

Abstract
Background & Aims
Celiac disease is an increasingly recognized disorder in Caucasian populations of European origin. Little is known about its prevalence in non-Caucasians. Although it is thought to be a cause of iron deficiency anemia, little is known about the extent to which celiac disease contributes to iron deficiency in Caucasians, and especially non-Caucasians. We analyzed samples collected from participants in the Hemochromatosis and Iron Overload Screening (HEIRS) study to identify individuals with iron deficiency and assess the frequency of celiac disease.
METHODS
We analyzed serum samples from white men (25 y old or older) and women (50 y old or older) who participated the HEIRS study; cases were defined as individuals with iron deficiency (serum level of ferritin ≤12 mg/L) and controls were those without (serum level of ferritin >100 mg/L in men and >50 mg/L in women). All samples were also analyzed for human recombinant tissue transglutaminase immunoglobulin A; positive results were confirmed by an assay for endomysial antibodies. Patients with positive results from both celiac disease tests were presumed to have untreated celiac disease, and those with a positive result from only 1 test were excluded from analysis. We analyzed HLA genotypes and frequencies of celiac disease between Caucasians and non-Caucasians with iron deficiency.
RESULTS
Celiac disease occurred in 14 of 567 of cases (2.5%) and in only 1 of 1136 controls (0.1%; Fisher’s exact test, P=1.92 × 10−6). Celiac disease was more common in Caucasian cases (14/363, 4%) than non-Caucasian cases (0/204; P=.003). Only 1 Caucasian control and no non-Caucasian controls had celiac disease. The odds of celiac disease in individuals with iron deficiency was 28-fold (95% confidence interval, 3.7–212.8) that of controls; 13/14 cases with celiac disease carried the DQ2.5 variant of the HLA genotype.
CONCLUSIONS
Celiac disease is associated with iron deficiency of Caucasians. Celiac disease is rare among non-Caucasians—even among individuals with features of celiac disease, such as iron deficiency. Celiac disease is also rare among individuals without iron deficiency. Men and post-menopausal women with iron deficiency should be tested for celiac disease.
Keywords: SNP, risk factor, gluten allergy, intestine, absorption
INTRODUCTON
Iron deficiency is one of the most common nutritional deficiencies in both the developed and developing populations (1). In those with adequate intake of iron, deficiency can occur either from loss of blood or via the failure of absorption of sufficient iron from the diet by the enterocytes of the proximal small intestine. Iron deficiency is the most common nutritional disorder in the world with an estimated four to five billion affected persons. Disorders of iron metabolism underlie some of the most prevalent diseases in humans and encompass a broad spectrum of clinical manifestations, ranging from anemia to iron overload and neurodegenerative diseases (2).
We hypothesized that as iron is exclusively absorbed via the proximal small intestinal mucosa, celiac disease (CD) should be more commonly found in those with iron deficiency and not often in iron replete individuals. Furthermore, we examined whether or not the extent of the occurrence of CD in Caucasians is replicated in non-Caucasians. A unique multiethnic population of iron-deficient individuals was identified in the Hemochromatosis and Iron Overload Screening (HEIRS) Study (3). In the HEIRS Study, 101,168 participants were screened with serum biochemical tests of iron status (4). As expected, the HEIRS Study identified not only those with biochemical evidence of iron overload, but also iron deficiency (5). To identify CD associated with iron deficiency, we performed a sequential serological strategy using sera collected from HEIRS Study participants who were iron deficient and iron replete. Case status was determined by serum ferritin concentration. The association between case status and CD serology was examined.
We further hypothesized that low serum ferritin and an appropriate Human Leukocyte Antigen (HLA) variant, may be predictive of CD. If so, it could be important for identification of asymptomatic cases that should be monitored for CD.
METHODS
Study population
The current study utilized a subset of subjects who had been enrolled in the initial screening phase of the HEIRS Study at five Field Centers encompassing six geographic locations including Alabama, California, District of Columbia, Hawaii, and Oregon in the United States, and Ontario, Canada (3, 4). Participants were eligible for the current study if they had not withdrawn consent and had agreed to blood storage. Selection criteria for this sub-study of the HEIRS population included self-report of white or Caucasian, African-American, Hispanic or Asian race/ethnicity, males at least 25 years of age and females at least 50 years. Females younger than 50 years were excluded because of the possibility of pre-menopausal iron depletion from blood loss. Cases of iron deficiency were defined as having a serum ferritin concentration (SF) ≤ 12 μg/L which is indicative of depletion of iron stores (6, 7). Controls (SF >100 μg/L in men, SF >50 μg/L in women) were frequency matched 2:1 to cases by sex and geographic location. Serum hemoglobin was not available.
Approval for the study was obtained from the following: Institutional Review Board of the University of California, Irvine; Institutional Review Board of the University of California, Berkeley; Institutional Review Board of the University of Minnesota; Howard University Institutional Review Board; Institutional Review Board of the University of Alabama at Birmingham; Institutional Review Board of Kaiser Permanente Center for Health Research; Institutional Review Board of Wake Forest University Health Sciences; the University of Western Ontario Research Ethics Board for Health; and the Institutional Review Board of the Department of Veterans Affairs Long Beach Healthcare System. Written informed consent was obtained from all participants.
Laboratory Methods
Celiac disease serology
The CD screen was performed using a sequential approach that we have used with high sensitivity and specificity for CD and is illustrated in Figure 1 (8, 9). First, for all samples, the IgA was measured against human recombinant tissue transglutaminase by ELISA (Inova Diagnostics, San Diego, CA). For those samples showing positive or borderline results for the TTG-IgA, an anti-endomysial antibody (ema-IgA) test was then performed. Double positives were presumed to have untreated CD. Single positives were considered to have indeterminate CD status and were excluded from analysis (9). C-reactive protein (CRP), alanine aminotransferase (ALT), and gamma-glutamyltransferase (GGT) were measured to identify acute phase protein elevations in serum ferritin.
Figure 1.
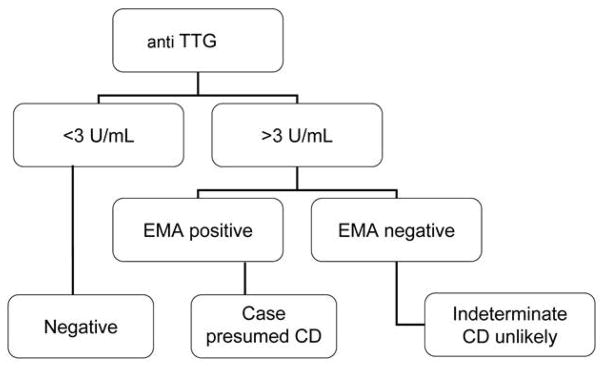
Schema for detection of celiac disease status
Genome-wide genotyping and quality control procedures
Buffy coat DNA was extracted and purified by SDS cell lysis followed by a salt precipitation method for protein removal using commercial Puregene® reagents (Gentra System, Inc., Minneapolis, MN, now Qiagen, Valencia, CA). Using a genome-wide association study (GWAS) approach, genotyping was performed on a subset of Caucasian samples including all available cases and controls frequency-matched 1:1 to cases by sex and geographic location (361 cases and 352 controls) using the Illumina HumanCNV370K BeadChip platform as reported elsewhere (5). Eight samples were excluded due to misclassification of sex with data from 706 samples (357 cases and 349 controls) remaining for further analysis.
Predicting HLA risk variants
For Caucasians, the genotypes that confer susceptibility to CD were predicted using the method described and validated in several populations of European origin (10). Briefly, we utilized the single nucleotide polymorphism (SNP) data from a larger genome-wide search undertaken in the subset of iron-deficient Caucasian cohort (5). Information was extracted using the specific six SNPs thought to be informative for HLA class 2 typing and to be able to distinguish between DQ2.2, DQ2.5, DQ7, and DQ8 risk variants based on strong linkage disequilibrium (LD) at the HLA loci. Out of six SNPs, rs2187668, rs2395182, rs7775228 and rs4639334 were available on the Illumina chip, while rs4639334 and rs4713586 were not measured. Therefore we were able to infer the DQ2.5 and DQ8 variants, but not the DQ7 variant, and we could not differentiate between DQ2.2 and DQ4 status. Details of HLA variant prediction can be found in Monsuur et al. (10) Briefly, predictive alleles are T on rs2187668 for DQ2.5, G on rs7454108 for DQ8, and T and G on rs2395182 and rs7775228, respectively, for DQ2.2 and DQ4, with rs4713586 further differentiating between these two variants.
Statistical analyses
Samples with indeterminate results for CD were excluded from analyses. From the remainder, the proportion of iron-deficient cases and iron-replete controls with CD was estimated for the Caucasian and non-Caucasian cohorts. Exact 95% confidence intervals for these proportions were computed. Fisher’s exact test was used to examine the association between iron deficient case control status and the presence of CD, in combined data from Caucasians and non-Caucasians. Log-binomial regression was applied to estimate the odds of CD in iron-deficient cases relative to that of controls. Models were examined to assess the effect of including covariates representing race-ethnicity and presence of an HFE mutation. For the subset of Caucasians, Fisher’s exact test was applied to examine the association between the presence of CD and HLA risk variants, represented as categorical variables (absent, heterozygous, homozygous).
RESULTS
Serological Testing
A total of 1713 subjects were tested for TTG IgA antibodies. Participants included 1100 Caucasians, 221 African Americans, 153 Asians, and 239 Hispanics. Of these, 25 subjects had TTG > 3 AU/mL and had EMA testing done; of these, 15 were positive (Fig. 2). Fourteen of 571 iron-deficient cases were positive for the double serology compared to just one control (Fig. 3). All of the seropositive tests were seen in Caucasians and none in the non-Caucasians (Fig. 4; Table 1). Only one Caucasian control was also positive, suggesting that CD is relatively rare in iron-replete people.
Figure 2.
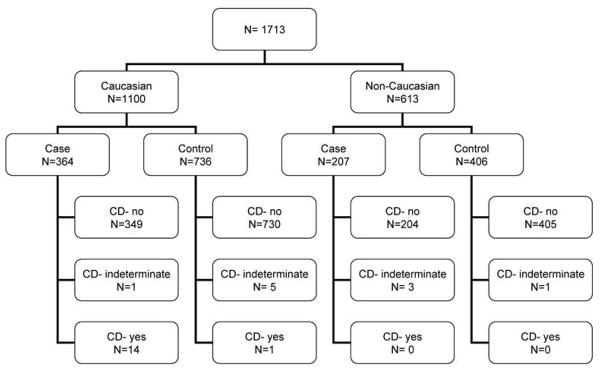
Results of serologic testing for celiac disease in Caucasian and non-Caucasians
Figure 3.

Percentage of positive celiac serology in iron-deficient cases versus iron-replete controls. Fisher’s exact test (p = 1.96×10−6).
Figure 4.

Positive celiac serology in Caucasians and non-Caucasians. All individuals with celiac disease were Caucasian (15/1094); celiac disease was absent in non-Caucasians (0/609, Fisher’s exact test p = 0.002).
TABLE 1.
Characteristics of Caucasian and non-Caucasian Cases and Controls
| Caucasian | non-Caucasian | |||
|---|---|---|---|---|
|
| ||||
| Characteristics | Iron-deficient cases (n=364) | Iron-replete controls (n=736) | Iron-deficient cases (n=207) | Iron-replete controls (n=406) |
| Age, mean (SD) | 59 (10.2) | 61 (10.7) | 57 (10.9) | 57 (11.5) |
| Male, n (%) | 91 (25.0%) | 179 (24.3%) | 64 (30.9%) | 131 (32.3%) |
| Celiac disease1, n (%) | ||||
| Yes | 14 (3.8%) | 1 (0.1%) | 0 | 0 |
| Indeterminate | 1 (0.3%) | 5 (0.7%) | 3 (1.4%) | 1 (0.2%) |
| No | 349 (95.9%) | 730 (99.2%) | 204 (98.6%) | 405 (99.8%) |
| HFE genotype, n (%) | ||||
| C282Y/C282Y | 4 (1.1%) | 8 (1.1%) | 0 | 0 |
| C282Y/H63D | 7 (1.9%) | 17 (2.3%) | 1 (0.5%) | 2 (0.5%) |
| H63D/H63D | 9 (2.5%) | 21 (2.9%) | 1 (0.5%) | 1 (0.3%) |
| C282Y/wt | 36 (9.9%) | 75 (10.2%) | 3 (1.5%) | 11 (2.7%) |
| H63D/wt | 90 (24.7%) | 169 (23.0%) | 18 (8.7%) | 47 (11.6%) |
| wt/wt | 218 (59.9%) | 446 (60.6%) | 184 (88.9%) | 345 (85.0%) |
| Laboratory measures | ||||
| CRP (mg/dL), mean (SD) | 3.95 (5.53) | 6.16 (12.27) | 3.85 (4.98) | 5.25 (7.47) |
| ALT (U/L), mean (SD) | 20.05 (8.04) | 21.18 (16.4) | 15.46 (10.08) | 18.99 (34.59) |
| GGT (U/L), mean (SD) | 24.9 (33.41) | 32.05 (46.93) | 28.83 (38.82) | 37.09 (64.94) |
| CEA (ng/mL), mean (SD) | 1.67 (1.46) | 1.51 (1.49) | 1.51 (1.49) | 1.46 (0.93) |
The CD screen was performed using a sequential approach. First, for all samples, the IgA was measured against human recombinant tissue transglutaminase by ELISA (Inova Diagnostics, San Diego, CA). For those samples showing positive or borderline results for the TTG-IgA, an anti-endomysial antibody (ema-IgA) test was then performed. Double positives were presumed to have untreated CD. Single positives were considered to have indeterminate CD status and were excluded from analysis.
Frequency of Celiac Disease
Excluding data from participants with indeterminate serological screen results (6 Caucasians, 4 non-Caucasians), evidence of CD was found in 14 of the remaining 567 iron–deficient cases (2.5%, 95% C.I., 2.4%, 4.1%) but in only 1 of 1136 (0.1%; 0.0%, 0.5%) iron–replete controls (Table 1, Fisher’s exact test, p=1.92×10−6). From log binomial regression, the odds of CD in individuals with iron deficiency was 28 (3.7, 212.8) times that in controls. The estimated odds of CD remained 28 (3.7, 214.3) when a variable was included in the model that represented the presence of any HFE genotype (Wald chi-square, p=0.054). All individuals with CD were Caucasian (15 of 1094); CD was absent in non-Caucasians (0 of 609, Fisher’s exact test p = 0.002). Of the 363 Caucasian iron deficient cases, 14 (3.9%; 2.1%, 6.4%) had CD. Of the 204 non–Caucasian cases, none had CD (Fisher’s exact test, p = 0.003). Only one Caucasian control and no non–Caucasian controls had CD.
Table 1 shows distributions of sex, HFE genotype mutations and self-reported clinical complications in iron-deficient and iron replete cases of iron deficiency and iron–replete controls for 364 Caucasians and 736 non-Caucasians as well as summary statistics for age, CRP, ALT, GGT, and CEA measures. The observed frequency of HFE genotypes was similar in iron-deficient and iron-replete cases and controls within Caucasians and non-Caucasians. Observed mean values for CRP, ALT, and GGT were lower in cases of iron deficiency than in controls for both race/ethnic groups. The observed proportion of subjects with self-reported arthritis was higher in Caucasians than in non-Caucasians, for both iron deficient cases and iron-replete controls, and lower in cases of iron deficiency than iron-replete controls for both race/ethnic groups.
Table 2 summarizes HFE genotype mutations, laboratory measures, and self-reported clinical complications in iron-deficient and iron replete Caucasians by categories of CD status (present, absent, indeterminate). There were no iron-deficient cases with homozygosity for either C282Y or H63D mutations in the HFE gene and no compound homozygotes for these mutations. Six of 14 cases of iron deficiency with CD (43%) had self-reported arthritis, but no other assessed clinical complications. The single iron-replete control with CD did not report any of the assessed clinical complications.
TABLE 2.
Assessment of HFE gene mutations, laboratory measures, and clinical outcomes in Caucasians.
| Characteristics | Celiac Disease Status in Iron-deficient Caucasian Cases (n=364) | Celiac Disease Status in Iron-replete Caucasian Controls (n=736) | ||
|---|---|---|---|---|
|
| ||||
| Yes 14 (3.8%) |
No 349 (95.9%) |
Yes 1 (0.1%) |
No 730 (99.2%) |
|
|
|
||||
| HFE genotype, n (%) | ||||
| C282Y/C282Y | 0 | 4 (1.2%) | 0 | 8 (1.1%) |
| C282Y/H63D | 0 | 7 (2.0%) | 0 | 17 (2.3%) |
| H63D/H63D | 0 | 9 (2.6%) | 0 | 21 (2.9%) |
| C282Y/wt | 3 (21.4%) | 33 (9.5%) | 0 | 75 (10.3%) |
| H63D/wt | 4 (28.6%) | 86 (24.6%) | 1 (100.0) | 165 (22.6%) |
| wt/wt | 7 (50.0%) | 210 (60.2%) | 0 | 444 (60.8%) |
| Laboratory measures | ||||
| CRP (mg/dL), mean (SD) | 2 (2.62) | 4.03 (5.61) | 0.40 | 6.2 (12.31) |
| ALT (U/L), mean (SD) | 27.21 (8.5) | 19.74 (7.91) | 12.00 | 21.24 (16.45) |
| GGT (U/L), mean (SD) | 14.21 (6.1) | 25.34 (34.03) | 19.00 | 32.13 (47.11) |
| CEA (ng/mL), mean (SD) | 1.38 (0.7) | 1.69 (1.49) | 1.20 | 1.52 (1.49) |
| Self report | ||||
| Arthritis (n, %) | 6 (42.9%) | 145 (41.6%) | 0 | 326 (44.7%) |
| Diabetes (n, %) | 0 | 48 (13.8%) | 0 | 119 (16.3%) |
| Liver Disease or Liver Cancer (n, %) | 0 | 1 (0.3%) | 0 | 21 (2.9%) |
| Heart Failure (n, %) | 0 | 26 (7.5%) | 0 | 39 (5.3%) |
| Fertility problems or impotence (n, %) | 0 | 23 (6.6%) | 0 | 30 (4.1%) |
For Caucasian males and females, Table 3 displays the distribution of iron deficient cases and controls and those excluded from the study. For 17,342 men at least 25 years old, there were 0.78% of eligible HEIRS participants with iron deficiency and 72.4% who were iron replete. There were 3.03% of 15, 289 females at least 50 years of age who were iron deficient and 74.8% who were iron replete. In this case-control study serum ferritin thresholds were used to determine a subsample of all eligible HEIRS participants that consisted of iron-deficient cases (SF ≤ 12 μg/L) and iron-replete controls (SF >100 μg/L in men, SF >50 μg/L in women). Celiac disease serology was not assessed in 4,658 (26.9%) of eligible males with 12 μg/L <SF ≤100 μg/L nor in 3,389 (22.2%) of eligible females with12 μg/L <SF ≤50 μg/L.
TABLE 3.
Distribution of Caucasian HEIRS participants by serum ferritin levels
| Iron Deficiency Status
| ||||
|---|---|---|---|---|
| Gender | Iron Deficient | Excluded from Study | Iron Replete | Total |
| 0 < SF < 12 | 12 < SF ≤ 100 for men, 12 < SF ≤ 50 for women | SF >100 for men SF > 50 for women |
||
| Male, age ≥ 25 y (N, %) | 136 (0.78%) | 4,658 (26.9%) | 12,548 (72.4%) | 17,342 |
| Female, age ≥ 50 y (N, %) | 464 (3.03%) | 3,389 (22.2%) | 11,436 (74.8%) | 15,289 |
|
| ||||
| Total | 600 | 8,047 | 23,984 | 32,631 |
Frequency of HLA risk variants
Using four SNPs from HLA type 2 genes genotyped in a subset of 706 Caucasians (357 cases and 349 controls) we were able to identify the DQ8 variant in 139 samples (135 heterozygous for the variant, and four homozygous), DQ 2.2 or DQ4 variants in 177 samples (165 heterozygous, 12 homozygous) and the DQ2.5 variant in 161 samples (147 heterozygous, 14 homozygous). None of the DQ8 variants were identified in 14 iron-deficient cases with CD; however, 5 of these cases had DQ 2.2 or DQ4 heterozygous variants and 13 had the DQ2.5 variant (12 heterozygous, 1 homozygous). Presence of DQ2.5 was highly significantly associated with CD status in iron-deficient cases compared to controls (Fisher’s exact test, p = 7.183 × 10−8), while the presence of other variants showed no significant association with CD.
DISCUSSION
In this case-control study of participants in the HEIRS Study, analyses of data from iron-deficient cases and iron-replete controls identified an increased rate of CD in those within the iron-deficient cases and just one case on the iron-replete controls. This finding is consistent with iron absorption in the part of the intestine that is the most affected in CD (11–13). It has been suggested that there may be loss of blood from the intestine in CD, particularly in children—but, this is a matter of some controversy (14–18).
Iron deficiency anemia is commonly observed in CD and can occur even in the absence of GI symptoms. Iron deficiency anemia without evidence of intestinal malabsorption is encountered in approximately 50% of patients with subclinical CD (19, 20). A prospective study of 300 consecutive patients referred for hematologic evaluation of obscure or refractory iron deficiency anemia, identified 13 new cases of adult CD (4%) in those with iron deficiency anemia only (21, 22). Additional studies have found that among patients who have unexplained iron deficiency anemia, CD is responsible for anemia in 5%–6% of cases (23–25). A characteristic of iron deficiency anemia associated with CD is that it is refractory to oral iron treatment (26). The prevalence of CD in those studies may be biased to those who have iron deficiency anemia. Other causes of iron deficiency anemia are possible in CD, including blood loss.
Looker and colleagues estimated the prevalence of iron deficiency and iron deficiency anemia in the United States based on data collected in third National Health and Nutrition Examination Survey (NANES III, 1988–1994) (27). In females from 50 to 69 years of age, the prevalence of iron deficiency was 5%, and the prevalence of iron deficiency anemia was 2%. For those at least 70 years old, these values were 7% and 2%, respectively. In males under age 50, there was less than 1% prevalence of iron deficiency and iron deficiency anemia. In those from 50 to 69 years of age, the prevalence of iron deficiency was 2%, and the prevalence of iron deficiency anemia was 1%. For those at least 70 years old, these values were 4% and 2%, respectively.
We examined CD in iron deficiency without a selection bias for the presence of anemia. There is little data on the frequency of CD in individuals with iron deficiency that have not been selected for investigation of anemia. Our data showing a low prevalence of CD in iron-replete people suggests a low likelihood of unrecognized CD in those who are iron replete.
While it has been well established practice to test for CD as part of the investigation of subjects with unexplained iron deficiency anemia, it has not been clear whether finding iron deficiency itself is enough to justify testing for CD. Our study indicated a significant number of individuals with unrecognized CD who could benefit from appropriate treatment among the iron-deficient Caucasian men and postmenopausal women. We suggest that evidence of iron deficiency warrants an assessment for CD. We note, however, that pre-menopausal women were excluded in this study and it will be important to assess the role of CD in the etiology of iron deficiency in this population.
Using only six tagging SNPs that predict the CD-associated HLA variants has been shown to be very effective, correctly identifying risk types for >95% of CD patients (10) The most important risk factor for CD is DQ2.5, with the highest risk of CD in people homozygous for this variant (28, 29). In our study, for the subset of Caucasian samples, this variant was present in 13 of 14 iron-deficient CD subjects, significantly greater than the frequency in non-CD subjects (p = 7.183 × 10−8). This confirms previous findings of usefulness of tagging SNP approach as an initial screening tool for CD in Caucasians since the number of subjects with DQ2.5 is overrepresented among subjects with CD. Our findings also raise the question of the need to not only serologically test the individuals carrying HLA risk variants to confirm CD, but also further test them for presence of iron deficiency.
While CD has become relatively common in Caucasians (30, 31), the occurrence of CD has been little studied in ethnic groups. A single study suggested that CD was quite rare in Type I diabetes in African Americans in New York City (32). In a second study, the prevalence of CD was low (0.33%) in an urban, predominately male, African-American population with iron deficiency (33). The present study examines CD in non-Caucasians, who would otherwise be considered at risk for CD due to iron deficiency anemia, and in iron-replete controls and found none in these subgroups. This suggests that CD is primarily a disease affecting Caucasians and, if replicated in larger datasets, would indicate that CD is not a significant clinical concern in non-Caucasian individuals. Thus, it is possible that non-Caucasians either lack genes that drive susceptibility for CD, such as DQ2, or have protective genes that protect them from the development of CD. Alternatively, it is possible that there may be some culturally-directed cuisine or that dietary habits may be substantially different in those non-Caucasians, reducing for example their intake of gluten. However, there are little data to suggest that this is true in residents of the United States as for the majority of participants in the HEIRS Study.
A limitation in the Non-Caucasian group is the likely low representation of south Asians who have been shown to have a prevalence of CD similar to Caucasians (34). It is likely, though not addressed in this study, that CD could contribute to Iron deficiency in South Asians.
A limitation to this study may be the relatively small number of cases with iron deficiency determined through screening of 101,168 adults. Nevertheless, we identified an enriched number of subjects with CD that met our robust serological criteria for CD (35). Of course, replication of this study in other groups is necessary. For example, testing for CD in larger cohorts of non-Caucasians is needed.
Our study design differs from previously reported studies in that the testing was conducted on samples from iron-deficient and control groups identified through population-based screening of participants. In summary, CD was found in 4% (14/364) of Caucasians with iron deficiency, and in only 0.1% (1/736) of Caucasians who were iron-replete. In contrast, there were no cases of CD found in 207 iron-deficient and 406 iron-replete non-Caucasians. Testing for CD should be considered in those with iron deficiency irrespective of the presence of anemia. In this study, CD was confined to those Caucasians with iron deficiency and did not appear to affect other races, even those with iron deficiency.
Acknowledgments
We sincerely thank the HEIRS Study participants for volunteering for this research study and all of the HEIRS Study investigators, a full listing of which can be found in reference 3. We thank Ms. Wen-Pin Chen for assistance with statistical analyses.
Financial Support: Support was provided by grant R01 HL083328 from the National Heart, Lung, and Blood Institute (NHLBI) (C.E.M.). The HEIRS Study was initiated and funded by NHLBI, in conjunction with National Human Genome Research Institute (NHGRI). Data collection for this study was supported by contracts N01-HC-05185 (University of Minnesota), N01-HC-05186 (Howard University), N01-HC-05188 (University of Alabama at Birmingham), N01-HC-05189 (Kaiser Permanente Center for Health Research), N01-HC-05190 (University of California, Irvine), N01-HC-05191 (London Health Sciences Centre), and N01-HC-05192 (Wake Forest University). Additional funding was provided by the University of Alabama at Birmingham General Clinical Research Center (GCRC) grant M01-RR00032, Southern Iron Disorders Center (J.C.B.), Howard University GCRC grant M01-RR10284, Howard University Research Scientist Award UH1-HL03679-05 from the NHLBI and the Office of Research on Minority Health (V.R.G.), grant UC Irvine M01RR 00827-29 from the General Clinical Research Centers Program of the National Center for Research Resources National Institutes of Health, and a Merit Review grant from the Department of Veterans Affairs (G.D.M.)..
Footnotes
CONFLICT OF INTEREST
Guarantor of the article: Joseph A. Murray, MD.
Substantial contributions to the study concept and design; conducted data analysis and data interpretation; critically revised the article: Stela McLachlan, Chad P. Garner
Substantial contributions to the concept and design, as well as data acquisition, critically revised the article: Ronald T. Acton, Paul C. Adams, John H. Eckfeldt, Victor R. Gordeuk, Christine E. McLaren, Gordon D. McLaren
Conducted data analysis; critically revised the article: Tricia Brantner, Catherine Leiendecker-Foster,
Participated in data interpretation; critically revised the article: Lisa F. Barcellos, Kenneth B. Beckman, Antony A. Killeen, Deborah A Nickerson, Chris D. Vulpe
All authors have approved the final revision
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. Turning the tide of malnutrition: Responding to the challenge of the 21st century (WHO/NHD/00.7) 2000. [Google Scholar]
- 2.Lieu PT, Heiskala M, Peterson PA, et al. The roles of iron in health and disease. Mol Aspects Med. 2001;22:1–87. doi: 10.1016/s0098-2997(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 3.Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. The New England journal of medicine. 2005;352:1769–1778. doi: 10.1056/NEJMoa041534. [DOI] [PubMed] [Google Scholar]
- 4.McLaren CE, Barton JC, Adams PC, et al. Hemochromatosis and Iron Overload Screening (HEIRS) study design for an evaluation of 100,000 primary care-based adults. Am J Med Sci. 2003;325:53–62. doi: 10.1097/00000441-200302000-00001. [DOI] [PubMed] [Google Scholar]
- 5.McLaren CE, Garner CP, Constantine CC, et al. Genome-wide association study identifies genetic loci associated with iron deficiency. PloS one. 2011;6:e17390. doi: 10.1371/journal.pone.0017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Expert Scientific Working Group. Summary of a report on assessment of the iron nutritional status of the United States population. American Journal of Clinical Nutrition. 1985;42:1318–1330. doi: 10.1093/ajcn/42.6.1318. [DOI] [PubMed] [Google Scholar]
- 7.Skikne BS, Flowers CH, Cook JD. Serum transferrin receptor: a quantitative measure of tissue iron deficiency. Blood. 1990;75:1870–1876. [PubMed] [Google Scholar]
- 8.Katz KD, Rashtak S, Lahr BD, et al. Screening for celiac disease in a North American population: Sequential serology and gastrointestinal symptoms. American Journal of Gastrenterology. 2011;107:1333–1339. doi: 10.1038/ajg.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker MM, Murray JA, Ronkainen J, et al. Detection of celiac disease and lymphocytic enteropathy by parallel serology and histopathology in a population-based study. Gastroenterology. 2010;139:112–119. doi: 10.1053/j.gastro.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monsuur AJ, de Bakker PI, Zhernakova A, et al. Effective detection of human leukocyte antigen risk alleles in celiac disease using tag single nucleotide polymorphisms. PloS one. 2008;3:e2270. doi: 10.1371/journal.pone.0002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atlas DS, Rubio-Tapia A, Van Dyke CT, et al. Capsule endoscopy in nonresponsive celiac disease. Gastrointestinal endoscopy. 2011;74:1315–1322. doi: 10.1016/j.gie.2011.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubio-Tapia A, Van Dyke CT, Lahr BD, et al. Predictors of family risk for celiac disease: a population-based study. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2008;6:983–987. doi: 10.1016/j.cgh.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farrell RJ, Kelly CP. Celiac sprue. The New England journal of medicine. 2002;346:180–188. doi: 10.1056/NEJMra010852. [DOI] [PubMed] [Google Scholar]
- 14.Kosnai I, Kuitunen P, Siimes MA. Iron deficiency in children with coeliac disease on treatment with gluten-free diet. Role of intestinal blood loss Archives of disease in childhood. 1979;54:375–378. doi: 10.1136/adc.54.5.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shamir R, Levine A, Yalon-Hacohen M, et al. Faecal occult blood in children with coeliac disease. European journal of pediatrics. 2000;159:832–834. doi: 10.1007/pl00008348. [DOI] [PubMed] [Google Scholar]
- 16.Fine KD. The prevalence of occult gastrointestinal bleeding in celiac sprue. The New England journal of medicine. 1996;334:1163–1167. doi: 10.1056/NEJM199605023341804. [DOI] [PubMed] [Google Scholar]
- 17.Logan RF, Howarth GF, West J, et al. How often is a positive faecal occult blood test the result of coeliac disease? European journal of gastroenterology & hepatology. 2003;15:1097–1100. doi: 10.1097/00042737-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Mant MJ, Bain VG, Maguire CG, et al. Prevalence of occult gastrointestinal bleeding in celiac disease. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2006;4:451–454. doi: 10.1016/j.cgh.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 19.Halfdanarson TR, Litzow MR, Murray JA. Hematologic manifestations of celiac disease. Blood. 2007;109:412–421. doi: 10.1182/blood-2006-07-031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bottaro G, Cataldo F, Rotolo N, et al. The clinical pattern of subclinical/silent celiac disease: an analysis on 1026 consecutive cases. The American journal of gastroenterology. 1999;94:691–696. doi: 10.1111/j.1572-0241.1999.00938.x. [DOI] [PubMed] [Google Scholar]
- 21.Hershko C, Hoffbrand AV, Keret D, et al. Role of autoimmune gastritis, Helicobacter pylori and celiac disease in refractory or unexplained iron deficiency anemia. Haematologica. 2005;90:585–595. [PubMed] [Google Scholar]
- 22.Skikne B, Chaim H. Iron Physiology and Pathophysiology in Humans. Humana Press; New York: 2012. [Google Scholar]
- 23.Corazza GR, Valentini RA, Andreani ML, et al. Subclinical coeliac disease is a frequent cause of iron-deficiency anaemia. Scandinavian journal of gastroenterology. 1995;30:153–156. doi: 10.3109/00365529509093254. [DOI] [PubMed] [Google Scholar]
- 24.Carroccio A, Iannitto E, Cavataio F, et al. Sideropenic anemia and celiac disease: one study, two points of view. Digestive diseases and sciences. 1998;43:673–678. doi: 10.1023/a:1018896015530. [DOI] [PubMed] [Google Scholar]
- 25.Howard MR, Turnbull AJ, Morley P, et al. A prospective study of the prevalence of undiagnosed coeliac disease in laboratory defined iron and folate deficiency. Journal of clinical pathology. 2002;55:754–757. doi: 10.1136/jcp.55.10.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershko C, Skikne B. Pathogenesis and management of iron deficiency anemia: emerging role of celiac disease, helicobacter pylori, and autoimmune gastritis. Seminars in hematology. 2009;46:339–350. doi: 10.1053/j.seminhematol.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 27.Looker AC, Dallman PR, Carroll MD, et al. Prevalence of iron deficiency in the United States. JAMA: the journal of the American Medical Association. 1997;277:973–976. doi: 10.1001/jama.1997.03540360041028. [DOI] [PubMed] [Google Scholar]
- 28.Koning F. Celiac disease: caught between a rock and a hard place. Gastroenterology. 2005;129:1294–1301. doi: 10.1053/j.gastro.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 29.Al-Toma A, Goerres MS, Meijer JW, et al. Human leukocyte antigen-DQ2 homozygosity and the development of refractory celiac disease and enteropathy-associated T-cell lymphoma. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2006;4:315–319. doi: 10.1016/j.cgh.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Rubio-Tapia A, Kyle RA, Kaplan EL, et al. Increased prevalence and mortality in undiagnosed celiac disease. Gastroenterology. 2009;137:88–93. doi: 10.1053/j.gastro.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Annals of medicine. 2010;42:530–538. doi: 10.3109/07853890.2010.514285. [DOI] [PubMed] [Google Scholar]
- 32.Kaistha A, Castells S. Celiac disease in African American children with type 1 diabetes mellitus in inner city Brooklyn. Pediatric endocrinology reviews: PER. 2008;5 (Suppl 4):994–998. [PubMed] [Google Scholar]
- 33.Abbass R, Hopkins M, Dufour DR, et al. Celiac disease in an urban VA population with iron deficiency: the case against routine duodenal biopsy. Digestive diseases and sciences. 2011;56:2037–2041. doi: 10.1007/s10620-010-1549-y. [DOI] [PubMed] [Google Scholar]
- 34.Nelson R, McNeish AS, Anderson CM. Coeliac disease in children of Asian immigrants. Lancet. 1973;1:348–50. doi: 10.1016/s0140-6736(73)90132-3. [DOI] [PubMed] [Google Scholar]
- 35.Walker MM, Murray JA. An update in the diagnosis of coeliac disease. Histopathology. 2011;59:166–179. doi: 10.1111/j.1365-2559.2010.03680.x. [DOI] [PubMed] [Google Scholar]