

Abstract
Perineurioma is a rare benign peripheral nerve sheath tumor featuring perineurial differentiation. Perineurioma occurs sporadically with only one reported case in the setting of neurofibromatosis type 1 (NF-1). We present a 6.7-cm soft tissue perineurioma of the lower leg in a 51-year-old man with proven NF-1. The tumor displayed whorled and fascicular pattern with infiltrative margins and expressed EMA, GLUT-1, claudin-1, and CD34. Electron microscopy confirmed diagnosis. Furthermore, lipomatosis, cutaneous angiomatous nodules, vasculopathy, and iliac spine lesion consistent with non-ossifying fibroma were observed. Tumor DNA revealed no NF2 mutations or chromosomal aberrations but a germline NF1-deletion (c.449_502delTGTT) was detected in his blood sample. His brother displayed neurofibromas, duodenal ganglioneuroma and colonic juvenile polyp, and his mother a neurofibroma, cutaneous squamous cell carcinoma, and jejunal gastrointestinal stromal tumor (GIST); both were affected by NF-1. In conclusion, perineurioma may rarely be NF-1 related and should be included in the spectrum of neoplasms occurring in this disorder.
Keywords: Perineurioma, soft tissue, neurofibromatosis, vasculopathy, NF1
Introduction
Perineurioma is a rare benign peripheral nerve sheath tumor displaying perineurial cell differentiation throughout. Since their first description by Lazarus and Trombetta in 1978 [1] several reports and a few large series appeared in the literature [2]. Perineurioma has been traditionally subdivided into intraneural (so-called localized hypertrophic neuropathy) and extraneural (soft tissue) subtypes. Soft tissue perineurioma typically arises in the soft tissue of adults with a mean age of 46 (range, 10-79) years and shows a predilection for the extremities and trunk without gender predilection [2]. Although the tumors are usually well defined but not encapsulated, microscopic infiltrative margins have been reported in 15% [2]. Although chromosomal aberrations affecting the neurofibromatosis type 2 locus (NF-2) at chromosome 22 have been reported in many cases of soft tissue perineurioma [3,4] and somatic NF2 mutations are well known to occur in sporadic perineuriomas [5,6], perineuriomas were to date not perceived to be associated with NF-1 or NF-2, with the exception of a single case reported by Ausmus et al. in a 20-year-old patient with NF-1 [5,6]. Immunohistochemically, soft tissue perineurioma stains with the perineurial cell markers epithelial membrane antigen (EMA), claudin-1, and human erythrocyte glucose transporter-1 (GLUT-1), as well as CD34 but are usually negative for S-100 protein [2]. Atypical features (increased mitotic rate, cellular pleomorphism, hypercellularity, and infiltrative growth) are infrequent and do not seem to be associated with aggressive course or increased risk of recurrence [2].
We here describe an unusual large soft tissue perineurioma grossly arising from the right sural nerve of a 51-year-old man with proven NF-1. In addition, the patient featured multiple lipomas of extremities and trunk, and cutaneous angiomatous nodules (not verified histologically). Besides the conventional histopathological and immunohistochemical findings, we present molecular features of the perineurioma and discuss its possible link to NF-1.
Case presentation
A 51-year-old male with clinical features of NF-1 was admitted to hospital because of a paramalleolar mass lesion of his right lower leg. His relevant medical history included a thoracic neurofibroma resected two years ago and a current nephrolithiasis. Clinical examination revealed several café-au-lait spots (Figure 1A) and multiple cutaneous/subcutaneous tumors of the trunk and extremities, involving his left little finger and index finger, right wrist, left upper leg, thoracic spine, left abdomen, and his head (Figure 1B). Magnetic resonance imaging (MRI) of the lower leg tumor revealed a hyperintense circumscribed mass surrounding the right sural nerve (Figure 1C). Surgical biopsy was obtained from the right leg tumor and was consistent with soft tissue perineurioma. The mass was then excised completely. Additionally, abdominal computed tomography (CT) scan revealed multiple subcutaneous and partly intramuscular mass lesions located in the thoracic and abdominal wall measuring up to 5 cm, as well as a lytic bone lesion in the right iliac crest, consistent with non-ossifying fibroma (Figure 1B inset). Some of these lesions were interpreted as consistent with lipomas, others as neurofibromas with soft tissue signal intensity. Furthermore, a large intramuscular mass of 7.6 x 6 x 3 cm was detected in the left adductor compartment displaying contrast enhancement in MRI. Biopsy of this mass was obtained and was consistent with lipoma. The patient recovered well and was discharged after 20 days. Currently the patient is well and there is no evidence of relapse after 9 months. Genetic counseling of the index patient confirmed that he fulfilled the criteria for the diagnosis of NF-1 as he presented 2 café-au-lait spots >1.5 cm, axillary freckling, multiple neurofibromas, and a positive family history. His mother had a spinal cord neurofibroma, a cutaneous squamous cell carcinoma, and a jejunal gastrointestinal stromal tumor (GIST) being wild-type for KIT and PDGFRA. His brother had neurofibromas located at his hand and chest wall, a duodenal ganglioneuroma positive for protein S-100, but negative for perineurial markers, and a juvenile polyp in the colon. A nephew showed similar cutaneous tumors at the extremities and a sister underwent renal surgery for unknown reason.
Figure 1.
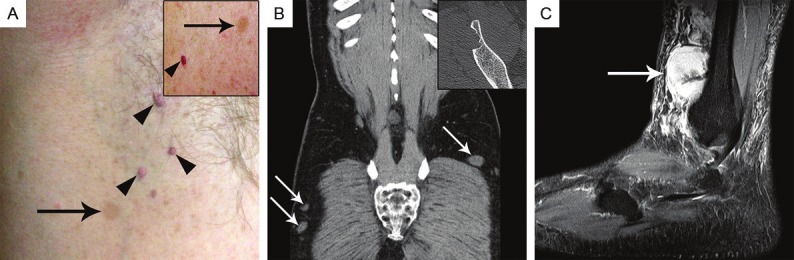
The index patient displayed axillary freckling (A, arrow), cutaneous neurofibromas (arrowheads) as well as café-au-lait spots >1.5 cm on his abdominal wall (A, inset, arrow) and small vascular lesions consistent with cutaneous hemangiomas or venous malformation (A, inset, arrowhead) (although not histopathologically confirmed). Abdominal CT detected multiple small tumors (probably neurofibromas) in the abdominal wall and lower extremity (B, arrows). A lytic bone lesion was detected in the right iliac crest (B, inset), consistent with non-ossifying fibroma. Magnetic resonance imaging of the perineurioma revealed a hyperintense circumscribed lesion located at the lateral lower right leg surrounding the intermediate dorsal cutaneous nerve in sagittal view (C, arrow).
Pathological findings
The tumor resection specimen from the right lower leg measured 9 x 5.4 x 1.5 cm with a 10 cm-nerve encased in the center. The tumor was lobulated and ill defined, partly covered by fascia, and displayed a pale fleshy, solid, white to yellow cut-surface (Figure 2A). The tumor surrounded the sural nerve but it was not embedded in the nerve substance as intraneural perineurioma. Histopathological evaluation revealed a moderately cellular mesenchymal tumor composed of slender bipolar spindle cells with slightly hyperchromatic, occasionally wavy nuclei with fine chromatin, inconspicuous nucleoli and pale eosinophilic cytoplasm with indistinct cell borders. The tumor cells formed short parallel oriented or irregular fascicles with storiform lamellar and focally whorled growth pattern (Figure 2B). Intervening small capillaries and variably prominent interspersed delicate collagen fibers were observed between tumor cells. In the central portion of the tumor, small twigs of peripheral nerves were seen encased by tumor tissue. Furthermore, a prominent involvement and entrapment of surrounding fatty tissue was seen that occasionally deceptively mimicked an integral lipomatous component of the tumor. The resection margins were involved by tumor tissue. No mitotic figures were detected. At the tumor margins several venous vessels showed remarkable vasculopathic changes with significant myointimal spindle cell proliferation, some accompanied by small venule-like capillary vessels (Figure 2C). Immunohistochemical staining for CD34, EMA, alpha-smooth-muscle actin (ASMA), desmin, GLUT-1, claudin-1, S-100 protein, glial fibrillary acidic protein (GFAP), pan-cytokeratin, and Ki67 was performed. The tumor cells expressed CD34, EMA (Figure 2B, inset), GLUT-1, and claudin-1, whereas protein S-100, ASMA, desmin, GFAP, and pan-cytokeratin were not expressed. The proliferative activity assessed by Ki67-staining was estimated at 2%. Sections of the sural nerve showed prominent perineurial proliferation of similar tumor cells coexpressing EMA, claudin-1 and GLUT-1 and extending into adjacent fatty tissue. Nests of perineurial cells were seen infiltrating along small venous vessels between fascicles of the nerve trunk. Interestingly, perineurial cell markers highlighted an abnormal network of intraneural perineurial cells that were arranged in a reticular pattern lacking the onion skin appearance of intraneural perineurioma. The latter involved only a few nerve fascicles of the sural nerve. Transmission electron microscopy (EM) performed on 3% glutaraldehyde-fixed, uranyl acetate and lead citrate stained ultrathin sections revealed spindled tumor cells with long tapering nuclei, inconspicuous nucleoli, bipolar cytoplasmic processes embedded in a collagen-rich matrix and foci of numerous intracytoplasmatic globular structures compatible with pinocytotic vesicles as well as a discontinuous external lamina – features previously reported for perineuriomas [2]. Mutation analysis of NF1 (from patient’s blood) revealed a known 4-bp deletion in the NF1 gene (c.449_502delTGTT) [7], confirming the germline mutation and diagnosis of NF-1. Chromosomal analysis from blood cells revealed a normal karyotype. Furthermore, mutation analysis of NF2 exon 1-15 was performed as described previously [5] and revealed no NF2 mutation in the perineurioma. Comparative genomic hybridization (CGH) performed as described before [8] detected no chromosomal imbalances.
Figure 2.
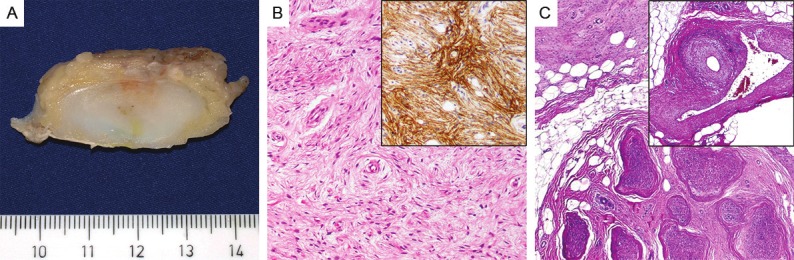
The resected tumor had ill-defined margins and a pale fleshy, solid cut-surface (A). On microscopic view, the tumor cells were arranged in parallel oriented fascicles with a focally whorled growth pattern and showed ovoid to elongated slender nuclei and pale cytoplasm (B, H&E, x100) with expression of EMA (B, inset). Perineurial proliferations were seen extending from the perineurium of the sural nerve into surrounding adipose tissue (C, H&E, x40), Striking vasculopathy was seen within surrounding fat with prominent thickening of arterial and venous vessels (C, inset, H&E, x40).
Discussion
Soft tissue perineurioma is a rare distinctive tumor entity with exclusive perineurial cell differentiation as evident from regular expression of perineurial cell markers and ultrastructural features of perineurial cells [2]. Recognition of perineurioma and its distinction from potential low-grade malignant mimics is of clinical importance as this tumor usual behaves in a benign fashion with a low recurrence rate (5%) [2]. In the present case, the tumor was recognizably originating from the sural nerve. It is very unusual for perineurioma to arise from a grossly identifiable nerve in contrast to several malignant peripheral nerve sheath tumors (MPNST). Thus, our case is unique and it is likely that this unusual gross presentation is related to the background NF-1 disease in this patient.
NF-1 (von Recklinghausen disease) is an autosomal-dominant inherited disorder with an estimated worldwide incidence of approximately 1:3,000 individuals which makes NF-1 the most common hereditary multitumor syndrome [9,10]. The NF1 gene (OMIM *613113) is located at 17q11.2 [9]. NF1 gene mutations leads to increased cell growth, unopposed Ras activity and constitutive downstream signaling [9]. As a phacomatosis, NF-1 presents various lesions and/or neoplasms of the skin and neural system, including café-au-lait spots, axillary or inguinal freckling, Lisch-nodules and cutaneous neurofibromas (60%). An increased susceptibility to other malignances in patients with NF-1 has been observed, including sarcomas (MPNST and others), but squamous cell and other types of carcinomas have been also observed occasionally [11]. Also gastrointestinal tumor manifestation has been described, such as small bowel and/or stomach neurofibromas and undifferentiated sarcomas of the upper gastrointestinal tract [10]. GISTs arising in the setting of NF-1 are well documented and represent 1.5% of all GISTs [10]. These tumors typically present as small asymptomatic lesions, histologically display spindle cell morphology and so-called skeinoid fibres, a low mitotic rate, and KIT/PDGFRA wild type.
In patients with NF-1, a positive family history is reported in approximately 50% of cases [9]. Increased morbidity of disease in individuals born to affected mothers than in those born to affected fathers or those who have new mutations suggested a possible maternal impact on the severity of NF-1 [12]. This mechanism may also play a role in the family reported herein. An association of perineuriomas with NF-1, NF-2 or other syndromes has not been observed in larger series. To our knowledge, only a single case of perineurioma associated with NF-1 has been documented [2,6]. However, in the present case, the patient, his brother, and mother had NF-1 and displayed associated lipomas, neurofibromas, a ganglioneuroma, a jejunal GIST, and a colonic juvenile polyp. Of note, an association of NF-1 and juvenile polyps, vascular anomalies (including “prominent veins” on the trunk), and horseshoe kidney have previously been reported by Oktenli et al in a patient with NF-1 and his relatives [13]. Although in our case the presence of horseshoe kidneys was not examined in the patient or his relatives, the patient and his mother suffered from nephrolithiasis, and a sister underwent renal surgery for unknown reason. Furthermore, our patient displayed cutaneous angiomatous nodules (Figure 1), a striking vasculopathy in the soft tissue of the lower extremity surrounding the perineurioma, a lytic bone lesion (consistent with non-ossifying fibroma), and his brother had a juvenile polyp, indicating that this family might have a similar phenotype as the family reported by Oktenli et al. [13]. Since an NF1 mutation was not detected in the other family reported by Oktenli et al, a phenotype-genotype correlation can not yet be made. Based on the National Institutes of Health Consensus Conference criteria developed in 1988, the diagnosis was made in our patient due to presence of multiple major criteria and positive family history [9]. In the present family, multiple neurofibromas and an affected relative were present as diagnostic criteria for all three patients.
Although chromosomal aberrations at chromosome 22 have been reported in perineuriomas previously, the perineurioma in this case displayed no chromosomal imbalances [3,4]. As previously reported by Lasota et al, the NF2 gene (OMIM *607379 at 22q12.2) may be mutated in sporadic perineuriomas [5]. In that study, point mutations in the 5’-untranslated region and in exons 3, 6, and 8 were detected in 4 of 8 perineuriomas examined [5]. In the present case the perineurioma did not harbor an NF2 mutation in all 15 examined exons and also lacked chromosomal aberrations at chromosome 22, thus supporting our hypothesis that the tumor is likely driven by NF1 inactivation in the setting of his genetic disease. Given the facts that the precise subtyping of many benign peripheral nerve sheath tumors might be difficult or even some times impossible without the use of immunohistochemistry, and that perineurial markers are not always applied for diagnosing benign neurogenic tumors in patients with a known NF-1, it is not excluded that perineuriomas be under-recognized in NF-1 setting. A recent study showed significant over-representation of hybrid peripheral nerve sheath tumors (61% hybrid schwannoma-neurofibroma) in patients with schwannomatosis, NF-2, and NF-1 [14]. Retrospective analysis of archival cases revealed that at least 9% of hybrid schwannoma-neurofibromas affected patients with NF-1 in the same study [14]. Although no clear-cut perineurial cell differentiation was stated in that study, the authors reported a mosaic GLUT-1 immunopositivity in the schwannoma-like areas in several cases which was associated with CD34 expression suggesting at least partial perineurial differentiation. During preparation of this manuscript, another study was published by Kacerovska et al. [15] describing 5 cases of hybrid nerve sheath tumors composed of plexiform neurofibroma with considerable perineurial component in patients with NF-1 (three from the same family). All 4 lesions were evident on immunohistochemistry. The fifth lesion was evident in HE stained sections and showed evidence of malignancy in the S100 positive neurofibromatous component. These recent studies suggest under-recognition of perineurial differentiation in benign peripheral nerve sheath tumors traditionally classified as neurofibromas in NF-1.
In conclusion, the present case highlights the rare occurrence of soft tissue perineurioma in a background of NF-1. This uncommon entity might be underreported in NF-1 patients. We therefore recommend using perineurial cell markers in classifying benign peripheral nerve sheath tumors in patients with NF-1 in order to assess the possibility of under-recognized NF-1-associated perineurial lesions.
Acknowledgements
Inga-Marie Schaefer is supported by a research grant from the Dr. Mildred Scheel Stiftung für Krebsforschung (No. 110822).
Written informed consent was obtained from the patient and his brother for the publication of this case report and any accompanying images. As next of kin they provided informed consent for their late mother.
Disclosure of conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823–1829. doi: 10.1002/1097-0142(197805)41:5<1823::aid-cncr2820410525>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 2.Hornick JL, Fletcher CD. Soft tissue perineurioma: clinicopathologic analysis of 81 cases including those with atypical histologic features. Am J Surg Pathol. 2005;29:845–858. doi: 10.1097/01.pas.0000155166.86409.d2. [DOI] [PubMed] [Google Scholar]
- 3.Lasota J, Wozniak A, Debiec-Rychter M. Loss of chromosome 22q and lack of NF2 mutations in perineuriomas [abstract 46] . Mod Pathol. 2000;13:11A. [Google Scholar]
- 4.Giannini C, Scheithauer BW, Jenkins RB, Erlandson RA, Perry A, Borell TJ, Hoda RS, Woodruff JM. Soft-tissue perineurioma. Evidence for an abnormality of chromosome 22, criteria for diagnosis, and review of the literature. Am J Surg Pathol. 1997;21:164–173. doi: 10.1097/00000478-199702000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Lasota J, Fetsch JF, Wozniak A, Wasag B, Sciot R, Miettinen M. The neurofibromatosis type 2 gene is mutated in perineurial cell tumors: a molecular genetic study of eight cases. Am J Pathol. 2001;158:1223–1229. doi: 10.1016/S0002-9440(10)64072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ausmus GG, Piliang MP, Bergfeld WF, Goldblum JR. Soft-tissue perineurioma in a 20-year-old patient with neurofibromatosis type 1 (NF1): report of a case and review of the literature. J Cutan Pathol. 2007;34:726–730. doi: 10.1111/j.1600-0560.2006.00702.x. [DOI] [PubMed] [Google Scholar]
- 7.Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S, Nurnberg P. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. 2000;66:790–818. doi: 10.1086/302809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunawan B, Schulten HJ, von Heydebreck A, Schmidt B, Enders C, Höer J, Langer C, Schüler P, Schindler CG, Kuhlgatz J, Füzesi L. Site-independent prognostic value of chromosome 9q loss in primary gastrointestinal stromal tumours. J Pathol. 2004;202:421–429. doi: 10.1002/path.1537. [DOI] [PubMed] [Google Scholar]
- 9.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–133. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 10.Agaimy A, Vassos N, Croner RS. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen's disease): clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol. 2012;5:852–862. [PMC free article] [PubMed] [Google Scholar]
- 11.Knight WA III, Murphy WK, Gottlieb JA. Neurofibromatosis associated with malignant neurofibromas. Arch Dermatol. 1973;107:747–750. [PubMed] [Google Scholar]
- 12.Miller M, Hall JG. Possible maternal effect on severity of neurofibromatosis. Lancet. 1978;2:1071–1073. doi: 10.1016/s0140-6736(78)91804-4. [DOI] [PubMed] [Google Scholar]
- 13.Oktenli C, Gul D, Deveci MS, Saglam M, Upadhyaya M, Thompson P, Consoli C, Kocar IH, Pilarski R, Zhou XP, Eng C. Unusual features in a patient with neurofibromatosis type 1: multiple subcutaneous lipomas, a juvenile polyp in ascending colon, congenital intrahepatic portosystemic venous shunt, and horseshoe kidney. Am J Med Genet A. 2004;127A:298–301. doi: 10.1002/ajmg.a.30008. [DOI] [PubMed] [Google Scholar]
- 14.Harder A, Wesemann M, Hagel C, Schittenhelm J, Fischer S, Tatagiba M, Nagel C, Jeibmann A, Bohring A, Mautner VF, Paulus W. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am J Surg Pathol. 2012;36:702–709. doi: 10.1097/PAS.0b013e31824d3155. [DOI] [PubMed] [Google Scholar]
- 15.Kacerovska D, Michal M, Kuroda N, Tanaka A, Sima R, Denisjuk N, Kreuzberg B, Ricarova R, Kazakov DV. Hybrid peripheral nerve sheath tumors, including a malignant variant in type 1 neurofibromatosis. Am J Dermatopathol. 2013;35:641–649. doi: 10.1097/DAD.0b013e31827e2917. [DOI] [PubMed] [Google Scholar]