of interest
None.
References
- 1.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–16. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 2.Gravallese EM. Bone destruction in arthritis. Ann Rheum Dis. 2002;61:ii84–6. doi: 10.1136/ard.61.suppl_2.ii84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. 2005;7:S4–14. doi: 10.1186/ar1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma Y, Pope RM. The role of macrophages in rheumatoid arthritis. Curr Pharm Des. 2005;11:569–580. doi: 10.2174/1381612053381927. [DOI] [PubMed] [Google Scholar]
- 5.Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci U S A. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roivainen A, Jalava J, Pirila L, Yli-Jama T, Tiusanen H, Toivanen P. H-ras oncogene point mutations in arthritic synovium. Arthritis Rheum. 1997;40:1636–1643. doi: 10.1002/art.1780400913. [DOI] [PubMed] [Google Scholar]
- 7.Muller-Ladner U, Pap T, Gay RE, Neidhart M, Gay S. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol. 2005;1:102–110. doi: 10.1038/ncprheum0047. [DOI] [PubMed] [Google Scholar]
- 8.Huber LC, Distler O, Tarner I, Gay RE, Gay S, Pap T. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology (Oxford) 2006;45:669–675. doi: 10.1093/rheumatology/kel065. [DOI] [PubMed] [Google Scholar]
- 9.Muller-Ladner U, Kriegsmann J, Franklin BN, Matsumoto S, Geiler T, Gay RE, Gay S. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- 10.Pap T, Meinecke I, Muller-Ladner U, Gay S. Are fibroblasts involved in joint destruction? Ann Rheum Dis. 2005;64:iv52–54. doi: 10.1136/ard.2005.042424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsai C, Diaz LA Jr, Singer NG, Li LL, Kirsch AH, Mitra R, Nickoloff BJ, Crofford LJ, Fox DA. Responsiveness of human T lymphocytes to bacterial superantigens presented by cultured rheumatoid arthritis synoviocytes. Arthritis Rheum. 1996;39:125–136. doi: 10.1002/art.1780390117. [DOI] [PubMed] [Google Scholar]
- 12.Tran CN, Davis MJ, Tesmer LA, Endres JL, Motyl CD, Smuda C, Somers EC, Chung KC, Urquhart AG, Lundy SK, Kovats S, Fox DA. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007;56:1497–1506. doi: 10.1002/art.22573. [DOI] [PubMed] [Google Scholar]
- 13.Kim WU, Cho ML, Jung YO, Min SY, Park SW, Min DJ, Yoon JH, Kim HY. Type II collagen autoimmunity in rheumatoid arthritis. Am J Med Sci. 2004;327:202–211. doi: 10.1097/00000441-200404000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Reichard P. Interactions between deoxyribonucleotide and DNA synthesis. Annu Rev Biochem. 1988;57:349–374. doi: 10.1146/annurev.bi.57.070188.002025. [DOI] [PubMed] [Google Scholar]
- 15.Eriksson S, Munch-Petersen B, Johansson K, Eklund H. Structure and function of cellular deoxyribonucleoside kinases. Cell Mol Life Sci. 2002;59:1327–1346. doi: 10.1007/s00018-002-8511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carson DA, Kaye J, Seegmiller JE. Lymphospecific toxicity in adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency: possible role of nucleoside kinase(s) Proc Natl Acad Sci U S A. 1977;74:5677–5681. doi: 10.1073/pnas.74.12.5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staub M, Eriksson S. Deoxynucleoside analogs in cancer therapy. 2007. The role of deoxycytidine kinase in DNA synthesis and nucleoside analog activation; pp. 29–52. [Google Scholar]
- 18.Carson DA, Kaye J, Wasson DB. The potential importance of soluble deoxynucleotidase activity in mediating deoxyadenosine toxicity in human lymphoblasts. J Immunol. 1981;126:348–352. [PubMed] [Google Scholar]
- 19.Toy G, Austin WR, Liao HI, Cheng D, Singh A, Campbell DO, Ishikawa TO, Lehmann LW, Satyamurthy N, Phelps ME, Herschman HR, Czernin J, Witte ON, Radu CG. Requirement for deoxycytidine kinase in T and B lymphocyte development. Proc Natl Acad Sci U S A. 2010;107:5551–6. doi: 10.1073/pnas.0913900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi O, Heathcote DA, Ho KK, Muller PJ, Ghani H, Lam EW, Ashton-Rickardt PG, Rutschmann S. A deficiency in nucleoside salvage impairs murine lymphocyte development, homeostasis, and survival. J Immunol. 2012;188:3920–3927. doi: 10.4049/jimmunol.1102587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tran CN, Thacker SG, Louie DM, Oliver J, White PT, Endres JL, Urquhart AG, Chung KC, Fox DA. Interactions of T cells with fibroblast-like synoviocytes: role of the B7 family costimulatory ligand B7-H3. J Immunol. 2008;180:2989–2998. doi: 10.4049/jimmunol.180.5.2989. [DOI] [PubMed] [Google Scholar]
- 22.Lefèvre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, Korb A, Schnäker EM, Tarner IH, Robbins PD, Evans CH, Stürz H, Steinmeyer J, Gay S, Schölmerich J, Pap T, Müller-Ladner U, Neumann E. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–1420. doi: 10.1038/nm.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 24.Du F, Lü LJ, Teng JL, Shen N, Ye P, Bao CD. T-614 alters the production of matrix metalloproteinases (MMP-1 andMMP-3) and inhibits the migratory expansion of rheumatoid synovial fibroblasts, in vitro. Int Immunopharmacol. 2012 May;13:54–60. doi: 10.1016/j.intimp.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 25.Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J Biol Chem. 2006;281:36443–36453. doi: 10.1074/jbc.M600788200. [DOI] [PubMed] [Google Scholar]
- 26.Higuchi M, Onishi K, Kikuchi C, Gotoh Y. Scaffolding function of PAK in the PDK1-Akt pathway. Nat Cell Biol. 2008;10:1356–1364. doi: 10.1038/ncb1795. [DOI] [PubMed] [Google Scholar]
- 27.Higuchi M, Kihara R, Okazaki T, Aoki I, Suetsugu S, Gotoh Y. Akt1 promotes focal adhesion disassembly and cell motility through phosphorylation of FAK in growth factor-stimulated cells. J Cell Sci. 2013;126:745–55. doi: 10.1242/jcs.112722. [DOI] [PubMed] [Google Scholar]
- 28.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 29.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Chen T, Zhang J, Mao Q, Li S, Xiong W, Qiu Y, Xie Q, Ge J. Notch1 promotes glioma cell migration and invasion by stimulating beta-catenin and NF-kappaB signaling via AKT activation. Cancer Sci. 2012;103:181–190. doi: 10.1111/j.1349-7006.2011.02154.x. [DOI] [PubMed] [Google Scholar]
- 31.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2:1067–1069. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- 32.Giblett ER, Ammann AJ, Wara DW, Sandman R, Diamond LK. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. 1975;1:1010–1013. doi: 10.1016/s0140-6736(75)91950-9. [DOI] [PubMed] [Google Scholar]
- 33.Cohen A, Gudas LJ, Ammann AJ, Staal GE, Martin DW Jr. Deoxyguanosine triphosphate as a possible toxic metabolite in the immunodeficiency associated with purine nucleoside phosphorylase deficiency. J Clin Invest. 1978;61:1405–1409. doi: 10.1172/JCI109058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu XC, Miranda M, Liu Z, Patel S, Nguyen N, Carson K, Liu Q, Swaffield JC. Novel potent inhibitors of deoxycytidine kinase identified and compared by multiple assays. J Biomol Screen. 2010;15:72–79. doi: 10.1177/1087057109353604. [DOI] [PubMed] [Google Scholar]
- 35.Volin MV, Huynh N, Klosowska K, Chong KK, Woods JM. Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 2007;56:2512–2522. doi: 10.1002/art.22806. [DOI] [PubMed] [Google Scholar]
- 36.Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 37.Higuchi M, Masuyama N, Fukui Y, Suzuki A, Gotoh Y. Akt mediates Rac/Cdc42-regulated cell motility in growth factor-stimulated cells and in invasive PTEN knockout cells. Curr Biol. 2001;11:1958–1962. doi: 10.1016/s0960-9822(01)00599-1. [DOI] [PubMed] [Google Scholar]
- 38.Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, Chung J. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- 39.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–166. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 40.van Zijl F, Zulehner G, Petz M, Schneller D, Kornauth C, Hau M, Machat G, Grubinger M, Huber H, Mikulits W. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169–1179. doi: 10.2217/fon.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polette M, Mestdagt M, Bindels S, Nawrocki-Raby B, Hunziker W, Foidart JM, Birembaut P, Gilles C. Beta-catenin and ZO-1: shuttle molecules involved in tumor invasion-associated epithelial-mesenchymal transition processes. Cells Tissues Organs. 2007;185:61–65. doi: 10.1159/000101304. [DOI] [PubMed] [Google Scholar]
- 42.Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther. 2010;10:219–222. doi: 10.4161/cbt.10.6312548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112:2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- 44.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6:154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 45.MacNaul KL, Chartrain N, Lark M, Tocci MJ, Hutchinson NI. Discoordinate expression of stromelysin, collagenase, and tissue inhibitor of metalloproteinases-1 in rheumatoid human synovial fibroblasts. Synergistic effects of interleukin- 1 and tumor necrosis factor-alpha on stromelysin expression. J Biol Chem. 1990;265:17238–17245. [PubMed] [Google Scholar]
- 46.Tchetverikov I, Ronday HK, Van El B, Kiers GH, Verzijl N, TeKoppele JM, Huizinga TW, DeGroot J, Hanemaaijer R. MMP profile in paired serum and synovial fluid samples of patients with rheumatoid arthritis. Ann Rheum Dis. 2004;63:881–883. doi: 10.1136/ard.2003.013243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westhoff CS, Freudiger D, Petrow P, Seyfert C, Zacher J, Kriegsmann J, Pap T, Gay S, Stiehl P, Gromnica-Ihle E, Wernicke D. Characterization of collagenase 3 (matrix metalloproteinase 13) messenger RNA expression in the synovial membrane and synovial fibroblasts of patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:1517–1527. doi: 10.1002/1529-0131(199907)42:7<1517::AID-ANR27>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 48.Posthumus MD, Limburg PC, Westra J, van Leeuwen MA, van Rijswijk MH. Serum matrix metalloproteinase 3 in early rheumatoid arthritis is correlated with disease activity and radiological progression. J Rheumatol. 2000;27:2761–2768. [PubMed] [Google Scholar]
- 49.Mori H, Nakanishi T. Signal transduction of inflammatory synoviocytes in rheumatoid arthritis. Yakugaku Zasshi. 2008;128:263–268. doi: 10.1248/yakushi.128.263. [DOI] [PubMed] [Google Scholar]