

Abstract
Ubiquitin-like proteins (UBLs) are activated, transferred and conjugated by E1-E2-E3 enzyme cascades. E2 enzymes for canonical UBLs such as ubiquitin, SUMO, and NEDD8 typically use common surfaces to bind to E1 and E3 enzymes. Thus, canonical E2s are required to disengage from E1 prior to E3-mediated UBL ligation. However, E1, E2, and E3 enzymes in the autophagy pathway are structurally and functionally distinct from canonical enzymes, and it has not been possible to predict whether autophagy UBL cascades are organized according to the same principles. Here, we address this question for the pathway mediating lipidation of the human autophagy UBL, LC3. We utilized bioinformatic and experimental approaches to identify a distinctive region in the autophagy E2, Atg3, that binds to the autophagy E3, Atg12∼Atg5-Atg16. Short peptides corresponding to this Atg3 sequence inhibit LC3 lipidation in vitro. Notably, the E3-binding site on Atg3 overlaps with the binding site for the E1, Atg7. Accordingly, the E3 competes with Atg7 for binding to Atg3, implying that Atg3 likely cycles back and forth between binding to Atg7 for loading with the UBL LC3 and binding to E3 to promote LC3 lipidation. The results show that common organizational principles underlie canonical and noncanonical UBL transfer cascades, but are established through distinct structural features.
Keywords: Atg3, Atg7, Atg12∼Atg5, E1-E2 binding, E2-E3 binding, autophagy, ubiquitin-like protein, E1 enzyme, E2 enzyme, E3 enzyme
Introduction
Macroautophagy is a lysosomal degradation pathway in which substrates are engulfed within a double-membrane structure called an autophagosome. Fusion of autophagosomes with lysosomes (or with the vacuole in yeast) enables lysosomal hydrolases to degrade the sequestered substrates.1,2 Macroautophagy is carried out by a large set of proteins that includes ∼35 so-called “Atg” proteins that were originally identified by genetic screens in yeast.3,4 Among these is the ubiquitin-like protein (UBL) Atg8 in yeast or LC3 in mammals (for simplification, the multiple mammalian orthologs of yeast Atg8 are referred to here as LC3, although similar properties were found for others including the GABARAP family). Atg8/LC3 is ultimately conjugated to lipid to dynamically recruit autophagy machinery and substrates to the membrane to promote autophagosome formation.5–7
Like other UBLs, LC3 is conjugated to substrates via the sequential action of a hierarchical cascade of E1, E2, and E3 enzymes [Fig. 1(A)]. The autophagy specific E1 enzyme Atg7 binds LC3 and uses a molecule of ATP to adenylate the LC3 C terminus by a mechanism that is similar to that of other E1s, such as those for the canonical UBLs ubiquitin, SUMO and NEDD8.8–11 Similar to canonical UBL transfer cascades, Atg7 next forms a covalent thioester-linked intermediate between the UBL C terminus and an E1 enzyme catalytic cysteine residue and transfers the UBL to the catalytic cysteine residue of an E2 enzyme (Atg3). Finally, an E3 enzyme Atg12∼Atg5-Atg16 (“∼” denotes a covalent complex and “-” denotes a non-covalent complex), containing an Atg12∼Atg5 isopeptide-linked covalent conjugate, promotes the lipidation of LC3.6,12–14
Figure 1.
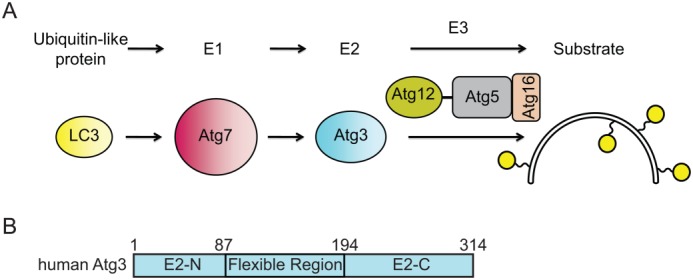
LC3 conjugation pathway. (A) Atg8 family members such as human LC3 are activated by the E1 Atg7 and then transferred to the E2 enzyme, Atg3. Atg12∼Atg5-Atg16 acts as an E3 enzyme that catalyzes lipid conjugation of LC3 and other Atg8 protein family members. (B) Domain diagram representing the primary structure of Atg3 and highlighting the Atg3 Flexible Region. E2-N and E2-C are the N- and C-terminal parts of an E2 fold.
Despite both using E1-E2-E3 cascades, there are no similarities between structures of E2 binding sites on the E1s for canonical and autophagy-specific UBL transfer enzymes, mechanisms of E1–E2 interactions, or structures of E3 enzymes in canonical and autophagy-specific UBL conjugation cascades.13–19 Thus, the extent to which common principles underlie both the canonical and autophagy-specific cascades remains incompletely understood. For instance, one important principle of canonical cascades is that E1 and E3 enzymes have overlapping binding sites on the E2 and an E2 must therefore disengage from an E3 before reloading with another activated UBL.20,21 Thus, we sought to evaluate whether an autophagy UBL cascade likewise displays this organization.
Prior structures of complexes with yeast Atg7 and biochemical studies show that a segment of the “flexible region” (FR) from yeast Atg3 binds to Atg7.15–18,22 Although mutation of the corresponding Atg3FR-binding region in mouse Atg7 led to impaired LC3 lipidation in mouse embryonic fibroblast cells,15 alignments of the Atg3FR sequences between yeast, animals, and plants show poor conservation and this region was not ordered in the electron density for a complex of Arabidopsis Atg3 with a domain from Atg7.19 We therefore generated hidden Markov models (HMMs) and used HMM-HMM comparison methods to detect protein homology among the flexible regions of Atg3 homologs and evaluated the importance of conserved regions by performing affinity capture experiments with human Atg3 deletion constructs. Binding studies and competition experiments demonstrate that overlapping sites in the Atg3FR are important for E3 binding and E1 binding.
Results and Discussion
To investigate whether a peptide-like Atg7-interacting motif within the Atg3FR (termed Atg3FRpep) is conserved in nonfungal proteins, we attempted to construct multiple sequence alignments of fungal, plant and animal homologs. Because the Atg3FR is highly divergent and difficult to align, we first constructed three separate alignments for representative Atg3 homologs from the fungal, animal and plant kingdoms23 (Supporting Information Fig. 1). Hidden Markov models derived from the three alignments were then compared to each other by the HHpred method.24 The result of this analysis suggests that the Atg7-binding motif conserved in the fungal proteins corresponds to a conserved motif in animals (purple bars in Supporting Information Fig. 1). This motif appears to be duplicated in plants. The alignment suggests that features of the Atg3FRpep that binds yeast Atg7 appear to be conserved and to encompass at least residues 156–167 of human Atg3.
Since multiple lines of evidence suggested the importance of the Atg3FR for Atg3 function,15,16,22 we performed affinity capture experiments with purified human Atg3 deletion mutants and GST-tagged versions of either Atg7 or Atg12∼Atg5-Atg16L1 to identify Atg3FR residues essential for binding to its cognate E1 and E3, respectively. Of the Atg3FR deletion mutants tested, Atg3(Δ161–176) and Atg3(Δ171–192) did not pull down with GST-Atg7 [Figs. 1(B) and 2(A)], consistent with prior work demonstrating the importance of the Atg3FR for binding to Atg7 in yeast.15,16,22 Notably, deletion mutants Atg3(Δ145–160) and Atg3(Δ161–176) prevented binding to GST-Atg12∼Atg5-Atg16L1 [Fig. 2(B)]. The common binding defect caused by the 16-residue deletion mutant Atg3(Δ161–176) raised the possibility that Atg3 may interact with its E1 and E3 enzymes via overlapping binding sites.
Figure 2.
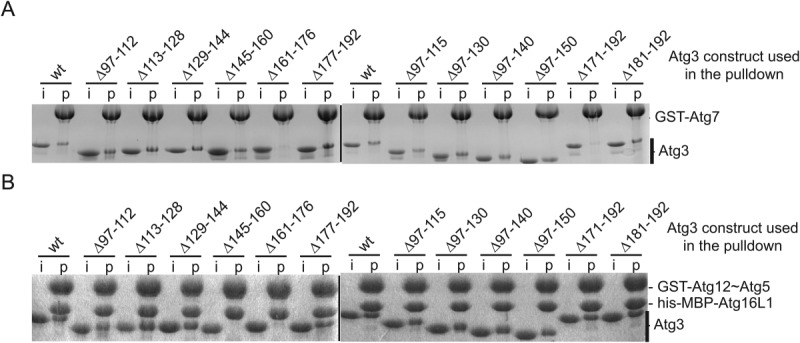
Atg7 and Atg12∼Atg5-Atg16L1 bind to an overlapping region in the Atg3 flexible region (FR). (A) Coomassie-stained SDS-PAGE gels of affinity pulldowns with purified recombinant GST-Atg7 and wild-type (wt) or deletion mutant versions of Atg3. Lanes showing input material are labeled “i” and lanes with affinity pulldowns are labeled “p.” (B) Affinity pulldown with purified recombinant GST-Atg12∼Atg5-his-MBP-Atg16L1 complex and wild-type or deletion mutant versions of Atg3.
To explore the potential overlap of Atg3 binding sites for E1 and E3 observed in our deletion analyses, we performed competitive binding experiments. We mixed Atg7 and Atg3 and observed complex formation in a nondenaturing gel mobility shift assay [Fig. 3(A)]. Similarly, Atg3 binding to Atg12∼Atg5-Atg16L1 can also be observed with this technique. When we titrated into the Atg7-Atg3 complex (20 μM) increasing amounts of Atg12∼Atg5-Atg16L1 (0, 4, 10, 20 μM), the Atg7-Atg3 complex was disrupted by Atg12∼Atg5-Atg16L1 and replaced by an Atg3-Atg12∼Atg5-Atg16L1 complex [Fig. 3(A)]. The results suggested that Atg3 binding to its E1 and E3 is mutually exclusive. In support of this conclusion, the Atg7 complex with Atg3(Δ145–160), which does not bind Atg12∼Atg5-Atg16L1 by affinity capture, was not disrupted in the same titration experiments [Fig. 3(B)]. Interestingly, in a reverse titration, increasing amounts of Atg7 had negligible effects on an Atg3-Atg12∼Atg5-Atg16L1 complex [Fig. 3(C)]. In sum, the data suggest that Atg3 has overlapping but nonidentical binding sites for its E1 and E3 enzymes, with E3 capable of disrupting the Atg3–Atg7 interaction but not vice-versa.
Figure 3.
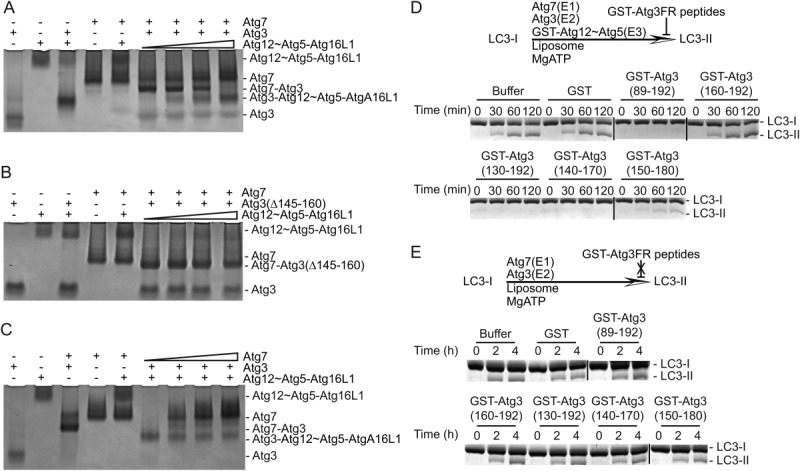
Competition experiments. (A,B) 20 μM Atg7-Atg3 or Atg7-Atg3(Δ145–160) complex was incubated with increasing amounts of Atg12∼Atg5-Atg16L1 complex (0, 4, 10, 20 μM) as described under “Materials and Methods.” Atg3, Atg12∼Atg5-Atg16L1 complex, the Atg7-Atg3 complex, and the Atg3-Atg12∼Atg5-Atg16L1 complex were identified by their different migrations on a Coomassie-stained nondenaturing polyacrylamide gel and are indicated at the right. (C) Binding competition assay performed as for A and B showing titration of Atg3-Atg12∼Atg5-Atg16L1 with Atg7. (D) LC3 lipidation competition assays. Coomassie-stained urea SDS-PAGE showing in vitro LC3 lipidation in multiple turnover assays containing Atg7, Atg3, Atg12∼Atg5, and LC3. GST or GST-Atg3 peptides were added at a 10-fold molar excess over Atg3. (E) LC3 lipidation competition assays performed in the absence of E3 enzyme. Sypro-stained urea SDS-PAGE showing in vitro LC3 lipidation in multiple turnover assays containing Atg7, Atg3, and LC3. GST or GST-Atg3 peptides were added at a 10-fold molar excess over Atg3.
To gain further insights into Atg3 binding to Atg7 and Atg12∼Atg5-Atg16L1, we quantified interactions by isothermal titration calorimetry (ITC). We measured an affinity for Atg3 binding to Atg12∼Atg5-Atg16L1 of 80 nM, which is similar to the previously reported value of 51 nM13 (Table I and Supporting Information Fig. 2). Atg3FR deletion mutants lacking residues ∼150–170 are deficient for Atg12∼Atg5-Atg16L1 binding while GST-Atg3 fragments or peptides containing residues ∼140–170 display high affinity for Atg12∼Atg5-Atg16L1 (Table I and Supporting Information Fig. 2). Indeed, functional significance of this region was confirmed by inhibition of LC3 lipidation in vitro: addition of GST-Atg3(140–170), but not GST or the non-binding GST-Atg3(160–192), blocked the reaction [Fig. 3(D)]. In similar reactions lacking E3 enzyme, lipidation is not inhibited by GST or GST-Atg3 peptides [Fig. 3(E)], indicating that the inhibition in Figure 3(D) is due to disruption of E2–E3 interaction rather than E1–E2 interaction.
Table I.
E1 and E3 Recognition of the Atg3FRpep Motif
| Molecule: titrant | Kd (µM) | ΔH × 103 (cal mol−1) | ΔS (cal mol−1 deg−1) | N |
|---|---|---|---|---|
| His-Atg3 : Atg12∼Atg5-Atg16L1 | 0.08 ± 0.02 | −13.20 ± 0.15 | −12.0 | 0.82 ± 0.01 |
| His-Atg3(Δ97–150) : Atg12∼Atg5-Atg16L1 | 0.27 ± 0.05 | −14.80 ± 0.25 | −19.5 | 0.80 ± 0.01 |
| His-Atg3(Δ97–192) : Atg12∼Atg5-Atg16L1 | NB | NB | NB | NB |
| Atg3(89–192) : Atg12∼Atg5-Atg16L1 | 0.09 ± 0.04 | −11.30 ± 0.25 | −5.5 | 0.99 ± 0.01 |
| Atg3(140–170) : Atg12∼Atg5-Atg16L1a | 0.07 ± 0.004 | −10.34 ± 0.03 | −1.9 | 1.00 ± 0.002 |
| Atg3(145–175) : Atg12∼Atg5-Atg16L1a | 0.05 ± 0.005 | −9.08 ± 0.05 | 3.0 | 1.04 ± 0.003 |
| Atg3(152–181) : Atg12∼Atg5-Atg16L1a,b | ≥52.36 ± 17.05 | −13.40 ± 5.63 | −25.3 | 0.99 ± 0.35 |
| GST-Atg3(89–192) : Atg12∼Atg5-Atg16L1 | 0.02 ± 0.002 | −11.00 ± 0.04 | −1.6 | 0.83 ± 0.002 |
| GST-Atg3(130–192): Atg12∼Atg5-Atg16L1 | 0.02 ± 0.005 | −10.50 ± 0.07 | −0.3 | 0.88 ± 0.003 |
| GST-Atg3(140–170) : Atg12∼Atg5-Atg16L1 | 0.02 ± 0.004 | −11.10 ± 0.09 | −1.9 | 0.84 ± 0.004 |
| GST-Atg3(150–180) : Atg12∼Atg5-Atg16L1 | 0.16 ± 0.02 | −12.30 ± 0.10 | −10.2 | 0.86 ± 0.01 |
| GST-Atg3(160–192) : Atg12∼Atg5-Atg16L1 | NB | NB | NB | NB |
| His-Atg3 : Atg7 | 2.22 ± 0.17 | 2.69 ± 0.03 | 34.9 | 0.62 ± 0.004 |
| Atg3(89–192) : Atg7a,b | ≥72.46 ± 17.01 | −2.65 ± 0.82 | 10.0 | 0.99 ± 0.25 |
| Atg3(152–181) : Atg7a,b | ≥101.32 ± 15.30 | −2.26 ± 0.42 | 10.7 | 0.99 ± 0.15 |
Thermodynamic parameters for Atg3, Atg3 mutants or Atg3 peptides binding to Atg7 or Atg12∼Atg5-Atg16L1 by ITC. NB = no quantifiable binding.
Concentration was measured by weighing.
The concentrations of the proteins used for ITC were higher than the others to observe the weak binding.
Importantly, the ITC data explain the competitive binding results in Figure 3(C): Atg3 binds Atg12∼Atg5-Atg16L1 with substantially higher affinity than Atg7. Interestingly, Atg7 is a dimer and titrations with full-length Atg3 give rise to an N-value that is potentially consistent with one Atg3 binding per Atg7 homodimer, although 1:1 binding is observed for the Atg3FR fragments. Although this differs from the stoichiometric binding measured for yeast Atg7 and Atg315–18 (Supporting Information Fig. 3), Atg7 is a multidomain homodimer, and it is conceivable that additional factors, such as LC3 in the Atg7 active site, may be required for both protomers within human Atg7 to achieve a conformation that is competent for Atg3 binding. Future studies will be required to understand factors modulating this interaction in detail.
Taken together with previous studies,15–19 our data demonstrate that the autophagy E2 Atg3 acts as a shuttle between E1, from which it receives the UBL to form a transient covalent E2∼UBL intermediate, and E3, which promotes delivery of the UBL to a lipid acceptor in autophagosomal membranes. In the case of canonical UBL cascades, such mutually exclusive binding of canonical E2s to their E1s and E3s is thought to control the nature of the reaction. For example, an E3-target complex with a relatively fast dissociation rate may be modified by a single canonical UBL molecule, whereas relatively long-lived E3-substrate complexes may enable processive modification such as polyubiquitination.20,21,25–28 In autophagy, it is thought that an individual lipid molecule such as phosphatidylethanolamine is modified with only a single molecule of LC3, although it is possible that processive modification of a segment of membrane could occur depending on the relative timing of Atg12∼Atg5-Atg16 anchoring to autophagosomes compared with Atg3 shuttling to and from Atg7. Interestingly, free Atg3 binds with a submicromolar affinity to its E3, which is the hallmark of one canonical E2 mediating processive polyubiquitination.27 Atg3's high affinity for its E3 enzyme also raises the question of how flux through the LC3 enzyme cascade is established. Notably, canonical UBL pathways may be driven forward by E1∼UBL intermediates displaying a relatively high affinity for free E2s, and E3s preferentially binding E2∼UBL intermediates and typically displaying weak affinities for free E2s.9,10,11,29–31 Thus, it seems likely that future studies will reveal that the substrates—LC3 and its target lipids—influence properties, interactions, dynamics, and organization of their conjugation enzymes to promote UBL modification in autophagy. Nonetheless, our data demonstrate that a segment of the Atg3FR provides a means of simultaneously regulating both E1–E2 and E2–E3 interactions, and raise the possibility that the inhibition of LC3 observed with short peptides could be recapitulated with small molecules.
Materials and Methods
Affinity capture assay
For affinity pulldowns, Atg3 deletion mutants were incubated with GST-Atg12∼Atg5-hisMBP-Atg16L1 or GST-Atg7 bound to 50 μL of glutathione-Sepharose beads (GE life sciences) in binding buffer (25 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM DTT) for 15 min at room temperature. The beads were subsequently washed two times with 500 µL binding buffer. Bound proteins were eluted with 2× SDS sample buffer, resolved by SDS-PAGE and visualized by Commassie staining.
Nondenaturing gel mobility shift assay
Nondenaturing gel mobility shift assays were performed with 20 μM Atg7, 20 μM Atg3, and 4–20 μM Atg12∼Atg5-Atg16L1 in 50 μL volumes in 20 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 5 mM DTT, for 15 min at room temperature. Free and Atg3-bound Atg7 and Atg12∼Atg5-Atg16L1 were separated on a 4.5% polyacrylamide gel (acrylamide:bis, 37.5:1) in a buffer of 90 mM Tris borate, 2% glycerol, pH 8.0, and were visualized with Coomassie staining.
Isothermal titration calorimetry (ITC)
ITC was performed with proteins or peptides in 50 mM HEPES (pH 7.5), 150 mM NaCl, and 1 mM β-mercaptoethanol (BME) at 25°C using a MicroCal ITC200 (GE life sciences). Atg7 and Atg12∼Atg5-Atg16 were placed in the sample cell at concentrations of ∼51–78 µM and 20–51 µM, respectively, and Atg3, Atg3ΔFR, GST-Atg3FRpep, or Atg3FRpep were titrated from the syringe. Data were evaluated using Origin (V 7.0) (OriginLab, Northampton, MA) to determine the values of the thermodynamic parameters. For Atg3(152–181):Atg12∼Atg5-Atg16L1, Atg3(89–192):Atg7, and Atg3(152–181):Atg7 experiments, 1.8, 1.18, and 1.8 mM titrant were used, respectively, in order to measure the weak binding.
In vitro LC3 lipidation assay
Lipidation assays were performed essentially as outlined32,33; E. coli polar extract lipids (Avanti Polar Lipids, Alabaster, AL) dissolved in chloroform was used to prepare the liposomes. After drying under nitrogen to remove the chloroform, lipids were hydrated in 20 mM HEPES 7.5 for 60 min with periodic vortexing, and then extruded (11 times) with a microextruder through a 400 nm polycarbonate filter (Avanti Polar Lipids, Alabaster, AL). Liposomes composed of 67% PE, 23.2% PG, 9.8% CA were added to a final concentration of 1 mM in reactions with 900 nM Atg7, 5 µM Atg3, 20 µM LC3B, 0 or 2.5 µM GST-Atg12(52–140)∼Atg5, 50 µM GST or GST-Atg3FR peptides, 50 mM HEPES 7.5, 200 mM NaCl, 10 mM ATP, 10 mM MgCl2. At the indicated times, reactions were quenched by adding an equal volume of SDS loading buffer containing 100 mM DTT, and analyzed on 15% PAGE gels containing 6M urea.
Acknowledgments
B.A.S. is an Investigator of the Howard Hughes Medical Institute. The authors are grateful to A. Taherbhoy for help in early stages of this project. They thank D. Scott, H. Kamadurai, J. Frye, D. King, and D.W. Miller for advice and/or assistance.
Glossary
- BME
β-mercaptoethanol
- DTT
dithiothreitol
- FR
flexible region
- HMM
hidden Markov model
- ITC
isothermal titration calorimetry
- LC3
microtubule-associated protein 1A/1B-light chain 3
- UBL
ubiquitin-like protein
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. Protein modifications: beyond the usual suspects review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 4.Devenish RJ, Klionsky DJ. Autophagy: mechanism and physiological relevance 'brewed' from yeast studies. Front Biosci (Schol Ed) 2012;4:1354–1363. doi: 10.2741/s337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 6.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 7.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 8.Hanzelmann P, Schafer A, Voller D, Schindelin H. Structural insights into functional modes of proteins involved in ubiquitin family pathways. Methods Mol Biol. 2012;832:547–576. doi: 10.1007/978-1-61779-474-2_39. [DOI] [PubMed] [Google Scholar]
- 9.Streich FC, Jr, Haas AL. Activation of ubiquitin and ubiquitin-like proteins. Subcell Biochem. 2010;54:1–16. doi: 10.1007/978-1-4419-6676-6_1. [DOI] [PubMed] [Google Scholar]
- 10.Capili AD, Lima CD. Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J Mol Biol. 2007;369:608–618. doi: 10.1016/j.jmb.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 13.Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20:59–66. doi: 10.1038/nsmb.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noda NN, Fujioka Y, Hanada T, Ohsumi Y, Inagaki F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013;14:206–211. doi: 10.1038/embor.2012.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taherbhoy AM, Tait SW, Kaiser SE, Williams AH, Deng A, Nourse A, Hammel M, Kurinov I, Rock CO, Green DR, Schulman BA. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell. 2011;44:451–461. doi: 10.1016/j.molcel.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noda NN, Satoo K, Fujioka Y, Kumeta H, Ogura K, Nakatogawa H, Ohsumi Y, Inagaki F. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell. 2011;44:462–475. doi: 10.1016/j.molcel.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 17.Hong SB, Kim BW, Lee KE, Kim SW, Jeon H, Kim J, Song HK. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol. 2011;18:1323–1330. doi: 10.1038/nsmb.2165. [DOI] [PubMed] [Google Scholar]
- 18.Kaiser SE, Mao K, Taherbhoy AM, Yu S, Olszewski JL, Duda DM, Kurinov I, Deng A, Fenn TD, Klionsky DJ, Schulman BA. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nat Struct Mol Biol. 2012;19:1242–1249. doi: 10.1038/nsmb.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaguchi M, Matoba K, Sawada R, Fujioka Y, Nakatogawa H, Yamamoto H, Kobashigawa Y, Hoshida H, Akada R, Ohsumi Y, Noda NN, Inagaki F. Noncanonical recognition and UBL loading of distinct E2s by autophagy-essential Atg7. Nat Struct Mol Biol. 2012;19:1250–1256. doi: 10.1038/nsmb.2451. [DOI] [PubMed] [Google Scholar]
- 20.Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8's E1. Mol Cell. 2005;17:341–350. doi: 10.1016/j.molcel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 21.Eletr ZM, Huang DT, Duda DM, Schulman BA, Kuhlman B. E2 conjugating enzymes must disengage from their E1 enzymes before E3-dependent ubiquitin and ubiquitin-like transfer. Nat Struct Mol Biol. 2005;12:933–934. doi: 10.1038/nsmb984. [DOI] [PubMed] [Google Scholar]
- 22.Yamada Y, Suzuki NN, Hanada T, Ichimura Y, Kumeta H, Fujioka Y, Ohsumi Y, Inagaki F. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem. 2007;282:8036–8043. doi: 10.1074/jbc.M611473200. [DOI] [PubMed] [Google Scholar]
- 23.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 25.Kurz T, Chou YC, Willems AR, Meyer-Schaller N, Hecht ML, Tyers M, Peter M, Sicheri F. Dcn1 functions as a scaffold-type E3 ligase for cullin neddylation. Mol Cell. 2008;29:23–35. doi: 10.1016/j.molcel.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Scott DC, Monda JK, Bennett EJ, Harper JW, Schulman BA. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science. 2011;334:674–678. doi: 10.1126/science.1209307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleiger G, Saha A, Lewis S, Kuhlman B, Deshaies RJ. Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell. 2009;139:957–968. doi: 10.1016/j.cell.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim AY, Bommelje CC, Lee BE, Yonekawa Y, Choi L, Morris LG, Huang G, Kaufman A, Ryan RJ, Hao B, Ramanathan Y, Singh B. SCCRO (DCUN1D1) is an essential component of the E3 complex for neddylation. J Biol Chem. 2008;283:33211–33220. doi: 10.1074/jbc.M804440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siepmann TJ, Bohnsack RN, Tokgoz Z, Baboshina OV, Haas AL. Protein interactions within the N-end rule ubiquitin ligation pathway. J Biol Chem. 2003;278:9448–9457. doi: 10.1074/jbc.M211240200. [DOI] [PubMed] [Google Scholar]
- 30.Kamadurai HB, Souphron J, Scott DC, Duda DM, Miller DJ, Stringer D, Piper RC, Schulman BA. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol Cell. 2009;36:1095–1102. doi: 10.1016/j.molcel.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spratt DE, Wu K, Kovacev J, Pan ZQ, Shaw GS. Selective recruitment of an E2∼ubiquitin complex by an E3 ubiquitin ligase. J Biol Chem. 2012;287:17374–17385. doi: 10.1074/jbc.M112.353748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichimura Y, Imamura Y, Emoto K, Umeda M, Noda T, Ohsumi Y. In vivo and in vitro reconstitution of Atg8 conjugation essential for autophagy. J Biol Chem. 2004;279:40584–40592. doi: 10.1074/jbc.M405860200. [DOI] [PubMed] [Google Scholar]
- 33.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.