

Abstract
As the technology of proteomics moves from a theoretical approach to a practical reality, neuroscientists will have to determine the most appropriate applications for this technology. Neuroscientists will have to surmount difficulties particular to their research, such as limited sample amounts, heterogeneous cellular compositions in samples, and the fact that many proteins of interest are rare, hydrophobic proteins. This review examines protein isolation and protein fractionation and separation using two-dimensional electrophoresis (2-DE) and mass spectrometry proteomic methods. Methods for quantifying relative protein expression between samples (e.g., 2-DIGE, and ICAT) are also described. The coverage of the proteome, ability to detect membrane proteins, resource requirements, and quantitative reliability of different approaches is also discussed. Although there are many challenges in proteomic neuroscience, this field promises many rewards in the future.
Keywords: Brain, drug abuse, mass spectrometry, neuronal, protein expression
INTRODUCTION
Research into gene expression within the brain and its role in health and disease is rapidly advancing. In the past, research technology allowed the examination of a few genes at a time; however, it is rapidly becoming the norm to examine simultaneously the expression of thousands of genes. The information gained by the sequencing of the genomes for humans and laboratory animals combined with ingenious technical advancements have made large-scale analysis of mRNA expression possible [e.g., microarrays, GeneChips, serial-analysis of gene expression (SAGE), and differential display]. Even as the neuroscience field is becoming familiar with and adopting these genomic approaches, another paradigm shift is occurring—proteomics, the simultaneous assessment of all, or many, of the proteins expressed in a cell.
The term “proteome” has been used for less than a decade (1), and the technical advancements that make proteomics feasible are swiftly evolving. The purpose of this review is to familiarize neuroscientists with the available tools for proteome research and their relative abilities and limitations. Quantitative methodologies for assessment of differences in protein expression are the primary focus of this review, but other nonquantitative techniques are also discussed.
Before delving any further, the importance to neuroscience research of examining thousands of proteins in a cell should be established. Though certain RNAs can act as effector molecules, proteins perform the majority of biological actions in the cell. To identify the thousands of different proteins in a cell, the modifications to these proteins, along with how the amounts of both of these change in different conditions would revolutionize biology and medicine. Though important strides are being made toward achieving the goal of global mRNA analysis, mRNA is not the functional endpoint of gene expression, and mRNA expression may not directly equate with protein expression (2,3).
For the purpose of this review, it is also important to provide an operational definition of the term “proteomics.” Proteomic research refers to the examination of protein expression, modification, or interaction on a large, parallel scale—hundreds to thousands of proteins simultaneously. Examination of protein expression, modification, and interaction is hardly novel; however, the scale on which this can now be performed warrants the terms “proteome” and “proteomics.” Rather than traditional approaches that examined one or a few proteins at a time in a few samples, proteomics attempts to concurrently examine large numbers (hundreds to thousandsplus) of proteins. There are many potential applications for proteomics in neuroscience: determination of the neuroproteome, comparative protein expression profiling, post-translational protein modification profiling, and mapping protein–protein interactions, to name but a few. The present review will comment on all of these applications with an emphasis on protein expression profiling.
In thinking about genomics and proteomics, it is necessary to emphasize the fundamental differences between nucleic acid research (genomics or functional genomics) and proteomics. Proteins have no equivalent to Watson–Crick base pairing, and there are no methods of protein amplification like those for nucleic acids [e.g., polymerase chain reaction (PCR) (4) or antisense RNA (aRNA) (5)]. Nucleic acid hybridization relies on base pairing and is a highly regular and specific event that can be controlled by temperature and solution composition, allowing the construction of probes that will recognize a specific nucleic acid sequence of interest. Probes can be made in unlimited quantities with the assistance of PCR and plasmid DNAs. For proteomic research, the recognition of specific proteins is more difficult. Traditionally, antibodies have been used to identify specific proteins, but the limitation of antibodies is that they are not as specific as DNA probes, usually require some preseparation of the protein of interest, and that antibodies are time consuming to produce. For nucleic acid research, very small amounts of sample are needed because samples can be quantitatively amplified by PCR and aRNA to such a degree that there is more than enough sample for expression profiling and further assays (e.g., sequencing). Proteomics is currently limited in its ability to detect rare proteins (those that are not expressed in many copies per cell), primarily due to the lack of viable protein amplification methodology. Technical improvements have therefore focused on signal amplification. [Some methods for protein amplification have been reported but have not gained wide acceptance (6,7).]
Steps in Proteomics Experiments
Different proteomic methods use a variety of technologies and approaches. All of these methods contain, in some form, protein isolation, separation/fractionation, quantitation, and identification (Fig. 1). Many of the procedures (Fig. 2) used for these individual methods can be combined interchangeably to optimize proteome coverage. The challenge to neuroscientists is find the best combination of techniques to achieve experimental goals (Table I).
Fig. 1.
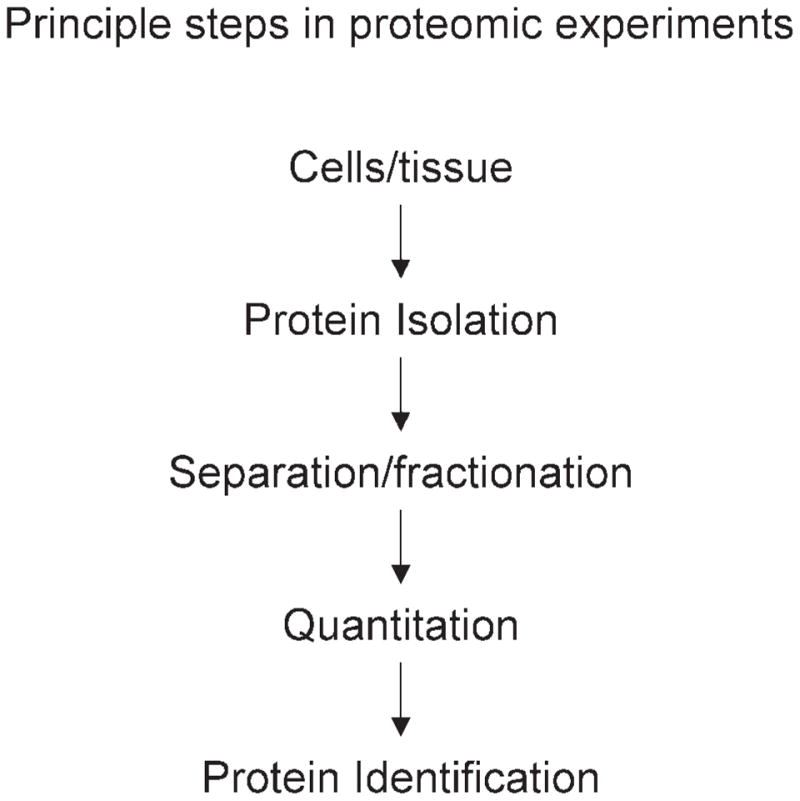
All proteomic experiments incorporate these experimental steps. Though most experiments occur in this order, some methods do proceed in a different order.
Fig. 2.

Common methods for protein separation, quantitation, and identification.
Table I.
Comparison of Different Proteomic Technologies
| 2-DE | 2-DIGE | MuDPIT | ICAT | SELDI | Protein arrays | |
|---|---|---|---|---|---|---|
| Separation | Electrophoresis: IEF PAGE | Electrophoresis: IEF PAGE | LC/LC of peptides | LC/LC of peptides | Binding characteristics of chips, MS | Binding to affinity reagent |
| Quantitation | Densitometry of stains | Imaging of Cy3 and Cy5 labeled proteins normalized to Cy2 | None | Through use of heavy and light tags | Comparison of MS peaks | Densitometry of binding |
| Identification | PMF | PMF | MS/MS | MS/MS | Difficult, requires serial washing of sample or coupling to second MS instrument | Binding to particular affinity reagent |
| No. of proteins | 100s to low 1000s | 100s to low 1000s | 100s to mid-1000s | 100s to low 1000s | 100s | 100s to 1000s (limited only by no. of antibodies) |
| Hydrophobic proteins | Moderate, dependent on detergents used | Moderate, dependent on detergents used | Moderate/good, theoretically better than electrophoresis, not systematically examined | Moderate, could be better than electrophoresis but has not been demonstrated | Moderate | Unknown |
| Low expressed proteins | Marginal | Moderate (especially with saturating labels) | Moderate, often used with large sample amounts | Moderate | Marginal to moderate | Unknown |
| Strength | Low cost, comparably easy techniques | High quality of quantitation of multiple samples | Excellent approach if no quantitation is desired | Theoretically, higher coverage of proteome | Easiest MS instrumentation | High throughput |
| Limitations | Limited quantitation, difficulty of spot matching, no. of proteins examined | Requires expensive dedicated instrumentation | No quantitation, requires high level of MS skill, complicated data compilation | Requires high level of MS skill, complicated data compilation | Difficult to identify proteins | Specificity of antigen/antibody binding, questions on quantitative accuracy |
Note: 2-DE, two-dimensional electrophoresis; 2-DIGE, two-dimensional difference in gel electrophoresis; MuDPIT, multidimensional protein identification technology; ICAT, isotope coded affinity tags; SELDI, spectrum enhanced laser desorption ionization; IEF, isoelectric focusing; PAGE, polyacrylamide gel electrophoresis; LC/LC, tandem liquid chromatography; PMF, peptide mass fingerprinting; MS/MS, tandem mass spectrometry.
SAMPLE PREPARATION
As with any experiment, the quality of the results is completely dependent on the quality of the starting material. Adequate coverage of the intricacies of protein isolation would require more space than is available in the current review; however, a few points of significance for proteomic experiments are included. Protein stability and purity are of critical importance to proteomic studies as are prevention of protein degradation and modification during sample preparation. Rapid removal of brain tissue, dissection, and freezing are obvious imperatives for the maintenance of the proteome state in the animal. Human postmortem studies pose unique challenges, but these can be addressed by careful documentation of postmortem interval, brain pH, and agonal state (8). Specific proteins have been shown to degrade in a time-dependent manner, highlighting the need for careful selection of controls in human brain postmortem studies (9). Specifically, dihydropyrimidinase related protein-2 has putatively been identified as a marker of postmortem interval and temperature in mice (10). Protease and phosphatase inhibitors are commonly used to help prevent degradation and dephosphorylation of proteins during protein preparation (11); however, care should be taken so that adducts and charge trains are not introduced by these inhibitors.
Purification of protein from other cellular substances is also necessary. Lipids are particularly abundant in the brain and along with nucleic acids must be eliminated from the protein sample for good-quality results. The most common methods of protein purification rely on selective precipitation. Acetone, trichloroacetic acid (TCA), and other precipitation methods can be performed, and a number of commercially available kits make this a routine procedure (12,13). In some instances though, pure protein may not be sufficient because proteins such as IgGs or albumin constitute the vast majority of protein concentrations in cells. Selective elimination of these proteins (14) improves detection of less highly expressed proteins.
PROTEIN FRACTIONATION
As depicted in Fig. 2, there are a number of approaches to reduce the complexity of a protein sample for analysis. Depending on the rationale for a study, analyzing the entire complement of cellular proteins in one experimental run may cause a loss of sensitivity and result in interpretational difficulties. In such cases, it is recommended that the proteome be fractionated in order to remove contaminating material from the sample and to enrich the proteins of interest for further analysis. Protein fractionation can be thought of as different from protein separation in that it isolates a subset of proteins from the entire proteome. Protein separation can be thought of as segregation of individual proteins. The starting material can be fractionated using a variety of approaches including centrifugation (e.g., soluble/insoluble, membrane/cytosolic/nuclear), salt precipitation, liquid chromatographic separation (e.g., ion exchange, affinity, gel filtration), and velocity or equilibrium sedimentation.
PROTEIN SEPARATION AND QUANTITATION
Proteins must be separated in some manner so that they can be quantitatively compared and identified based on intrinsic properties of proteins such as charge, isoelectric point, size, or some other parameter. In some cases, proteins are separated by more than one parameter to allow an even more detailed portrait of the proteome [e.g., two-dimensional electrophoresis (2-DE) and tandem liquid chromatography (LC/LC)].
Electrophoresis Methods
Electrophoresic Separation Methods
One-Dimensional Electrophoresis
Separation of proteins by size is the traditional method for analyzing one protein at a time. A number of common techniques give comparative quantitation between samples; immunoblotting/Western blotting being the most popular [enzyme-linked immunoabsorbent assay (ELISA) and immunocytochemistry being others]. Western blotting allows the visualization of antibodies directed against specific proteins of interest. In Western blotting, proteins are electrophoresed into a gel containing sodium dodecyl sulfate (SDS), where proteins migrate based on size and charge with smaller proteins migrating through the gel faster than larger proteins. Once the proteins have been separated, the proteins in the gel are transferred to a solid membrane (nylon or PVDF) for analysis by applying an electric current to the gel so that the separated proteins transfer through the gel and onto the membrane in the same pattern as they separate on the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). To detect a specific protein on the membrane, a primary antibody is added at an appropriate dilution and incubated with the membrane such that the antibody will bind to the protein(s). In order to detect bound antibodies, secondary antibodies are added (immunoglobulin antibodies conju gated to a reporter group such as the enzyme alkaline phosphatase). Finally, a substrate is added to react with the secondary antibody resulting in a visible band(s) where the primary antibody is bound to the protein.
Western blot analysis and the aforementioned procedures are serial approaches that enable the quantitation of one or few proteins at a time. The primary limitation of these approaches is that they require a selective antibody for each protein of interest. Recent technical innovations have been made, though, to increase the number of proteins that can be examined by Western blotting, such as large-scale immunoblotting [e.g., PowerBlot (15)]. This requires that the protein(s) of interest be defined beforehand and that an antibody for each protein be available. Unfortunately, the reality is that many proteins have still not been identified, and the majority of those that have been identified lack high-quality antibodies. As it currently exists, proteomics will not replace these methods, but rather these methods will serve as excellent tools for confirmation and further experimentation on targets found by proteomics. One-dimensional electrophoresis as a separation technique before tandem mass spectrometry (MS/MS) separation and identification has been used for neuroscience studies of receptor complexes (16).
Isolelectric Focusing
Isoelectric focusing (IEF) separates proteins according to their isoelectric point (pI). The pI of a protein is primarily a function of its amino acid sequence although post-translational modifications can also contribute to the pI. Proteins are amphoteric molecules, capable of acting as either an acid or a base. The side chains of the amino acids in proteins have acidic or basic buffering groups that are protonated or deprotonated, depending on the pH of the solution in which the protein is present. At a particular pH, the sum of the charges on the amino acids will equal zero. Isoelectric focusing takes advantage of this property by placing proteins in a pH gradient and applying a potential. The protein will migrate toward the anode or cathode, depending on its net charge. Eventually, the protein will reach the point in the pH gradient where the net charge of the protein is zero, at the pI, and stop migrating.
Initially, the preparation and use of the pH gradients needed for isoelectric focusing was very difficult and inconsistent. These pH gradients were often in the form of tube gels with carrier ampholytes. The introduction of chemistries (17) to immobilize the pH gradient into the gel matrix and the construction of the gel on a solid backing (18,19) was a significant step in making IEF accessible to more than a handful of researchers. Most current applications of IEF use these immobilized gradients in some sort of dedicated instrument that controls both gel current and temperature (20). Exceedingly high currents (e.g., 8000 V) are needed to focus proteins, and consistent focusing requires close control of the temperature (21). Commercial suppliers are now producing IEF gels that are of narrow pH ranges, which, when used in an overlapping fashion (pH 4–5, 4.5–5.5, 5–6, 5.5–6.7, and 6–9), enable the separation and detection of thousands of proteins (22). Isoelectric focusing is rarely used on its own and is usually followed by applying the IEP strip to SDS-PAGE gel to make two-dimensional electrophoresis (described below).
Solution IEF
Solution IEF operates on the same principle as normal IEF except that proteins are separated, in solution, into pI range bins (23,24). These proteins can then be run on IEF gels with an appropriate pI range, the same as the pI range of the bin. One of the reasons for performing solution IEF is that when loading a whole cell lysate onto a narrow pI range IEF gel (using normal IEF), proteins outside the IEF range precipitate and can pull out proteins from within the range of the IEF gel, and those proteins outside of the range of the gel are wasted. Solution IEF requires more development, but with improvements this technology has great potential.
Second-Dimension SDS-PAGE
Two-dimensional electrophoresis is one of the most commonly used techniques in proteomics. The basic principles of 2-DE remain the same since its introduction in 1972 by O’Farrell (25) and Klose (26), namely, the separation of proteins by IEF (first dimension, separation by pI) followed by SDS-PAGE (second dimension, separation by molecular weight), which involves the separation by molecular weight of proteins that have already been separated by IEF. In general, the IEF gel or strip is equilibrated to SDS and then placed on top of the SDS gel. This equilibration step is necessary to allow the SDS molecules to associate with the proteins and produce the anionic complexes that have a net negative charge roughly equal to the molecular weight of the protein. The SDS gel is then run, and the proteins migrate out of the IEF gel and into the SDS gel, where they separate according to molecular weight. Though most applications use denaturing SDS-PAGE, approaches to assess native protein conformation have also been used. Both conventional SDS-PAGE instruments, such as those used for Western blotting, and special purpose apparatuses can be used for this step.
Detection/Quantitation for Electrophoresis
Stains
After electrophoresis, the proteins in the gel must be detected in some manner. Traditionally, this is accomplished by the use of a visible stain, whereas newer approaches use fluorescent dyes or stains (27). The most common visible stain has traditionally been silver stain. This highly sensitive stain is the “gold standard” for sensitivity. However, silver staining has decreased in popularity due to complications with background, quantitation, reproducibility, and mass spectrometry compatibility. Most silver stains will also stain DNA and lipopolysaccharides resulting in increased background signal. The quantitative limitations of silver staining stem from a limited dynamic range (28) and the dependency of signal intensity on the length of developing time. Another limitation is that the gluteraldehyde present in most silver staining protocols is incompatible with mass spectrometry; however, the procedure can be modified for compatibility (29). For these reasons, other visible stains such as zinc imidazol (30) and Coomassie blue (31) have been developed. Though both are more compatible with mass spectrometry, zinc imidazol has limited quantitation abilities and Coomassie staining is much less sensitive than silver staining. All of these visual stains can be converted to digital images by either scanning or camera densitometry.
Because of the aforementioned limitations, fluorescent stains and dyes have been developed that attempt to combine sensitivity, quantitation, and mass spectrometry compatibility. Fluorescent stains such as ruthenium bathophenanthroline disulfonate (Sypro Ruby) (32) are simple to use, quantitative, and compatible with mass spectrometry. The sensitivity of Sypro Ruby approaches that of silver staining but does not equal it. Some of these fluorescent stains have been developed such that they are selective for post-translational modifications of proteins (phosphorylation and glycosylation) (33,34). Fluorescent dyes can be visualized with fluorescent scanners or ultraviolet light boxes. With these developments, another possibility emerged: to label the proteins before electrophoresis with fluorescent dyes. This is discussed in detail below. In total, staining methods continue to improve but no single stain has been found to be the most sensitive, quantitative, and compatible with mass spectrometry.
Dyes
As described above, conventional 2-DE consists of IEF, SDS-PAGE, and visualization. The problem that has bedeviled the use of 2-DE for applications like protein expression profiling in neuroscience research has been reproducibility and quantitation. To address some of these problems a new method, two-dimensional difference in 2-D Fluorescence Difference Gel Electrophoresis (2-DIGE) has been developed that uses direct labeling of proteins with cyanine dyes prior to IEF (35,36) (see Ref. 37 for detailed methodological discussion). This method relies on cyanine dyes that react with lysine groups on proteins (38). Dye concentrations are kept low, such that approximately one dye is added per protein. The critical aspect of the use of 2-DIGE technology is the ability to label two or more samples with different dyes and run them on the same IEF and SDS-PAGE gels—this makes spot matching and quantitation much more simple and accurate. As it has been popularized, 2-DIGE uses Cy3, Cy5, and Cy2 dyes, with Cy2 being used for a normalization pool created from a mix of all samples in the experiment (Fig. 3). The normalization pool is created by combining equal amounts of protein from all the samples in the study. This Cy2 pool is run on all gels, facilitating spot matching and normalization of signal from different gels (Figs. 4A and 4B). The 2-DIGE approach offers great promise to researchers using 2-DE. The dyes are comparable in sensitivity to silver staining methods, are compatible with mass spectrometry, and offer the best quantitation of any 2-DE method. The major drawback of this technique is that it is proprietary to Amersham Biosciences and requires expensive and specific equipment such as a three-laser fluorescent scanner and dedicated software. A modification of 2-DIGE in which cyanine dyes that label all of the cystine residues of proteins are labeled has recently been introduced (39). This is intended to reduce the amount of protein sample required, and currently, only two saturation dyes (Cy3 and Cy5) are available.
Fig. 3.

Difference in gel electrophoresis (DIGE) departs from standard 2-DE in that the samples are labeled with fluorescent dyes and mixed together before being run on the same IEF and second-dimension gel. A pool, created by mixing equal amounts of each sample analyzed, is run to both normalize quantitation across gels and for spiking in to increase the amount of protein to be picked for MS protein identification.
Fig. 4.

Spot matching and quantitation with different two-dimensional electrophoresis approaches. (A) Spot matching is the process of determining which spot is the same across multiple gels. Two-dimensional DIGE aids matching by using an internal control channel present on each gel and fewer gels in total. (B) Quantitation of traditional 2-DE uses densitometry of matched spots. 2-DIGE improves quantitation by assessing abundance in relation to a normalization pool on each gel, thereby normalizing for gel to gel technical differences.
Two-Dimensional Electrophoresis Data Analysis
Once proteins have been visualized, image analysis is required. Image analysis can be segregated into spot detection, spot matching, and data analysis. Manually detecting the hundreds to thousands of spots on each 2-DE gel for the number of gels in an experiment would take far too long and be inconsistent. A number of companies now offer software that, using a digital image of a gel, detects the spots and provide a quantitative value (40). For any experiment that requires multiple gels to look at different conditions, samples, and so forth, the different gels must be matched to each other (Fig. 4A). In other words, a spot at a certain location on gel 1 must be matched with the same spot on gel 2, gel 3, and so forth. In reality, proteins do not migrate to exactly the same point on each gel (IEF or SDS-PAGE). Complex algorithms have been developed that attempt to match these spots across gels using the pattern spots on the whole gel and correcting for shifts in these patterns (41). The Cy2 pool used in 2-DIGE methodology is useful in this respect as it provides a consistent spot map on all gels in an experiment, facilitating spot matching (Fig. 4A). Also, with 2-DIGE the number of gels that need to be matched is reduced because two samples are run on each gel, and those two samples will have identical spot patterns. The need for computer-based spot matching cannot be underestimated, as matching by hand would take days. A good deal of manual intervention to correct for false matches and omissions of the software is still needed, though. These matching algorithms continue to improve, but the best thing that can be done to aid in spot matching is to develop reproducible laboratory technique. Rates of matching can range from 40%–80% of spots across gels in an experiment. With spots matched and with signal intensities known, comparisons can be made for changes in protein expression. The 2-DIGE method uses the Cy2 pool to normalize the signal abundances between gels, correcting for any differences in overall signal intensity. This provides for a more consistent expression measurement across gels (Fig. 4B).
A number of different approaches can be used to determine differential expression, from simple fold-change cutoffs to statistical tests. The challenges here are similar to those found in analyzing microarray data. Though the best insurance of accurate quantitation is confirmation by another method like immunoblotting, the proteomic field has yet to establish standard practices for data analysis. A common concern with 2-DE is that with a long experimental protocol of protein preparation, IEF, SDS-PAGE, visualization, and data analysis, small errors compound thus inhibiting meaningful quantitation. To address this very legitimate concern new methods have been developed, such as the normalization pool in 2-DIGE, that attempt to limit sources of error or normalize for them. Nonetheless, as the proteomic field matures, consistent data analysis methods and more post hoc confirmation of observed changes will be needed.
Liquid Chromatography
Liquid chromatographic (LC) methods provide a powerful means of protein/peptide separation of protein mixtures. Primary advantages of LC separation are the versatility of the methods and the ability to link LC directly to analysis methods such as MS. Proteins/peptides can be separated based on their physical properties, including affinity (interactions of specific moieties), ion exchange (anion or cation), reverse phase (hydrophobicity), and size exclusion (not as useful for peptide separation). LC has also been used to separate fractionated proteins prior to IEF to isolate specific populations of proteins in the mixture prior to 2-DE, allowing more discrete separation and identification (42). Of particular interest in the neuroscience community is the development of proteomics procedures to evaluate hydrophobic proteins (e.g., receptors). For example, the use of reverse phase separation prior to IEF would enhance the separation of hydrophobic proteins that could then be subjected to gradient IEF, enabling increased detection of this protein pool and lower abundance proteins within this pool. The development of LC that feeds directly into a mass spectrometer has been furthered by the use of LC in series/tandem (LC/LC) to separate peptides based on different properties (e.g., reverse phase coupled to strong cation exchange) [i.e., MuDPIT (43)]. Advantages of this approach include increased separation and ultimately identification of peptide mixtures in a single run and the increased potential to detect low abundance peptides.
Capillary electrophoresis (CE) offers several advantages over slab gel electrophoresis, including faster separations and better resolution. Though the number of samples running on a CE may be limited to one sample at a time, the development of capillary arrays can overcome this disadvantage. While LC and CE methods are very similar, the principal advantage of CE is the dramatic enhancement in resolution.
Mass Spectrometry
Mass Spectrometry for Protein Separation
Mass spectrometry (MS) is another principle method of protein separation used in proteomics. A number of different MS platforms exist, each with advantages and drawbacks. Before describing some of these platforms, a basic description of how mass spectrometry works is necessary because most neuroscientists are not familiar with this technology (Fig. 5). This is intended as only the most cursory of descriptions, and for more in-depth descriptions there are several excellent books available, written with the biologist in mind (37,44,45).
Fig. 5.

Schematics of common ionization and mass analysis technologies (see text for details).
Mass spectrometry is based on ionizing proteins or peptides and then measuring the mass to charge ratio (m/z). Though a variety of MS instruments exist, they all contain three parts: ionization, separation (mass analyzer), and detection. The sample is ionized, and the resulting ions are separated by m/z and detected. These three processes allow the calculation of the mass of a particular protein or peptide (see Refs. 37, 44, and 45 for detailed descriptions of how mass is calculated from m/z.)
A number of ionization technologies exist including fast ion bombardment (FAB) (46), matrix assisted laser desorption ionization (MALDI) (47), and electrospray ionization (ESI) (48–50). MALDI and ESI are the techniques of choice for most proteomic applications of neuroscience research. MALDI (Fig. 5A) works through mixing the protein sample with a light absorbing matrix that forms a crystal. This is usually done on some form of plate with multiple positions for different samples. When the plate is pulsed with a laser of a particular wavelength, the energy from the laser is absorbed by the crystal matrix. Most often in MALDI, the proteins accept a single proton from the matrix and are desorbed (ejected) from the plate in the gas phase and into the mass analyzer.
In ESI (Fig. 5B) (and nanospray ionization), ions are produced in a liquid phase (51–53). The protein sample, in a solvent solution, is ejected as a mist of droplets from a charged capillary tip. As the solvent in the droplets evaporates, the total charges of the proteins in the droplet remain but with a reduced surface area of the droplet. This continues to a point at which individual ions leave the droplet. Individual ions then pass on into the mass analyzer. Unlike MALDI where the samples are crystallized, the ionization state of the protein ions in ESI is determined by the pH of the solvent solution. The end result is that ESI produces a range of multiply charged ions, which can be useful for some MS analyses.
Whichever method of ionization is used, once the ions are created, they must be separated before being detected in such a way as to provide information on the m/z ratio. Mass analyzers do not actually detect the ions or measure ion mass; they are only used to separate ions according to their m/z ratio. A number of mass analyzer types exist: time-of-flight (ToF), quadrapole, ion trap, and Fourier transform-ion cyclotron resonance (FT-ICR).
Time-of-flight (Fig. 5C) mass analyzers can be thought of as a tube (54). The ionized proteins enter the tube by passing through a high-voltage accelerator. The speed at which the ion travels is proportional to its mass (m). If a number of ions are simultaneously produced, then passed through the ToF tube and to a detector, the ions with a higher m/z ratio will travel faster and reach the detector first. As the distance traveled and time are all known, the m/z ratio can be calculated and from that the mass.
Quadrapole mass analyzers (Fig. 5D) also involve ions traveling down what can be thought of as a tube. In this case though, the tube consists of four parallel rods. The rods are two pairs of two that can be tuned to different currents and radio frequencies. The two pairs of rods have opposite currents and shifted radio frequencies allowing a form of tuning in which only ions of a particular m/z ratio pass through the tube. A range of m/z ratios can be scanned, generating a m/z profile of the sample. Quadrapole mass analyzers are often used with an ESI (55) ion source.
Ion trap mass analyzers (56) (Fig. 5E) use the same principles as the quadrapole in that specific combinations of current and radio frequencies are used to select particular m/z ratios. The ion trap can be thought of as a small ball with one electrode around the equator and two more electrodes at the poles. Ions are introduced into the center of the ball and are kept in orbits within the trap. By changing current and radio-frequency combinations, particular m/z ratio ions are ejected from the ion trap through a port to the detector. By scanning voltages and radio frequencies, a complete m/z profile can made.
A number of hybrids of these separation strategies exist, all of which are generally designed to increase the accuracy of m/z measurements and sensitivity to low abundance ions. ToF analyzers can be placed in series (ToF/ToF) with a reflectron or collision cell (57) between them; quadrapoles and ToF can be placed in series (Q-ToF)(58); and extremely powerful magnets and Fourier transform algorithms (FT-ICR) can be used to determine the m/z ratios of all ions within an ion trap (58,59). With ions generated and separated, instruments like electron multipliers and scintillation counters detect the ions. Detectors change the kinetic energy of the ions into an electrical current that can be measured and passed along to a computer. Though these detectors give information on abundance of ions, quantitation of protein abundance differences between samples by MS is limited unless samples are linked to isotopes (see ICAT, below).
All of these MS techniques can be applied to complex protein samples (i.e., mixtures of hundreds or thousands of proteins. It is important to separate the use of MS instruments to separate proteins from the MS used for protein identification as will be described later. For separation, MS has great capabilities but also limitations. As described below, quantitative analysis by MS is limited to techniques like ICAT. For researchers looking to profile the expression of proteins in a large number of samples, MS can be problematic and requires a great deal of time on expensive instruments.
MS Quantitation
Isotope coded affinity tags (ICAT) developed by Aebersold and colleagues (60) was the first quantitative proteomic method to be based solely on using mass spectrometry. The method uses ICAT reagents that contain a reactive group that derivitizes with cysteinyl residues. The ICAT reagents also contain a biotin label and a linker segment. This linker segment either includes eight deuterium isotopes (heavy tag) or none (light tag). ICAT experiments take the form of two experimental conditions; protein from one is labeled with the heavy version of the ICAT reagents and the other with the light version. The samples are then combined, and the proteins are digested with an enzyme such as trypsin. The resulting peptides that contain ICAT reagent are isolated by avidin affinity for biotin. These peptides are then run through LC-MS, and the relative amounts of different peptides are determined. This quantitation can be performed because the heavy- and light-labeled peptides have slightly different masses. By comparing light ICAT peptide MS peak sizes with the peak sizes that are exactly eight daltons larger (because of the heavy reagent), relative quantities can be determined. The proteins from which these peptides originate are then identified by a second round of MS (tandem MS), as described later. This method has been improved to provide better ease of use and sensitivity by use of solid-phase chemistry (61).
ICAT methodology has the potential to detect more lowly expressed proteins and is more likely to detect hydrophobic and large proteins than 2-DE. One study (62) using both 2-DE and ICAT found that they provided complimentary sets of data, with some differentially expressed proteins found by both methods and other proteins detected by only one method or the other. In a recent, more comprehensive study, the methodologies were again found to complement each other (63). Two-dimensional electrophoresis was found be preferential for small-molecular-weight proteins and those proteins with few to no cysteines (which ICAT requires for labeling). ICAT was better with higher molecular weight proteins. Interestingly, in contrast to the assumed superiority of ICAT, the 2-DE approach identified more hydrophobic proteins. ICAT and similar approaches will be valuable tools for proteomics with the limitations of requiring MS/MS instrumentation and high levels of MS knowledge and skill.
One mass spectrometry method that does possess quantitative abilities without the use of tags is spectrum enhanced laser desorption ionization (SELDI) (64,65). This technique, also known by its trade name ProteinChip, is hindered by the difficulty in identifying proteins of interest (66). This platform has been coupled to Q-ToF mass spectrometers to help overcome this limitation (67).
Other Separation/Fractionation Methodologies
Approaches to reducing the complexity of proteins in a sample that are not strictly proteomic technologies can also be used to great advantage. The most obvious examples would be separation of the cell lysates into sub-cellular fractions (membrane, cytosolic, nuclear) before any proteomic analysis. This approach has been used in studies of the brain (42). Another possibility is to increase anatomical resolution through laser capture microdissection (LCM), which has been used successfully in 2-DE (68,69) and mass spectrometry (70) neuroproteome studies. Though LCM increases anatomical specificity, the amount of sample is very small, and the studies published so far have detected only the most abundant cellular proteins. Increased sensitivity of detection and identification methods are necessary to make this a truly viable approach. Another anatomically specific technique has been developed by Caprioli and collegues, where MALDI-ToF MS is performed directly from tissue sections (71,72). These anatomical techniques can even be applied to the cellular anatomy, such as identifying the proteins present in isolated postsynaptic densities (73).
PROTEIN IDENTIFICATION
Regardless of the separation and quantitation methods used, at the end of the experiment the proteins must be identified. Most approaches use mass spectrometry. Peptide mass fingerprinting (PMF) and tandem mass spectrometry (MS/MS) are the main methods for determining protein identities.
Peptide Mass Fingerprinting
PMF was developed by a number of research groups (74–76) and begins with digestion of a protein with an enzyme, typically trypsin. Trypsin cleaves proteins at very specific locations, resulting in a series of peptides. If this mixture of peptides is analyzed by MS, a series of peptide masses is created. These masses are searched against databases using one of a number of programs [e.g., Pro-Found (77) and MASCOT (78)]. These programs take DNA sequence databases translated into protein sequence and calculate the resulting peptide masses if these protein sequences were digested with trypsin. The peptide masses generated from the MS of the digested protein of interest is then compared against these databases and the protein can be identified. PMF of spots from 2-DE gels is one very common application. Gel plugs are either excised by hand or robot. Proteins are then digested with trypsin in the gel plug. With visual stains, the plug must often be destained, and some stains work better than others (79). Silver stains that use gluteraldehyde are not compatible with MS. The peptides from the digested proteins are then extracted from the gel plugs and are analyzed by MS.
Tandem Mass Spectrometry
Even if MS instead of 2-DE was chosen as the method of protein separation, MS is also used for protein identification through a process called tandem mass spectrometry (MS/MS). A number of different strategies exist for MS/MS (37). In general, the process entails the selection of one ion/protein generated during initial MS and then fragmenting this ion/protein into smaller pieces and measuring the mass of the resulting ions. These secondary ions can be decoded into protein sequence information that is searched against protein sequence databases to identify the protein (80,81). Almost all of the ionization and mass analyzer types can all be used for MS/MS provided that the instrument is appropriately configured. One MS/MS method that is particularly suited for proteome determination, but not quantitation, is multidimensional protein identification technology (MudPIT) (43,82). In this method, all the proteins in a sample are digested and loaded onto LC columns (see previous explanation). After fractionation of the peptides, the peptides are fed into a MS/MS instrument for protein identification. This method has identified thousands of proteins, can detect membrane proteins, and is similar in concept to shotgun sequencing of DNA.
Whichever protein identification method is used, not all proteins are confidently identified. Rates of identification range from 0% to over 80% depending on the method, instrument, and algorithm used, as well as the abundance of the protein. Careful handling of samples to limit contamination by substances like keratin and thoughtful consideration of the identification approach used will aid in success.
Other Methods
Some of the more traditional methods for identifying proteins are still used for proteomic experiments. Edman protein sequencing can be performed on proteins or peptides from gels transferred to membranes or extracted from gel plugs. This method is limited by low throughput and requires a comparatively large amount of protein. Another technique is the Far Western blot, where a 2-DE gel is blotted and the blot is probed with an antibody against a specific protein. This approach does not offer much progress over conventional immunoblotting.
PROTEIN ARRAYS
Because of some of the limitations of electrophoresis and mass spectrometry methods, selected research groups are attempting to create proteomic chips/arrays. The basic approach is very similar to that of microarrays (83–85). Antibodies or other affinity reagents (e.g., aptamers, peptides) are spotted onto some sort of matrix. Hundreds to thousands of spots are on a single array. A labeled protein sample is then washed across the array, and proteins bind to their specific antibody. The process can also be reversed whereby the protein samples of interest are spotted onto the matrix and then probed with different affinity reagents (86). Though these array or chip approaches have potential for greatly increasing the throughput of proteomic experiments, the use of affinity reagents as the separation method is a severely limiting factor and cannot be ignored. A high-quality antibody is needed for each protein of interest and each modification of that protein. Generation of antibodies remains a laborious task that is almost as much art as science. Separate antibodies also have to be generated for different organisms. In order to generate quantitative data from antibody arrays, and because association kinetics between different antibodies and antigens can vary tremendously, relative concentrations of each antibody and antigen have to be optimized for each protein in order to have quantitative information (87). Lastly, it should not be forgotten that sequence/structure knowledge is needed of any protein to be analyzed by protein microarrays in order to generate the affinity reagent, thus limiting this approach to known protein sequences and modifications. Though there seems to be a number of pitfalls to proteomic chips/arrays as an open screen technique, they do hold promise for routine examination of a small group of proteins. Well-known pathways or protein families could easily be examined by such an approach.
APPLICATIONS
There are a wide variety of applications for proteomic technology (see Ref. 45 for review) in neuroscience. These applications range from defining the proteome of a particular cell type, identifying changes in brain protein expression under different experimental or disease conditions, profiling protein modifications (e.g., phosphorylation) and mapping protein–protein interactions. Studies with all of these goals have been published and, with some examples, are available for neuroscience applications. Determining the proteome of a particular cell, or organism in the case of unicellular organisms, represents an extension of genomic and functional genomics. Yeast, as a simple organism, offers the best opportunity for a first comprehensive proteomic map of an organism and was analyzed using MudPIT by Washburn et al. (43). One of the larger studies of protein from a complex tissue was of the fetal brain, with 1700 proteins identified (88). With the incredible cellular heterogeneity of the brain, there are a number of defined cell types awaiting proteomic characterization. The possibility of an international collaborative effort, along the lines of the human genome project, to catalog the brain proteome is being discussed to take on this Herculean task (89).
Perhaps the proteomic application of greatest interest to neuroscience is protein expression profiling. Experiments such as these are the ones most likely to be undertaken by researchers who are neuroscientists first and foremost and proteomic researchers second. The number of such studies is still small but growing rapidly. Differential expression of alphaB-crystalline in Huntington’s disease patients (90) as well as a number of alterations seen in the rat brain after administration of kanaic acid (91) have been found by 2-DE. Several studies have examined protein expression by 2-DE in brains of Alzheimer’s disease patients (92–95). Methodology development must continue in this area to increase the sensitivity, quantitation, and ability to detect membrane proteins for these studies to reach their full potential. An advantage of profiling by those proteomic methods is that they offer an open and unbiased screen. mRNA-based techniques like microarrays are closed screens, limited to examining those genes that are deposited on the chip being used in the experiment. Techniques like 2-DE and MS/MS can, theoretically, detect any protein in an unbiased fashion. In this sense, most proteomic methods are open screens like the mRNA techniques of differential display and serial analysis of gene expression (SAGE).
While much of this review has focused on determining the proteins present in a cell and their relative expression levels, profiling of protein modifications is just as important. Many important cellular processes in the nervous system do not rely on changes in mRNA or protein abundance but on modifying proteins present in a cell to modify their function. A prime example is phosphorylation cascades in signal transduction (96). The technology needed to examine these changes is beginning to become available either in the form of fluorescent dyes (33,34) or mass spectrometry techniques (97).
Just as protein modifications are often required for cellular events to take place, protein–protein interactions are critical to the function and activity of many proteins. Though more standardized methods such as the yeast two hybrid exist to identify protein–protein interactions, analysis of protein complexes isolated from in vivo brain samples are also being analyzed in a proteomic manner. This can be accomplished by combining common techniques like immunoprecipitation and affinity chromatography with 2-DE and MS to determine the proteins bound in complexes to neurotransmitter receptors isolated from the brain (16,98).
LIMITATIONS
Though the proteomic field has advanced tremendously over the past few years, researchers must remain fully cognizant of the limitations that still remain. All of the methods described above tend to identify preferentially the most highly expressed proteins. Even when an experiment identifies thousands of proteins in a sample, they are only the most abundant of the proteins present in the sample. For neuroscientists, this represents a challenge because many proteins of interest in the brain are expressed at relatively low levels and are hydrophobic proteins. The effectiveness of 2-DE to examine membrane proteins is limited due to poor solubilization of hydrophobic proteins (99,100). New detergents and electrophoresis methods can aid in examining hydrophobic proteins, but more improvements are needed (101). MS technology is not as limited in this regard. Two-dimensional electrophoresis is also limited in its ability to detect very basic proteins (pI > 10)(102). The assumptions have been challenged by a recent study (63). In this instance, 2-DE was found to be better for small-molecular-weight, hydrophobic, and cysteinelacking proteins whereas ICAT was found to be superior in examining high-molecular-weight proteins.
Neuroscientists also face complications specific to the brain and neural tissue. The most pressing in our experience is that of sample requirements. 2-DE requires ≥100 μg of protein for an experiment, and many MS experiments require even greater amounts of sample. When planning proteomic research, careful attention must be paid to the sample amount available relative to the sample needs of techniques being used. The temptation to use large amounts of brain tissue must be weighed against the need for regional and cellular specificity. Similarly, animal models must be carefully selected such that the results will provide meaningful insight into human health and disease and not simply reflect laboratory animal manipulation.
Another challenge that, while not scientific, is a very real one, is the cost of proteomic research. Almost all of the methods described here require expensive instrumentation that does not exist at many research institutions. Mass spectrometers cost hundreds of thousands of dollars and 2-DIGE requires costly fluorescent scanners. Institutions and funding agencies will have to make substantial commitments of resources for researchers to exploit the full potential of proteomics. Symbolic investments will only garner token rewards.
CONCLUSIONS
The potential for proteomic neuroscience is so great that it will become more and more popular. Thoughtful science will be needed to combine technological advances with sound, relevant experimental designs. Though many neuroscientists will adopt proteomic approaches to their research, the true advances will come from those that use these new tools to create not just data but discoveries.
Acknowledgments
The authors would like to thank Wendy Fasulo for valuable comments on the manuscript. This work is supported by NIH Grants F32DA015945 (to W. M. F.) and R01DA013772 (to S. E. H.).
Footnotes
Special issue on Expression Profiling Within the Central Nervous System II.
References
- 1.Wilkins MR, Sanchez JC, Gooley AA, Appel RD, Humphery-Smith I, Hochstrasser DF, Williams KL. Progress with proteome projects: why all proteins expressed by a genome should be identified and how to do it. Biotechnol Genet Eng Rev. 1996;13:19–50. doi: 10.1080/02648725.1996.10647923. [DOI] [PubMed] [Google Scholar]
- 2.Anderson L, Seilhamer J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 1997;18:533–537. doi: 10.1002/elps.1150180333. [DOI] [PubMed] [Google Scholar]
- 3.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mullis KB. Target amplification for DNA analysis by the polymerase chain reaction. Ann Biol Clin (Paris) 1990;48:579–582. [PubMed] [Google Scholar]
- 5.Van Gelder RN, von Zastrow ME, Yool A, Dement WC, Barchas JD, Eberwine JH. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc Natl Acad Sci USA. 1990;87:1663–1667. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang HT, Kacharmina JE, Miyashiro K, Greene MI, Eberwine J. Protein quantification from complex protein mixtures using a proteomics methodology with single-cell resolution. Proc Natl Acad Sci USA. 2001;98:5497–5502. doi: 10.1073/pnas.101124598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sano T, Smith CL, Cantor CR. Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science. 1992;258:120–122. doi: 10.1126/science.1439758. [DOI] [PubMed] [Google Scholar]
- 8.Hynd MR, Lewohl JM, Scott HL, Dodd PR. Biochemical and molecular studies using human autopsy brain tissue. J Neurochem. 2003;85:543–562. doi: 10.1046/j.1471-4159.2003.01747.x. [DOI] [PubMed] [Google Scholar]
- 9.Fountoulakis M, Hardmeier R, Hoger H, Lubec G. Postmortem changes in the level of brain proteins. Exp Neurol. 2001;167:86–94. doi: 10.1006/exnr.2000.7529. [DOI] [PubMed] [Google Scholar]
- 10.Franzen B, Yang Y, Sunnemark D, Wickman M, Ottervald J, Oppermann M, Sandberg K. Dihydropyrimidinase related protein-2 as a biomarker for temperature and time dependent post mortem changes in the mouse brain proteome. Proteomics. 2003;3:1920–1929. doi: 10.1002/pmic.200300535. [DOI] [PubMed] [Google Scholar]
- 11.Olivieri E, Herbert B, Righetti PG. The effect of protease inhibitors on the two-dimensional electrophoresis pattern of red blood cell membranes. Electrophoresis. 2001;22:560–565. doi: 10.1002/1522-2683(200102)22:3<560::AID-ELPS560>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 12.Polson C, Sarkar P, Incledon B, Raguvaran V, Grant R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;785:263–275. doi: 10.1016/s1570-0232(02)00914-5. [DOI] [PubMed] [Google Scholar]
- 13.Chan LL, Lo SC, Hodgkiss IJ. Proteomic study of a model causative agent of harmful red tide, Prorocentrum triestinum I: optimization of sample preparation methodologies for analyzing with two-dimensional electrophoresis. Proteomics. 2002;2:1169–1186. doi: 10.1002/1615-9861(200209)2:9<1169::AID-PROT1169>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 14.Lollo BA, Harvey S, Liao J, Stevens AC, Wagenknecht R, Sayen R, Whaley J, Sajjadi FG. Improved two-dimensional gel electrophoresis representation of serum proteins by using ProtoClear. Electrophoresis. 1999;20:854–859. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<854::AID-ELPS854>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 15.Lorenz P, Ruschpler P, Koczan D, Stiehl P, Thiesen HJ. From transcriptome to proteome: differentially expressed proteins identified in synovial tissue of patients suffering from rheumatoid arthritis and osteoarthritis by an initial screen with a panel of 791 antibodies. Proteomics. 2003;3:991–1002. doi: 10.1002/pmic.200300412. [DOI] [PubMed] [Google Scholar]
- 16.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 17.Bjellqvist B, Ek K, Righetti PG, Gianazza E, Gorg A, Westermeier R, Postel W. Isoelectric focusing in immobilized pH gradients: principle, methodology and some applications. J Biochem Biophys Methods. 1982;6:317–339. doi: 10.1016/0165-022x(82)90013-6. [DOI] [PubMed] [Google Scholar]
- 18.Corbett JM, Dunn MJ, Posch A, Gorg A. Positional reproducibility of protein spots in two-dimensional polyacrylamide gel electrophoresis using immobilized pH gradient isoelectric focusing in the first dimension: an interlaboratory comparison. Electrophoresis. 1994;15:1205–1211. doi: 10.1002/elps.11501501182. [DOI] [PubMed] [Google Scholar]
- 19.Gorg A, Boguth G, Obermaier C, Posch A, Weiss W. Two-dimensional polyacrylamide gel electrophoresis with immobilized pH gradients in the first dimension (IPG-Dalt): the state of the art and the controversy of vertical versus horizontal systems. Electrophoresis. 1995;16:1079–1086. doi: 10.1002/elps.11501601183. [DOI] [PubMed] [Google Scholar]
- 20.Gorg A, Obermaier C, Boguth G, Harder A, Scheibe B, Wildgruber R, Weiss W. The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis. 2000;21:1037–1053. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1037::AID-ELPS1037>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 21.Gorg A, Postel W, Friedrich C, Kuick R, Strahler JR, Hanash SM. Temperature-dependent spot positional variability in two-dimensional polypeptide patterns. Electrophoresis. 1991;12:653–658. doi: 10.1002/elps.1150120910. [DOI] [PubMed] [Google Scholar]
- 22.Wildgruber R, Harder A, Obermaier C, Boguth G, Weiss W, Fey SJ, Larsen PM, Gorg A. Towards higher resolution: Two-dimensional electrophoresis of Saccharomyces cerevisiae proteins using overlapping narrow immobilized pH gradients. Electrophoresis. 2000;21:2610–2616. doi: 10.1002/1522-2683(20000701)21:13<2610::AID-ELPS2610>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 23.Righetti PG, Wenisch E, Jungbauer A, Katinger H, Faupel M. Preparative purification of human monoclonal antibody isoforms in a multicompartment electrolyzer with immobiline membranes. J Chromatogr. 1990;500:681–696. doi: 10.1016/s0021-9673(00)96103-x. [DOI] [PubMed] [Google Scholar]
- 24.Herbert B, Righetti PG. A turning point in proteome analysis: sample prefractionation via multicompartment electrolyzers with isoelectric membranes. Electrophoresis. 2000;21:3639–3648. doi: 10.1002/1522-2683(200011)21:17<3639::AID-ELPS3639>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 25.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 26.Klose J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik. 1975;26:231–243. doi: 10.1007/BF00281458. [DOI] [PubMed] [Google Scholar]
- 27.Rabilloud T. Detecting proteins separated by 2-D gel electrophoresis. Anal Chem. 2000;72:48A–55A. doi: 10.1021/ac002709u. [DOI] [PubMed] [Google Scholar]
- 28.Nishihara JC, Champion KM. Quantitative evaluation of proteins in one- and two-dimensional polyacrylamide gels using a fluorescent stain. Electrophoresis. 2002;23:2203–2215. doi: 10.1002/1522-2683(200207)23:14<2203::AID-ELPS2203>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 29.Yan JX, Wait R, Berkelman T, Harry RA, Westbrook JA, Wheeler CH, Dunn MJ. A modified silver staining protocol for visualization of proteins compatible with matrixassisted laser desorption/ionization and electrospray ionizationmass spectrometry. Electrophoresis. 2000;21:3666–3672. doi: 10.1002/1522-2683(200011)21:17<3666::AID-ELPS3666>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Patron C, Castellanos-Serra L, Hardy E, Guerra M, Estevez E, Mehl E, Frank RW. Understanding the mechanism of the zinc-ion stains of biomacromolecules in electrophoresis gels: generalization of the reverse-staining technique. Electrophoresis. 1998;19:2398–2406. doi: 10.1002/elps.1150191407. [DOI] [PubMed] [Google Scholar]
- 31.Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988;9:255–262. doi: 10.1002/elps.1150090603. [DOI] [PubMed] [Google Scholar]
- 32.Rabilloud T, Strub JM, Luche S, van Dorsselaer A, Lunardi J. A comparison between Sypro Ruby and ruthenium II tris (bathophenanthroline disulfonate) as fluorescent stains for protein detection in gels. Proteomics. 2001;1:699–704. doi: 10.1002/1615-9861(200104)1:5<699::AID-PROT699>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 33.Steinberg TH, Top KPO, Berggren KN, Kemper C, Jones L, Diwu ZJ, Haugland RP, Patton WF. Rapid and simple single nanogram detection of glycoproteins in polyacrylamide gels and on electroblots. Proteomics. 2001;1:841–855. doi: 10.1002/1615-9861(200107)1:7<841::AID-PROT841>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 34.Steinberg TH, Agnew BJ, Gee KR, Leung WY, Goodman T, Schulenberg B, Hendrickson J, Beechem JM, Haugland RP, Patton WF. Global quantitative phosphoprotein analysis using multiplexed proteomics technology. Proteomics. 2003;3:1128–1144. doi: 10.1002/pmic.200300434. [DOI] [PubMed] [Google Scholar]
- 35.Alban A, David SO, Bjorkesten L, Andersson C, Sloge E, Lewis S, Currie I. A novel experimental design for comparative two-dimensional gel analysis: two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics. 2003;3:36–44. doi: 10.1002/pmic.200390006. [DOI] [PubMed] [Google Scholar]
- 36.Tonge R, Shaw J, Middleton B, Rowlinson R, Rayner S, Young J, Pognan F, Hawkins E, Currie I, Davison M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 37.Westermeier R, Naven T. Proteomics in practice. Wiley-VCH; Weinheim: 2002. [Google Scholar]
- 38.Unlu M, Morgan ME, Minden JS. Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis. 1997;18:2071–2077. doi: 10.1002/elps.1150181133. [DOI] [PubMed] [Google Scholar]
- 39.Shaw J, Rowlinson R, Nickson J, Stone T, Sweet A, Williams K, Tonge R. Evaluation of saturation labelling two-dimensional difference gel electrophoresis fluorescent dyes. Proteomics. 2003;3:1181–1195. doi: 10.1002/pmic.200300439. [DOI] [PubMed] [Google Scholar]
- 40.Cutler P, Heald G, White IR, Ruan J. A novel approach to spot detection for two-dimensional gel electrophoresis images using pixel value collection. Proteomics. 2003;3:392–401. doi: 10.1002/pmic.200390054. [DOI] [PubMed] [Google Scholar]
- 41.Panek J, Vohradsky J. Point pattern matching in the analysis of two-dimensional gel electropherograms. Electrophoresis. 1999;20:3483–3491. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3483::AID-ELPS3483>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 42.Krapfenbauer K, Fountoulakis M, Lubec G. A rat brain protein expression map including cytosolic and enriched mitochondrial and microsomal fractions. Electrophoresis. 2003;24:1847–1870. doi: 10.1002/elps.200305401. [DOI] [PubMed] [Google Scholar]
- 43.Washburn MP, Wolters D, Yates JR., III Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 44.Siuzdak G. Mass spectrometry for biotechnology. Academic Press; San Diego: 1996. [Google Scholar]
- 45.Liebler DC. Introduction to proteomics. Humana Press; Totowa, NJ: 2002. [Google Scholar]
- 46.Barber M, Bordoli RS, Sedgwick RD, Tyler AN, Bycroft BW. Fast atom bombardment mass spectrometry of bleomycin A2 and B2 and their metal complexes. Biochem Biophys Res Commun. 1981;101:632–638. doi: 10.1016/0006-291x(81)91305-x. [DOI] [PubMed] [Google Scholar]
- 47.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 48.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 49.Emmett MR, Andren PE, Caprioli RM. Specific molecular mass detection of endogenously released neuropeptides using in vivo microdialysis/mass spectrometry. J Neurosci Methods. 1995;62:141–147. doi: 10.1016/0165-0270(95)00070-4. [DOI] [PubMed] [Google Scholar]
- 50.Wilm M, Mann M. Analytical properties of the nanoelectrospray ion source. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 51.Witzmann FA, Fultz CD, Grant RA, Wright LS, Kornguth SE, Siegel FL. Regional protein alterations in rat kidneys induced by lead exposure. Electrophoresis. 1999;20:943–951. doi: 10.1002/(SICI)1522-2683(19990101)20:4/5<943::AID-ELPS943>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 52.Premstaller A, Oberacher H, Walcher W, Timperio AM, Zolla L, Chervet JP, Cavusoglu N, van Dorsselaer A, Huber CG. High-performance liquid chromatography-electrospray ionization mass spectrometry using monolithic capillary columns for proteomic studies. Anal Chem. 2001;73:2390–2396. doi: 10.1021/ac010046q. [DOI] [PubMed] [Google Scholar]
- 53.Beck JL, Colgrave ML, Ralph SF, Sheil MM. Electrospray ionization mass spectrometry of oligonucleotide complexes with drugs, metals, and proteins. Mass Spectrom Rev. 2001;20:61–87. doi: 10.1002/mas.1003. [DOI] [PubMed] [Google Scholar]
- 54.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 55.von Haller PD, Donohoe S, Goodlett DR, Aebersold R, Watts JD. Mass spectrometric characterization of proteins extracted from Jurkat T cell detergent-resistant membrane domains. Proteomics. 2001;1:1010–1021. doi: 10.1002/1615-9861(200108)1:8<1010::AID-PROT1010>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 56.Jonscher KR, Yates JR., III the quadrupole ion trap mass spectrometer—a small solution to a big challenge. Anal Biochem. 1997;244:1–15. doi: 10.1006/abio.1996.9877. [DOI] [PubMed] [Google Scholar]
- 57.Bienvenut WV, Deon C, Pasquarello C, Campbell JM, Sanchez JC, Vestal ML, Hochstrasser DF. Matrix-assisted laser desorption/ionization-tandem mass spectrometry with high resolution and sensitivity for identification and characterization of proteins. Proteomics. 2002;2:868–876. doi: 10.1002/1615-9861(200207)2:7<868::AID-PROT868>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 58.Morris HR, Paxton T, Panico M, McDowell R, Dell A. A novel geometry mass spectrometer, the Q-TOF, for low-femtomole/attomole-range biopolymer sequencing. J Protein Chem. 1997;16:469–479. doi: 10.1023/a:1026309410737. [DOI] [PubMed] [Google Scholar]
- 59.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 60.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 61.Zhou H, Ranish JA, Watts JD, Aebersold R. Quantitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nat Biotechnol. 2002;20:512–515. doi: 10.1038/nbt0502-512. [DOI] [PubMed] [Google Scholar]
- 62.Kubota K, Wakabayashi K, Matsuoka T. Proteome analysis of secreted proteins during osteoclast differentiation using two different methods: Two-dimensional electrophoresis and isotope-coded affinity tags analysis with two-dimensional chromatography. Proteomics. 2003;3:616–626. doi: 10.1002/pmic.200300410. [DOI] [PubMed] [Google Scholar]
- 63.Schmidt F, Donahoe S, Hagens K, Mattow J, Schaible UE, Kaufmann SH, Aebersold R, Jungblut PR. Complementary analysis of the Mycobacterium tuberculosis proteome by two-dimensional electrophoresis and isotope coded affinity tag technology. Mol Cell Proteomics. 2003;3:24–42. doi: 10.1074/mcp.M300074-MCP200. [DOI] [PubMed] [Google Scholar]
- 64.Hutchins TW, Yip TT. New desorption strategies for the mass spectrometric analysis of macromolecules. Rapid Common Mass Spectrom. 1993;7:576–580. [Google Scholar]
- 65.Merchant M, Weinberger SR. Recent advancements in surface-enhanced laser desorption/ionization-time of flight-mass spectrometry. Electrophoresis. 2000;21:1164–1177. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1164::AID-ELPS1164>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 66.Fung ET, Thulasiraman V, Weinberger SR, Dalmasso EA. Protein biochips for differential profiling. Curr Opin Biotechnol. 2001;12:65–69. doi: 10.1016/s0958-1669(00)00167-1. [DOI] [PubMed] [Google Scholar]
- 67.Reid G, Gan BS, She YM, Ens W, Weinberger S, Howard JC. Rapid identification of probiotic lactobacillus biosurfactant proteins by ProteinChip tandem mass spectrometry tryptic peptide sequencing. Appl Environ Microbiol. 2002;68:977–980. doi: 10.1128/AEM.68.2.977-980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mouledous L, Hunt S, Harcourt R, Harry J, Williams KL, Gutstein HB. Navigated laser capture microdissection as an alternative to direct histological staining for proteomic analysis of brain samples. Proteomics. 2003;3:610–615. doi: 10.1002/pmic.200300398. [DOI] [PubMed] [Google Scholar]
- 69.Mouledous L, Hunt S, Harcourt R, Harry JL, Williams KL, Gutstein HB. Proteomic analysis of immunostained, laser-capture microdissected brain samples. Electrophoresis. 2003;24:296–302. doi: 10.1002/elps.200390026. [DOI] [PubMed] [Google Scholar]
- 70.Xu BJ, Caprioli RM, Sanders ME, Jensen RA. Direct analysis of laser capture microdissected cells by MALDI mass spectrometry. J Am Soc Mass Spectrom. 2002;13:1292–1297. doi: 10.1016/S1044-0305(02)00644-X. [DOI] [PubMed] [Google Scholar]
- 71.Schwartz SA, Reyzer ML, Caprioli RM. Direct tissue analysis using matrix-assisted laser desorption/ionization mass spectrometry: practical aspects of sample preparation. J Mass Spectrom. 2003;38:699–708. doi: 10.1002/jms.505. [DOI] [PubMed] [Google Scholar]
- 72.Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Imaging mass spectrometry: a new technology for the analysis of protein expression in mammalian tissues. Nat Med. 2001;7:493–496. doi: 10.1038/86573. [DOI] [PubMed] [Google Scholar]
- 73.Satoh K, Takeuchi M, Oda Y, Deguchi-Tawarada M, Sakamoto Y, Matsubara K, Nagasu T, Takai Y. Identification of activity-regulated proteins in the postsynaptic density fraction. Genes Cells. 2002;7:187–197. doi: 10.1046/j.1356-9597.2001.00505.x. [DOI] [PubMed] [Google Scholar]
- 74.Pappin DJ. Peptide mass fingerprinting using MALDITOF mass spectrometry. Methods Mol Biol. 2003;211:211–219. doi: 10.1385/1-59259-342-9:211. [DOI] [PubMed] [Google Scholar]
- 75.Mann M, Hojrup P, Roepstorff P. Use of mass spectrometric molecular weight information to identify proteins in sequence databases. Biol Mass Spectrom. 1993;22:338–345. doi: 10.1002/bms.1200220605. [DOI] [PubMed] [Google Scholar]
- 76.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang W, Chait BT. ProFound: an expert system for protein identification using mass spectrometric peptide mapping information. Anal Chem. 2000;72:2482–2489. doi: 10.1021/ac991363o. [DOI] [PubMed] [Google Scholar]
- 78.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 79.Scheler C, Lamer S, Pan Z, Li XP, Salnikow J, Jungblut P. Peptide mass fingerprint sequence coverage from differently stained proteins on two-dimensional electrophoresis patterns by matrix assisted laser desorption/ionization-mass spectrometry (MALDI-MS) Electrophoresis. 1998;19:918–927. doi: 10.1002/elps.1150190607. [DOI] [PubMed] [Google Scholar]
- 80.Spahr CS, Susin SA, Bures EJ, Robinson JH, Davis MT, McGinley MD, Kroemer G, Patterson SD. Simplification of complex peptide mixtures for proteomic analysis: reversible biotinylation of cysteinyl peptides. Electrophoresis. 2000;21:1635–1650. doi: 10.1002/(SICI)1522-2683(20000501)21:9<1635::AID-ELPS1635>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 81.Griffiths WJ, Jonsson AP, Liu S, Rai DK, Wang Y. Electrospray and tandem mass spectrometry in biochemistry. Biochem J. 2001;355:545–561. doi: 10.1042/bj3550545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu CC, MacCoss MJ, Howell KE, Yates JR. A method for the comprehensive proteomic analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 83.Petricoin EF, Zoon KC, Kohn EC, Barrett JC, Liotta LA. Clinical proteomics: translating benchside promise into bedside reality. Nat Rev Drug Discov. 2002;1:683–695. doi: 10.1038/nrd891. [DOI] [PubMed] [Google Scholar]
- 84.Lopez MF, Pluskal MG. Protein micro- and macroarrays: digitizing the proteome. J Chromatogr B: Analyt Technol Biomed and Life Sci. 2003;787:19–27. doi: 10.1016/s1570-0232(02)00336-7. [DOI] [PubMed] [Google Scholar]
- 85.Wilson DS, Nock S. Recent developments in protein microarray technology. Angew Chem Int Ed Engl. 2003;42:494–500. doi: 10.1002/anie.200390150. [DOI] [PubMed] [Google Scholar]
- 86.Paweletz CP, Charboneau L, Bichsel VE, Simone NL, Chen T, Gillespie JW, Emmert-Buck MR, Roth MJ, Petricoin EF, III, Liotta LA. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20:1981–1989. doi: 10.1038/sj.onc.1204265. [DOI] [PubMed] [Google Scholar]
- 87.Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:RESEARCH0004.1–0004.13. doi: 10.1186/gb-2001-2-2-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fountoulakis M, Juranville JF, Dierssen M, Lubec G. Proteomic analysis of the fetal brain. Proteomics. 2002;2:1547–1576. doi: 10.1002/1615-9861(200211)2:11<1547::AID-PROT1547>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 89.Habeck M. Brain proteome project launched. Nat Med. 2003;9:631. [Google Scholar]
- 90.Zabel C, Chamrad DC, Priller J, Woodman B, Meyer HE, Bates GP, Klose J. Alterations in the mouse and human proteome caused by Huntington’s disease. Mol Cell Proteomics. 2002;1:366–375. doi: 10.1074/mcp.m200016-mcp200. [DOI] [PubMed] [Google Scholar]
- 91.Krapfenbauer K, Berger M, Lubec G, Fountoulakis M. Changes in the brain protein levels following administration of kainic acid. Electrophoresis. 2001;22:2086–2091. doi: 10.1002/1522-2683(200106)22:10<2086::AID-ELPS2086>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 92.Schonberger SJ, Edgar PF, Kydd R, Faull RLM, Cooper GJS. Proteomic analysis of the brain in Alzheimer’s disease: molecular phenotype of a complex disease process. Proteomics. 2001;1:1519–1528. doi: 10.1002/1615-9861(200111)1:12<1519::aid-prot1519>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 93.Castegna A, Aksenov M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002;82:1524–1532. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- 94.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxyterminal hydrolase L-1. Free Radic Biol Med. 2002 Aug 15;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 95.Korolainen MA, Goldsteins G, Alafuzoff I, Koistinaho J, Pirttila T. Proteomic analysis of protein oxidation in Alzheimer’s disease brain. Electrophoresis. 2002;23:3428–3433. doi: 10.1002/1522-2683(200210)23:19<3428::AID-ELPS3428>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 96.Graves PR, Haystead TA. A functional proteomics approach to signal transduction. Recent Prog Horm Res. 2003;58:1–24. doi: 10.1210/rp.58.1.1. [DOI] [PubMed] [Google Scholar]
- 97.Loughrey Chen S, Huddleston MJ, Shou W, Deshaies RJ, Annan RS, Carr SA. Mass spectrometry-based methods for phosphorylation site mapping of hyperphosphorylated proteins applied to Net1, a regulator of exit from mitosis in yeast. Mol Cell Proteomics. 2002;1:186–196. doi: 10.1074/mcp.m100032-mcp200. [DOI] [PubMed] [Google Scholar]
- 98.Becamel C, Alonso G, Galeotti N, Demey E, Jouin P, Ullmer C, Dumuis A, Bockaert J, Marin P. Synaptic multiprotein complexes associated with 5-HT(2C) receptors: a proteomic approach. EMBO J. 2002;21:2332–2342. doi: 10.1093/emboj/21.10.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Santoni V, Molloy M, Rabilloud T. Membrane proteins and proteomics: un amour impossible? Electrophoresis. 2000;21:1054–1070. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1054::AID-ELPS1054>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 100.Santoni V, Kieffer S, Desclaux D, Masson F, Rabilloud T. Membrane proteomics: use of additive main effects with multiplicative interaction model to classify plasma membrane proteins according to their solubility and electrophoretic properties. Electrophoresis. 2000;21:3329–3344. doi: 10.1002/1522-2683(20001001)21:16<3329::AID-ELPS3329>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 101.Taylor CM, Pfeiffer SE. Enhanced resolution of glycosylphosphatidylinositol-anchored and transmembrane proteins from the lipid-rich myelin membrane by two-dimensional gel electrophoresis. Proteomics. 2003;3:1303–1312. doi: 10.1002/pmic.200300451. [DOI] [PubMed] [Google Scholar]
- 102.Bae SH, Harris AG, Hains PG, Chen H, Garfin DE, Hazell SL, Paik YK, Walsh BJ, Cordwell SJ. Strategies for the enrichment and identification of basic proteins in proteome projects. Proteomics. 2003;3:569–579. doi: 10.1002/pmic.200300392. [DOI] [PubMed] [Google Scholar]