

Abstract
Iron is an essential element to all living organisms and is an important determinant of bacterial virulence. Bacteria have evolved specialized systems to sequester and transport iron from the environment or host. Pseudomonas aeruginosa, an opportunistic pathogen, uses two outer membrane receptor mediated systems (Phu and Has) to utilize host heme as a source of iron. PhuS is a 39 kDa soluble cytoplasmic heme binding protein which interacts and transports heme from the inner membrane heme transporter to the cytoplasm where it is degraded by heme oxygenase thus releasing iron. PhuS is unique among other cytoplasmic heme transporter proteins owing to the presence of three histidines in the heme binding pocket which can potentially serve as heme ligands. Out of the three histidine residues on the heme binding helix, His 209 is conserved among heme trafficking proteins while His 210 and His 212 are unique to PhuS. Here we report the crystal structure of PhuS at 1.98 Å resolution which shows a unique heme binding pocket and oligomeric structure compared to other known cytoplasmic heme transporter and accounts for some of the unusual biochemical properties of PhuS.
Keywords: PhuS, iron, heme, transport, Pseudomonas aeruginosa
1. Introduction
Pathogenic bacteria have evolved elaborate transport systems that enable the acquisition of nutrients from the host. One such nutrient is iron. Since the most abundant source of host iron is in heme, these bacteria have a complex system that transports host heme across the inner and outer membrane and into the cytoplasm where heme is degraded and iron released [1, 2]. The opportunistic pathogen, Pseudomonas aeruginosa, a gram-negative bacterium that causes infections in immune-compromised individuals, encodes two heme uptake systems: the phu (Pseudomonas heme uptake) and has (heme assimilation system) to utilize host heme and hemoproteins as a source of iron [3]. The Phu operon encodes inner and outer membranes proteins and soluble periplasmic space and cytoplasmic proteins required for heme transport and degradation [4]. The first step of heme transport involves a TonB dependent cell surface receptor that acquires heme. Heme transport across the periplasmic space involves an active transport system, composed of a soluble periplasmic heme binding protein, an inner membrane heme permease, and an ATPase. Upon crossing the inner membrane, the heme is broken down by heme oxygenase thus releasing iron.
The fate of heme once in the cytoplasm is not well understood and it remains unclear whether all pathogens use similar heme degradation pathways. In previous work we determined the crystal structures of the heme oxygenase from both Pseudomonas aeruginosa [5] and Neisseriae meningitidis [6, 7] and the soluble periplasmic heme binding protein that shuttles heme from the outer membrane to the inner membrane from both Shigella dysenteriae and Pseudomonas aeruginosa [8]. The Pseudomonas cytoplasmic heme binding protein, called PhuS, has been well characterized biochemically [4, 9-11]. PhuS is a monomeric 39 kDa protein that binds one heme per monomer of protein. A recent study [11] has shown that PhuS can form a 1:1 complex with heme oxygenase only when heme is bound to PhuS.
Pathogenic strains of E. coli use a heme transport system similar to P. aeruginosa and ChuS [12] from E. coli is a homologue of PhuS. While E. coli does not have a similar heme oxygenase, Suits et al. [12] reported that ChuS exhibits heme oxygenase activity [13] while we found that PhuS does not [14]. Resonance Raman spectroscopy coupled with site directed mutagenesis has identified His 209 as the proximal ligand to heme in PhuS. However, spectroscopic studies on holo-PhuS-H209A mutant shows an alternative heme binding site provided by co-ordination to His 212 [9]. Another histidine, His 210, does not directly co-ordinate to heme, but in the absence of His 209, His 210 was shown to stabilize ligand coordination through His 212. Out of three histidine residues on the heme binding helix, His 209 is conserved among heme trafficking proteins while His 210 and His 212 are unique to PhuS. O'Neill et al. [11] suggested a model in which heme binding to different histidine residues favors different functions (Scheme1).
Scheme 1. Mechanism of heme transfer from holo-PhuS to apo-HO (adopted from [11]). Heme binding to His 212 favors a conformational change required for interacting with and transferring heme to heme oxygenase.

In addition to PhuS, several other cytoplasmic heme-trafficking homologs have beeninvestigated; HemS from Yersinia enterocolitica [15], ChuS from Escherichia coli [12, 13], and ShuS from Shigella dysenteriae [16, 17]. To better understand and correlate the wealth of biochemical data on PhuS with structure we have determined the crystal structure of apo-PhuS.
2.0 Experimental Procedures
2.1 Protein Expression and Purification
PhuS was cloned into pET28a with a C-terminal His–tag for over-expression and purification. Protein was over-expressed in E. coli BL21 (plysS) cells by inducing with isopropyl β-D-thiogalactopyranoside (IPTG) at 0.6 OD and grown further for 12 h at 22°C. The IPTG (Isopropyl β-D-thiogalactopyranoside)-induced cells were harvested, resuspended in 50 mM Tris–HCl pH 7.8 and 10 mM Imidazole (buffer A) and lysed by sonication. The crude lysate was centrifuged at 17,000 rpm for 60 min. The supernatant was applied on to Ni2+-NTA column pre-equilibrated with buffer A. The column was washed with buffer containing 20 mM imidazole and protein was eluted using the same buffer supplemented with 200 mM Imidazole. Fractions containing protein were pooled and concentrated by using a 10 kDa centricon. PhuS was further purified and buffer exchanged by Gel filtration using Superdex 75 16/60 column equilibrated with 50 mM Tris (7.8). Size exclusion chromatography shows that the apo-protein exists as a moomer in solution. Protein concentrations were determined by using a Bio-Rad assay kit. The purity of the protein was confirmed using 12% SDS–PAGE.
2.2 Crystallization and Data Collection
We made many unsuccessful attempts to crystallize heme-bound holo-PhuS. Since PhuS has the unique property of using one of two heme pocket His residues as a ligand, it seemed possible that heme ligation heterogeneity was preventing crystallization. However, various His mutants which should enable only one His to coordinate also failed to crystallize. In contrast, apo-PhuS readily crystallized using the hanging drop method at 22°C. Crystals were obtained by mixing 2 μL of 50mg/ml protein and 1 μL of reservoir solution containing 200 mM MgCl2.6H2O, 100 mM HEPES (pH 7.5) and 20-30% Polyacrylic acid 5100. Crystals belong to the tetragonal space group I4. The final structure of PhuS contains two molecules in the asymmetric unit. Twenty % glycerol was used as the cryo-protectant and X-ray diffraction data collected from single crystal at SSRL beamline 7-1. Diffraction images were indexed, integrated and scaled using the HKL-2000 package [18].
2.3 Structure Solution and Refinement
Amino acid sequence alignment (Fig. 1) shows 41% identity with Escherichia coli, ChuS [13] (PDB; 2HQ2). Therefore, a search model derived from this structure was used in the molecular replacement calculations using the program Phaser [19] implemented in the CCP4 package [20]. The best solution gave a LLG of 454 and TFZ 10.3 using data between 15 and 3 Å. The transformed model was subjected to rigid body refinement using REFMAC5 [21]. Phases were improved and extended to 1.98 Å incrementally. The 2Fo-Fc and Fo-Fc electron density maps were visualized using the program Coot [22] which was also used for model building. The crystallographic R-factor and R-free were monitored at each stage to avoid any bias. Final refinement and water molecules were added by using Phenix [23]. In the final stages of refinement, TLS refinement was also performed. A few additional waters were manually identified on the basis of electron density maps. PhuS crystallizes as a dimer and each chain consists of residues from 8-354. Eight amino acids from N-terminal of each chain exhibit relatively weaker or no electron density. The final refinement statistics are given in Table 1.
Figure 1.

Amino acids sequence alignment between known structures of cytoplasmic heme transporter proteins Chus and HemS. Residues at the PhuS dimeric interface are marked with star while histidine residues in the active site marked with triangle. The figure was generated using ClustalW and Espript [25].
Table 1. Data Collection and Refinement statistics.
| Apo-PhuS | |
|---|---|
| PDB code | 4IMH |
| Space group | I4 |
| Unit cell dimensions (a,b,c) (Å), (α, β, γ (°) | 187.7, 187.7 42.4 90, 90, 90 |
| Resolution Range (Å) (highest shell) | 46.93-1.98 (2.08-1.98) |
| Wavelength (Å) | 0.98 |
| Total observations | 274132 |
| Unique reflections (highest shell) | 51820 (7551) |
| Completeness (%) (highest shell) | 98.9 (100) |
| Rmerge1 (highest shell) | 0.066 (0.266) |
| <I/σ> (highest shell) | 15.7 (5.6) |
| Redundancy (highest shell) | 5.3 (5.4) |
| Refinement | |
| Rwork %2/Rfree %3 | 17.5/22.2 |
| Number of atoms | |
| Protein | 5453 |
| Water | 371 |
| RMSD Bond length (Å) | 0.013 |
| RMSD Bond angle (°) | 1.45 |
Rsym =Σ |I - <I>|/Σ I, where I is the observed intensity and <I> the averaged intensity of multiple symmetry related observations of the reflection.
R factor = Σ‖Fol-|Fc‖/Σ|Fo|, Fo and Fc are the observed and calculated structure factors, respectively.
R-free was calculated with the 5% of reflections set aside randomly throughout the refinement.
2.4 Structural Analysis
The geometry of the refined model was checked using PROCHECK [24]. All structural superimposition and rmsd calculations were carried out using the CCP4 program LSQKAB [20]. The most favored and additionally allowed regions of the Ramachandran plot contained more than 98% of non-glycine and non-proline residues. B-factor analysis was carried out by using the BAVERAGE program from CCP4 package [20]. Interactions were calculated using CONTACTS module of the CCP4 package. Surface calculations were carried out using NACCESS [20]. All structural figures were prepared using PyMOL (http://pymol.org/). Modeling the heme in an alternate position to coordinate His 212 was carried out as follows. Using Pymol His 212 was modeled in the various favored rotamers and the heme manually positioned in place to check if a linear ≈2.0Å Fe-N bond could be formed without significant steric clashes. This was readily achieved with no requirement that any protein move except for a new His 212 rotamer. We also assumed that the heme propionates would be oriented toward the PhuS protein rather than solvent which would enable lateral transfer of the heme to heme oxygenase without the requirement that the heme flip over.
3.0 Result and Discussion
3.1 Tertiary Structure
Each PhuS monomer is made of two almost identical units of nine anti-parallel β-sheets which are flanked by α-helices on both ends (Fig 2). Including side chains, the two halves of each monomer share structural homology with a rmsd of 2.9 Å although they share only 20% sequence identity. Superimposition of each half shows a very well aligned central core of β-sheets while terminal α-helices exhibit major variations (Fig 3).
Figure 2.
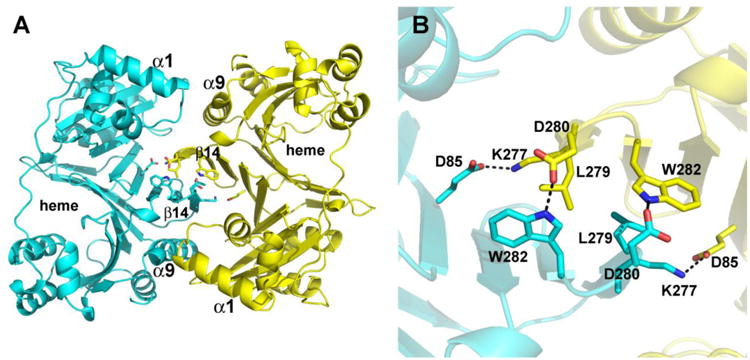
Structure of the apo-PhuS dimer showing a few limited key interactions at the dimer interface. A) Overall view of the dimer with key elements of secondary structure labeled. The central interaction involves the β14 segment. B) Close up view of the dimer interface in the same orientation as in panel A.
Figure 3.
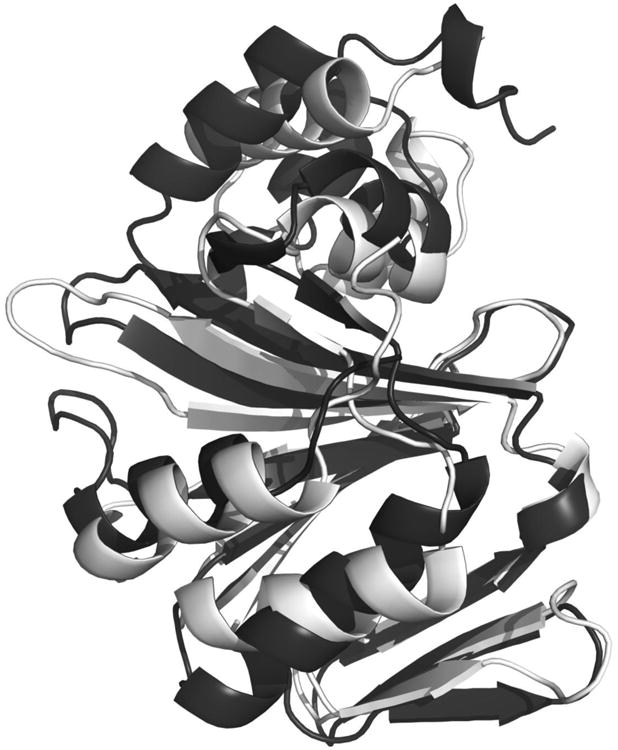
Ribbon diagram of two halves of individual subunit superimposed showing core antiparallel β-sheet decorated by α-helices on both sides. Residues 10-173 are light gray and 186-354 dark gray.
3.2 Dimeric Structure
A structure-similarity search using the PDBeFold protein-structure comparison service at the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm/) revealed that PhuS is a close structural homologue of HemS (rmsd=1.31 Å) and ChuS (rmsd=1.38 Å). PhuS crystallizes as a dimer while ChuS and HemS crystallized as monomers in both the apo and holo forms and HemS has been shown to behave as a monomer in solution [15]. As shown in Fig. 2, the dimeric interaction involves primarily the β14-15 antiparallel sheet structure on the surface of each monomer. Near the center of the dimeric interface symmetry related Leu 279 residues form nonbonded contacts with additional symmetry related ion pairs and H-bonds assisting in dimer stabilization. The total surface area of both chains in apo-PhuS is 31,890 Å2 out of which 2,236 Å2 is buried. About 40% of the buried surface on dimerization is polar in nature. There are 18 polar interactions inviolving 12 residues less than 3.6 Å apart indicating a strong dimeric association (Table 2). Of the 12 residues involved in these dimeric interactions only three residues (Glu85, Trp282 and His293) are conserved among the three known structures of cytoplasmic heme transporters (Fig 1)
Table 2. Polar interactions between two subunits in PhuS.
| Atom in subunit A | Atom in subunit B | Distance (Å) | ||
|---|---|---|---|---|
| Gln27 | NE2 | Asp281 | OD1 | 3.1 |
| Arg29 | NH2 | Trp278 | N | 3.4 |
| Trp278 | O | 3.0 | ||
| Glu85 | OE1 | Lys277 | NZ | 2.7 |
| OE2 | Lys277 | NZ | 3.3 | |
| Lys277 | NZ | Glu85 | OE1 | 2.6 |
| Glu85 | OE2 | 3.1 | ||
| Asn291 | OD1 | 2.8 | ||
| His293 | NE2 | 3.2 | ||
| Trp278 | O | Arg29 | NH2 | 2.9 |
| Asp280 | OD2 | Trp282 | NE1 | 2.7 |
| Asp281 | OD1 | Arg25 | NE | 3.0 |
| OD1 | Arg25 | NH2 | 2.8 | |
| OD1 | Gln27 | NE2 | 3.1 | |
| Trp282 | NE1 | Asp280 | OD2 | 2.9 |
| Asn291 | OD1 | Lys277 | NZ | 3.1 |
| His293 | NE2 | Lys277 | NZ | 3.0 |
| Lys295 | NZ | Asp280 | OD2 | 3.0 |
The functional relevance of the dimer, however, remains questionable. Lansky et al [14] found that purified holo-PhuS behaves as a mix of monomers and dimers and while the monomer is the dominant form in solution, the dimer is quite stable once purified away from the monomer [9]. Since the heme pocket is not involved in forming the dimer, PhuS should be able to bind heme either as a dimer or a monomer and thus serves its heme trafficking function in either form. Analysis of structure by PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver) shows that apo-PhuS dimer as seen in the crystal structure should not be stable in solution.
3.3 Mechanistic Insights
A superposition of the apo-PhuS structure with heme bound HemS [15] and ChuS [12, 13] did not show any large differences in the heme pocket which clearly indicates that heme binding does not require any major structural changes (Fig. 4). Moreover, the ChuS holo and apo structures are the same with little change in the heme pocket [12, 13]. Given the close similarity of PhuS and Chus it was fairly straightforward to model the ChuS heme into the PhuS structure (Fig. 5). His 209 (His 193 in ChuS) is perfectly positioned to serve as a heme ligand which is fully consistent with previous biochemical studies.
Figure 4.

Superimposition of apo-PhuS (light gray) with holo-ChuS (dark gray) structures. The heme is represented as the stick models. The coordinates used for the superposition were those of ChuS from E. coli (pdb id. 2HQ2).
Figure 5.
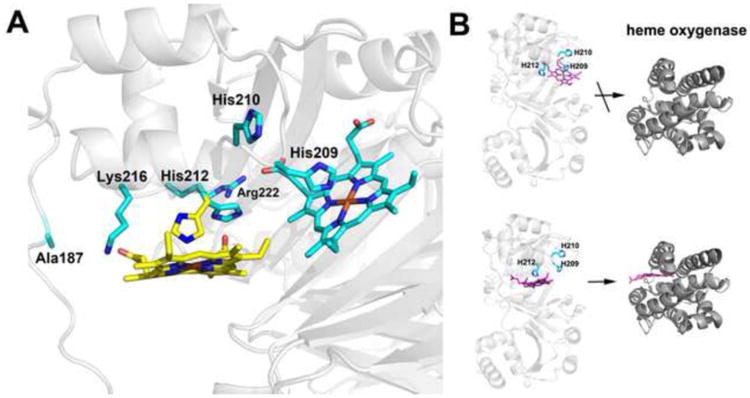
A) Modeling of two possible heme binding modes. Heme is modeled at both His 209 and His 212. His 209 is perfectly positioned to bind to heme while His 212 must adopt an alternative rotamer conformation to bind to heme. The rotamers observed in the crystal structure are cyan while the new rotamer conformation of His 212 required for heme ligation is yellow. We also checked to see if alternate rotamers of His 210 might be able to bind heme but His 210 extends out into solution and there is no rotamer position that orients His 210 into the pocket for heme coordination. B) Models emphasizing that heme transfer to heme oxygenase occurs only when His 212 is the ligand.
The unique feature of the PhuS heme pocket is the presence of His 210 and His 212 which correspond to Phe 196 and Gln 218 in ChuS, respectively. Previous work has shown that either His 209 or His 212 can serve as a heme ligand while His 210 does not [9]. This is fully consistent with the structure. As shown in Fig. 5 repositioning of the heme in the binding pocket will enable His 212 to ligate the heme with the main requirement being that His 212 adopt an alternative rotamer conformation. Interestingly, both Lys 216 and Arg 222 are in position to interact with the heme propionates thus providing some additional stability. Only moderate adjustments of side chains would be required with no changes in the polypeptide backbone. Of critical importance is the role that each of these His residues plays in the interaction with heme oxygenase, a requirement for heme transfer. The His209Ala mutant can bind heme and form a complex with heme oxygenase while the His212Ala mutant also binds heme but cannot bind to heme oxygenase [11]. This means that either His 212 must be the ligand for heme transfer or His 209 is the ligand but that His 212 is required for interaction with heme oxygenase. The structure supports the His 212 ligation model. If His 212 were required for direct interaction with heme oxygenase it should extend out to the surface where it can directly contact heme oxygenase. His 212 is locked into a helical segment and unless this region experiences an unlikely large conformational change, His 212 cannot extend out toward the surface where it could directly interact with heme oxygenase. In addition, heme coordination to His 212 places the heme in a more exposed environment and near a long segment centered near Ala186 that exhibits weak electron density indicative of high mobility. Finally, we need to take into account the finding that the His210Ala mutant does not bind heme oxygenase even though His 210 is never a heme ligand [11]. His 210 extends out toward the surface and if heme oxygenase approaches close to the PhuS heme pocket as it must, then His 210 will closely approach heme oxygenase and thus may play a role in direct binding. His 210 also would be more centrally located to the heme oxygenase docking site if the PhuS heme were to be transferred from His 212. It also is noteworthy that the PhuS homolog, ChuS, has not been implicated in heme transfer but instead is thought to be a heme degrading protein [12]. This raises the possibility that the unique His 212 ligation in PhuS evolved for the purpose of heme transfer to heme oxygenase while those homologs to PhuS without the unique His 212 are directly involved in heme degradation and/or participate in some other aspect of the heme trafficking process.
Highlights.
The crystal structure of the P. aeruginosa cytoplasmic heme binding protein has been determined to 1.98Å.
The structure is very similar to other heme binding proteins from other pathogenic bacteria.
Modeling studies suggest two binding modes for heme.
The structure and multiple heme binding modes helps to explain some of the biochemical/biophysical properties of PhuS.
Acknowledgments
This work was supported by NIH Grants GM42614 (TLP) and AI85535 (AW). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
This work was supported by NIH grant GM41614 (TLP) and AI85535 (AW)
Abbreviations
- IPTG
Isopropyl β-D-thiogalactopyranoside
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- rmsd
root mean square deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wandersman C, Delepelaire P. Annu Rev Microbiol. 2004;58:611–647. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 2.Wilks A, Burkhard KA. Nat Prod Rep. 2007;24:511–522. doi: 10.1039/b604193k. [DOI] [PubMed] [Google Scholar]
- 3.Ochsner UA, Johnson Z, Vasil ML. Microbiol. 2000;146(Pt 1):185–198. doi: 10.1099/00221287-146-1-185. [DOI] [PubMed] [Google Scholar]
- 4.Bhakta MN, Wilks A. Biochemistry. 2006;45:11642–11649. doi: 10.1021/bi060980l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman J, Lad L, Li H, Wilks A, Poulos TL. Biochemistry. 2004;43:5239–5245. doi: 10.1021/bi049687g. [DOI] [PubMed] [Google Scholar]
- 6.Friedman J, Lad L, Deshmukh R, Li H, Wilks A, Poulos TL. J Biol Chem. 2003;278:34654–34659. doi: 10.1074/jbc.M302985200. [DOI] [PubMed] [Google Scholar]
- 7.Friedman J, Meharenna YT, Wilks A, Poulos TL. J Biol Chem. 2007;282:1066–1071. doi: 10.1074/jbc.M609112200. [DOI] [PubMed] [Google Scholar]
- 8.Ho WW, Li H, Eakanunkul S, Tong Y, Wilks A, Guo M, Poulos TL. J Biol Chem. 2007;282:35796–35802. doi: 10.1074/jbc.M706761200. [DOI] [PubMed] [Google Scholar]
- 9.Block DR, Lukat-Rodgers GS, Rodgers KR, Wilks A, Bhakta MN, Lansky LB. Biochemistry. 2007;46:14391–14402. doi: 10.1021/bi701509n. [DOI] [PubMed] [Google Scholar]
- 10.Kaur AP, Lansky LB, Wilks A. J Biol Chem. 2009;284:56–66. doi: 10.1074/jbc.M806068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Neill MJ, Bhakta MN, Fleming KG, Wilks A. Proc Natl Acad Sci U S A. 2012;109:5639–5644. doi: 10.1073/pnas.1121549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suits MD, Pal GP, Nakatsu K, Matte A, Cygler M, Jia Z. Proc Natl Acad Sci U S A. 2005;102:16955–16960. doi: 10.1073/pnas.0504289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suits MD, Jaffer N, Jia Z. J Biol Chem. 2006;281:36776–36782. doi: 10.1074/jbc.M607684200. [DOI] [PubMed] [Google Scholar]
- 14.Lansky LB, Lukat-Rodgers GS, Block D, Rodgers KR, Ratliff M, Wilks A. J Biol Chem. 2006;281:13652–13662. doi: 10.1074/jbc.M600824200. [DOI] [PubMed] [Google Scholar]
- 15.Schneider S, Sharp KH, Barker PD, Paoli M. J Biol Chem. 2006;281:32606–32610. doi: 10.1074/jbc.M607516200. [DOI] [PubMed] [Google Scholar]
- 16.Wilks A. Arch Biochem Biophys. 2001;387:137–142. doi: 10.1006/abbi.2000.2250. [DOI] [PubMed] [Google Scholar]
- 17.Wyckoff EE, Lopreato GF, Tipton KA, Payne SM. J Bacteriol. 2005;187:5658–5664. doi: 10.1128/JB.187.16.5658-5664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otwinowski ZaM, Wladek Methods in Enzymology. 1997;276 A:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Acta Crystallogr. 2011;D67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 23.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 25.Gouet P, Robert X, Courcelle E. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]