

Abstract
Im7 folds via an on-pathway intermediate that contains three of the four native α-helices. The missing helix, helix III, is the shortest and its failure to be formed until late in the pathway is related to frustration in the structure. Im7H3M3, a 94-residue variant of the 87-residue Im7 in which helix III is the longest of the four native helices, also folds via an intermediate. To investigate the structural basis for this we calculated the frustration in the structure of Im7H3M3 and used NMR to investigate its dynamics. We found that the native state of Im7H3M3 is highly frustrated and in equilibrium with an intermediate state that lacks helix III, similar to Im7. Model-free analysis identified residues with chemical exchange contributions to their relaxation that aligned with the residues predicted to have highly frustrated interactions, also like Im7. Finally, we determined properties of urea-denatured Im7H3M3 and identified four clusters of interacting residues that corresponded to the α-helices of the native protein. In Im7 the cluster sizes were related to the lengths of the α-helices with cluster III being the smallest but in Im7H3M3 cluster III was also the smallest, despite this region forming the longest helix in the native state. These results suggest that the conformational properties of the urea-denatured states promote formation of a three-helix intermediate in which the residues that form helix III remain non-helical. Thus it appears that features of the native structure are formed early in folding linked to collapse of the unfolded state.
Keywords: protein folding, frustration, Im7, NMR
INTRODUCTION
The influence of α-helical propensity on how proteins fold has been well explored.1–5 In some cases it appears that the unfolded state of the protein contains nascent helical structure that favours folding by the diffusion-collision mechanism,6 in which marginally stable elements of secondary structure dock together aiding their stabilization and promoting formation of the native state. Other helical proteins fold via a hydrophobic collapse mechanism7 in which collapse of the chain precedes helix formation,8 thus again linking the propensity of an amino acid sequence to form secondary structure with its mechanism of folding. Daggett and Fersht9 view the hydrophobic collapse and diffusion-collision models as extremes of the nucleation-condensation mechanism in which secondary and tertiary structure form concomitantly and note that the position a protein occupies along the continuum is determined by the conformational preferences of the residues in the amino acid sequence.
The groundwork that led to current understanding of protein folding mechanisms has involved kinetic, thermodynamic and computational studies of many small proteins. Amongst these the colicin immunity proteins,10,11 which are inhibitors of DNase bacteriocins and provide immunity to the producing cells,12 have played an important part. The family of immunity proteins are highly homologous, with Im7 and Im9 having 57% sequence identity and sharing a common distorted four α-helical structure.13,14 Despite their high structural similarity, Im9 folds via a two-state mechanism from its urea-denatured state,10 while Im7 folds in a three-state manner [Fig. 1(A)] via an on-pathway kinetic intermediate.15 Φ-analysis,16 NMR spectroscopy,17–19 and MD simulations11,20 have revealed this intermediate to be a compact structure that contains helices I, II, and IV of the native state, arranged in a manner that allows both native and non-native inter-helical contacts. Building on these observations, Sutto et al.21 showed that Im7 has a native structure that is not minimally frustrated and hence has an energy landscape that is rough22,23 and gives rise to a low-lying excited state close to the native state which is populated during folding and is manifested as the intermediate. Even in the absence of chaotropes this excited state is populated at equilibrium which has allowed it to be probed by equilibrium NH exchange17 and relaxation-dispersion NMR.19
Figure 1.
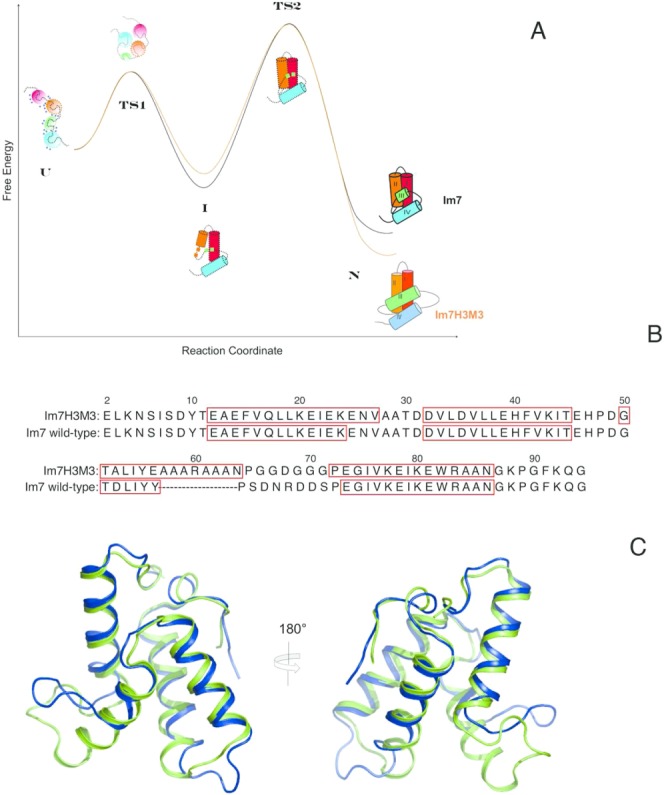
(A) Schematic comparison of the energetics of folding of Im7 and Im7H3M3 based on the thermodynamic and kinetic parameters reported by Knowling et al.24 for Im7H3M3. Except for the cartoon of the native structure of folded Im7H3M3 all other structure cartoons represent Im7 along the folding path. The first is the urea-unfolded state of Im7 which is extended, contains four independent clusters of residues with each cluster containing interacting hydrophobic side chains and has urea molecules (blue circles) associated with it. In the absence of urea the unfolded state (U) rapidly collapses towards the native state (N), traversing transition state 1 (TS1), which lies between the extended (U) and intermediate (I) states, and transition state 2 (TS2) which lies between I and N. (TS1) is almost devoid of secondary structure and lacks a stable hydrophobic core whilst TS2 is a three-helix bundle. The four helices of the native structures are coloured differently, with the important helix III coloured green. Even though helix III has been elongated in Im7H3M3 this protein folds via an on-pathway intermediate24 similarly to wild-type Im7 demonstrating that the formation of an intermediate is an integral feature of the folding mechanism of Im7 that does not result from the short length and low helical propensity of the native helix III. Overall, the elongation of helix III of Im7 to create Im7H3M3 has marginally destabilized the intermediate state by less than 2.5 kJ mol−1 and stabilized the native state by the same amount.24 (B) The Im7 and Im7H3M3 amino acid sequences. The redesigned helix III in Im7H3M3 is longer than its counterpart in Im7. Residues in boxes correspond to α-helices in the native structures. (C) Overlay of the most representative conformer (closest to the average model judged by global RMSDs) from the NMR solution structure ensemble of Im7H3M3 (green)24 and the X-ray crystal structure of Im7 (blue).14 The NMR ensemble of 30 conformers of Im7H3M3 was superimposed onto the crystal structure of Im7 using the SuperPose web server25 global RMSDs over all residues 1.3 Å, heavy atoms 1.1 Å, and backbone atoms 0.8 Å, respectively. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
A significant feature of the Im7 folding pathway is that the final step in folding is the formation of helix III of the native state [Fig. 1(A)]. This is the smallest of the four helices of Im7 and comprises only six residues in the native structure compared with the 13 or 14 residues of the other helices I, II, and IV. Helix III exhibits the lowest helical propensity of all the helices and this led to considerations of whether it is the last helical element to form because it has the lowest helix propensity, or because there are specific features of the amino acid sequence that promote formation of the three helix intermediate. To explore this question Knowling et al.24 engineered Im7 to create a variant in which helix III was lengthened via insertion of a polyalanine helix, designed to extend into the original residues of helix III and be stabilized via an internal solvent exposed salt bridge. Upon redesign helix III had the highest predicted helical propensity. Knowling et al.24 showed that the resulting variant, Im7H3M3 [Fig. 1(B,C)], had a three-dimensional structure little altered from that of native Im7 in the common areas despite the increased length of helix III, and also that this variant folds via the same on-pathway intermediate as the wild-type protein. Thus, it appears that folding of Im7 via a three-helical intermediate is independent of the helical propensity of helix III. This leads on to the question addressed by this paper: what are the specific features of the amino acid sequence that promote formation of the three-helix intermediate?
NMR studies of both urea-denatured Im726 and a triple-variant of Im7 that is unfolded under nondenaturing conditions27 have revealed that the unfolded states of these proteins contain four noninteracting hydrophobic clusters that align with the helices of the native state. To explore whether urea-denatured Im7H3M3 has similar clusters and to determine whether there is frustration in the structure of Im7H3M3 similar to that of native Im7, we here report NMR studies of the folded and urea-denatured states of Im7H3M3. Our results confirm that the presence of the on-pathway folding intermediate is connected with the presence of frustration in the structure of Im7H3M3, and show that the conformational properties of the denatured protein play a key role in determining the details of the folding landscape and the topology of the native state.
RESULTS AND DISCUSSION
Frustration in the structure of Im7H3M3
The structure of Im7H3M3 was determined by NMR as described in Knowling et al.24 The core region of Im7H3M3 that has the same amino acid sequence as Im7 [Fig. 1(B)], residues 2 to 55 and 72 to 93, has an identical fold to Im7 [Fig. 1(C)], as revealed by backbone RMSDs of 0.8 Å, and, importantly, the long helix III of Im7H3M3 overlays well with the shorter helix III of native Im7, as shown by the backbone RMSDs for the common residues, 50 to 55, of 0.4 Å. Having demonstrated that the structures of Im7 and Im7H3M3 are strikingly similar we carried out an analysis of frustration in the structure of Im7H3M3 using the approach of Sutto et al.21 with the protein Frustratometer Server (http://www.frustratometer.tk/),28 an algorithm that spatially localises and quantifies the energetics of frustration present in a protein structure. Energy landscape theory states that sites of minimal frustration are associated with stable folding cores of proteins and that these minimal frustrations result when inter-residue interactions in a polypeptide chain are not in conflict with each other and cooperatively lead to a low-energy conformation.23 In such cases the protein's statistical energy landscape may have a roughness reflecting the occurrence of favourable non-native interactions during the folding process but the consequences of this are not likely to be significant to the folding pathway. However, where the landscape is more rugged due to considerable roughness a relatively long-lived non-native state may arise that acts as a kinetic folding intermediate, as with Im7. Since analysis of frustration in protein structures highlights apparently unfavourable inter-atomic contacts, which might also come about through errors in the structure coordinates, in what follows it is pertinent to note that the structures of Im7 and Im7H3M3 were determined entirely independently, the former by X-ray crystallography14 and the latter by NMR spectroscopy.24
As Sutto et al. reported for Im721 we found that frustration is considerable and not randomly scattered across the Im7H3M3 primary sequence [Fig. 2(A)]. However, residues with minimal frustration are found within all four of the α-helices, generally located in inter-helix contacts so that the core of Im7H3M3 itself is largely minimally frustrated [Fig. 2(B)]. Importantly, residues 51 to 55 of Im7H3M3, which are equivalent to the same residues of Im7 and contribute to helix III in both proteins, show the same degree of frustration. Overall, the engineered helix III has the same number of minimally frustrated sites as helix III of Im7. However, despite the similarity between Im7 and Im7H3M3, the engineered protein has an increased number of highly frustrated interactions, particularly at the C-terminus of helix III and the adjacent loop [red lines in Fig. 2(B)].
Figure 2.
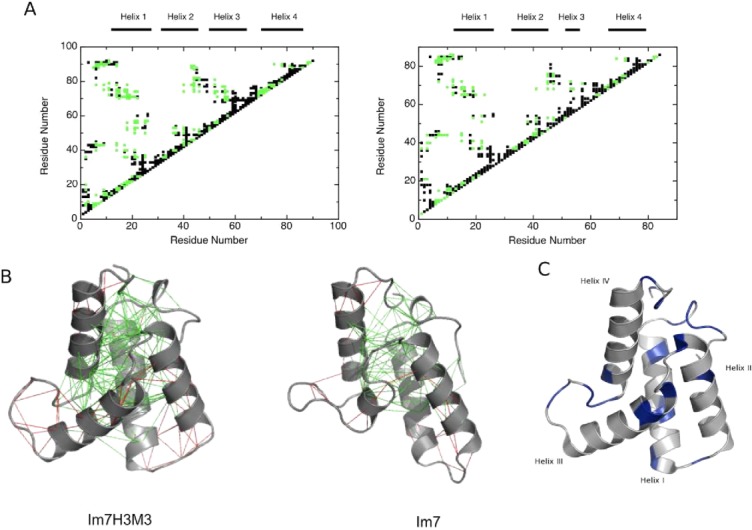
(A) Frustration contact maps of Im7H3M3 (left) and wild-type Im7 (right), with highly frustrated contacts coloured black and minimally frustrated coloured green. The frustration index (Fij)21 gives the energetic fitness for a given set of residues to interact. An interaction is minimally frustrated when Fij > 0.78 and highly frustrated when Fij < −1. (B) Configurational frustration network interactions represented on atomic structures of Im7H3M3 (PDB code: 2K0D) and wild-type Im7 (PDB code: 1AYI). A cluster of minimally frustrated contacts (green) defines the core of the protein, which involves all the α-helices, with highly frustrated contacts shown in red. (C) Backbone fold of Im7H3M3 with the residues exhibiting Rex terms derived from model-free analyses (see below) shown in blue. Residues without Rex terms are coloured grey. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
NMR relaxation studies of Im7 and Im7H3M3
Backbone NH groups of 15N-labeled proteins act as isolated IS spin systems with the relaxation of the 15N nuclei dominated by dipole-dipole interaction with their attached 1H and the chemical shift anisotropy, both of which are modulated by changes in orientation of the NH bond vector with time, making them good probes of protein dynamics.29,30 Detailed relaxation analyses have been reported previously for Im7 and its His-tagged variant, Im7*, leading to the identification of residues undergoing chemical exchange on a time scale that impacts the measured R2 rates.18 Whittaker et al.19 reported a direct correlation between residues involved in this conformational exchange and those that Sutto et al.21 reported as experiencing frustration, and showed that the correlation resulted from exchange between the native state of Im7 and a low-lying excited state that is populated as a consequence of frustration. To explore whether a similar excited state is present for Im7H3M3 we undertook 15N relaxation analyses (Table I). Data for 86 of the 94 residues of Im7H3M3 expected to have a detectable 1H-15N HSQC resonance (i.e. excluding the N-terminal and Pro residues) were obtained (Supporting Information Fig. S1), with residues for which relaxation data are not reported excluded because their resonances were too overlapped with others for accurate determination of signal intensities. Surprisingly, the average relaxation parameters for Im7H3M3 suggest the protein is behaving as if it were smaller than Im7* (Table I) despite its additional seven residues. Consistent with this, the hydrodynamic radii of Im7*18 and Im7H3M3, determined as described in Materials and Methods, 19.3 ± 0.4 Å and 17.8 ± 0.3 Å, respectively, indicates that Im7H3M3 is more compact than Im7*. This must largely be caused by a difference in the properties of the His-tags, with Im7H3M3 having a more restricted His-tag than Im7*.
Table I.
Parameters Describing Global Dynamics of Im7* and Im7H3M3 from 15N Relaxation Analyses at 600 MHz
| Im7* (Whittaker et al.18) | Im7H3M3 (this work) | |
|---|---|---|
| Average 15N R1 (s−1) | 1.71 ± 0.03 | 1.89± 0.04 |
| Average 15N R2 (s−1) | 10.18 ± 0.13 | 8.59 ± 0.08 |
| Average {1H}-15N NOE | 0.68 ± 0.09 | 0.71 ± 0.02 |
| τm (ns) | 5.27 | 5.98 |
| D||/D⊥ | 0.784 | 1.22 |
| Average S2 | 0.87 ± 0.09 | 0.88 ± 0.02 |
The sequence variations of 15N R1, 15N R2, and {1H}-15N NOE values (Supporting Information Fig. S1) are consistent with Im7H3M3 being a well-structured globular protein. With the exception of the residues close to the termini and the inter-helix loop regions, particularly the Gly-rich linker between helices III and IV the profiles are largely featureless. The sequence variation of the R2/R1 ratios for the backbone 15N resonances (Fig. 3) highlights those residues, which have relaxation properties significantly different from the majority of residues. The same plot for Im7 (Fig. 4 of Ref. 19 also identifies the corresponding residues to have heightened R2/R1 ratios, suggestive of similar motions in both proteins.
Figure 3.
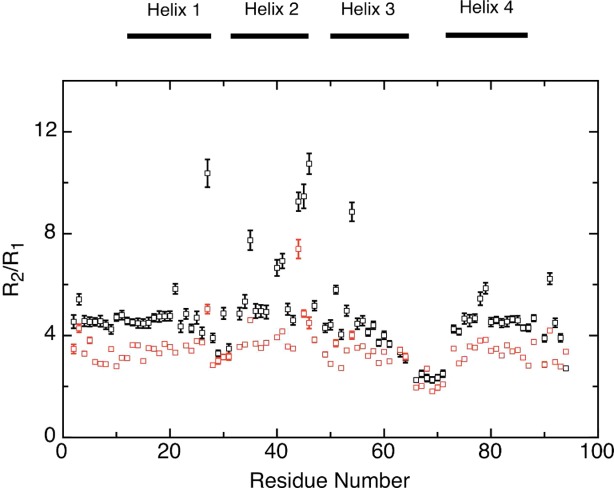
The sequence dependence of the 15N R2/R1 ratios of Im7H3M3 at 25°C, in 50 mM phosphate buffer, pH 7, 10% 2H2O, at a 1H frequency of 600 MHz (black) and 400 MHz (red). Horizontal bars at the top of the figure represent the secondary structure elements. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 4.
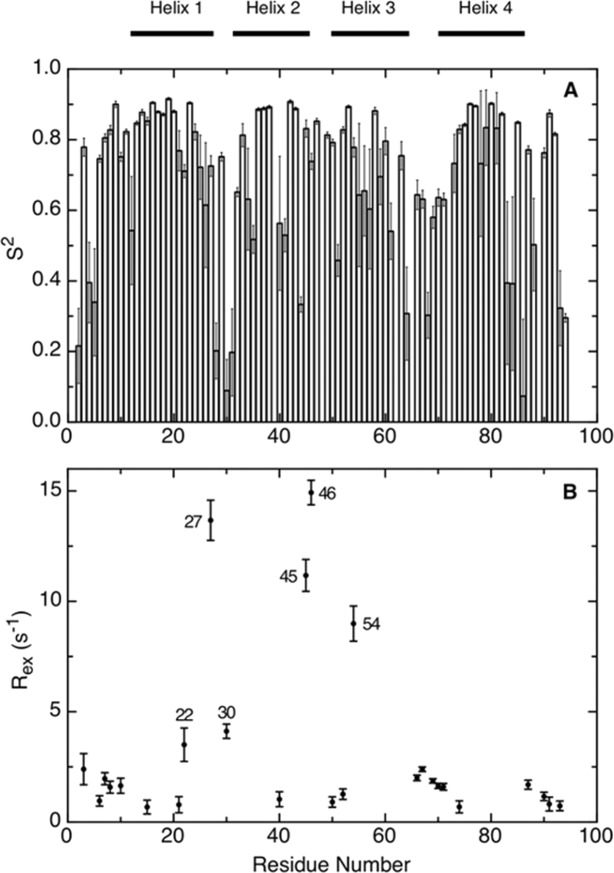
Model-free relaxation analysis of Im7H3M3 at 25°C using data measured at 400 and 600 MHz. The generalized order parameter S2 (A) provides a measure of atomic flexibility of the 1H-15N bond vector on the ps-ns time scale. The chemical exchange terms Rex at 600 MHz (B) describe residues affected by motions occurring on the μs-ms time scale. Residues with Rex values > 3 s−1 are identified. The horizontal bars at the top of the figure indicate secondary structure elements.
In order to proceed with a model-free analysis31–33 of the Im7H3M3 relaxation data the diffusion tensor was determined. This was performed by calculating the relative lengths of the principal axes of the inertia tensor using the program pdbinertia.34 These were 1.00:0.82:0.70, which indicates that the rotational diffusion tensor is either axially symmetric, as it is for Im7, or fully anisotropic. When the 15N R2/R1 ratio is independent of the rapid internal motions and magnitude of the chemical shift anisotropy, it can be used to derive the rotational diffusion tensor. Excluding relaxation parameters for residues Val 27, Asp 35, His 40, Phe 41, Ile 44, Thr 45, Glu 46, and Ile 54, which have 15N R2/R1 ratios indicating significant contributions from internal motions, the data gave an estimate of the correlation time (τc) of 5.98 ns calculated with Modelfree 4.2.29,35 This value is consistent with the 6.06 ns calculated from the structure coordinates with HYDRONMR.36 Since the fully anisotropic model does not provide an improvement relative to the axially symmetric model (Table S1 in Supporting Information), the axially symmetric diffusion tensor model was chosen to best represent the motion of Im7H3M3 in solution, which we assign to be rotation as a prolate ellipsoid. The description of the rotational diffusion tensor of Im7H3M3 as a prolate ellipsoid is consistent with the distribution of R2/R1 ratios according to the method developed by Clore37 (Supporting Information Fig. S2).
The backbone model-free parameters, S2 and Rex (Fig. 4), were determined from the relaxation data (Supporting Information Fig. S1) using the axially symmetric diffusion parameters as described in Materials and Methods; a full listing of the model-free parameters are given in Table S2 in Supporting Information. High values are seen for the average S2 giving a picture of a largely rigid protein, except for the termini and Gly-rich linker between helices III and IV. Of most significance in terms of the concept of frustration, the analysis reveals a considerable number of residues that exhibit sizable Rex terms, which align with the residues predicted to have highly frustrated interactions [Fig. 2(B,C)].
Equilibrium peptide N1H/N2H exchange rates of Im7 and Im7H3M3
Peptide hydrogen exchange experiments are routinely used to investigate conformational dynamics of proteins,38 including determining structural features of transiently populated intermediates39 such as that formed in Im7.17 Provided that hydrogen exchange occurs by an EX2 mechanism, free energies of exchange can be extracted from the observed rates of exchange and it is these free energies, which provide the key structural insights. Experimental exchange rates, kex and free energies of exchange, ΔGHX for Im7M3H3 are summarized in Table II (with the sequence dependence of ΔGHX given in Fig. 5) and compared with similar data obtained previously for Im7.17
Table II.
Amide Hydrogen Exchange Stabilities for Im7H3M3 Measured at 10°C
| Residue | Position in the native structure | kex (s−1) | ΔGHX (kJ mol−1) |
|---|---|---|---|
| Ser 6 | N-terminus | 2.42 × 10−3 | 18.80 |
| Asp 9 | N-terminus | 1.85 × 10−4 | 21.12 |
| Tyr 10 | N-terminus | 3.60 × 10−5 | 22.54 |
| Thr 11 | N-terminus | 3.97 × 10−5 | 24.64 |
| Phe 15 | Helix I | 1.33 × 10−5 | 25.21 |
| Val 16 | Helix I | 8.26 × 10−6 | 24.97 |
| Gln 17 | Helix I | 1.58 × 10−5 | 26.48 |
| Leu 18 | Helix I | 2.25 × 10−5 | 24.01 |
| Leu 19 | Helix I | 7.86 × 10−6 | 24.27 |
| Lys 20 | Helix I | 1.91 × 10−5 | 25.11 |
| Glu 21 | Helix I | 9.47 × 10−5 | 20.59 |
| Ile 22 | Helix I | 4.69 × 10−5 | 19.58 |
| Glu 23 | Helix I | 4.38 × 10−4 | 15.08 |
| Val 33 | Helix II | 3.97 × 10−4 | 14.55 |
| Leu 37 | Helix II | 4.03 × 10−4 | 15.38 |
| Leu 38 | Helix II | 1.22 × 10−4 | 17.81 |
| Phe 41 | Helix II | 2.20 × 10−3 | 15.43 |
| Val 42 | Helix II | 9.02 × 10−4 | 13.92 |
| Lys 43 | Helix II | 2.39 × 10−3 | 14.11 |
| Leu 53 | Helix III | 2.27 × 10−3 | 12.07 |
| Ile 54 | Helix III | 3.43 × 10−4 | 14.57 |
| Tyr 55 | Helix III | 4.84 × 10−4 | 16.14 |
| Glu 56 | Helix III | 4.52 × 10−3 | 11.10 |
| Ile 75 | Helix IV | 5.42 × 10−3 | 10.13 |
| Val 76 | Helix IV | 1.76 × 10−4 | 16.19 |
| Lys 77 | Helix IV | 2.82 × 10−4 | 19.14 |
| Glu 78 | Helix IV | 2.59 × 10−4 | 18.21 |
| Ile 79 | Helix IV | 3.79 × 10−5 | 20.09 |
| Lys 80 | Helix IV | 1.10 × 10−4 | 20.87 |
| Glu 81 | Helix IV | 1.30 × 10−4 | 19.85 |
| Trp 82 | Helix IV | 8.39 × 10−5 | 19.94 |
| Arg 83 | Helix IV | 1.12 × 10−4 | 22.13 |
| Ala 84 | Helix IV | 1.98 × 10−4 | 22.14 |
| Ala 85 | Helix IV | 4.78 × 10−4 | 18.88 |
| Lys 88 | C-terminus | 1.78 × 10−4 | 21.91 |
| Lys 92 | C-terminus | 5.43 × 10−4 | 18.69 |
Figure 5.

Gibbs free energy of exchange (ΔGHX) for residues of wild type Im7 (grey) and Im7H3M3 (blue) measured at 10°C, in 50 mM phosphate buffer, pH 7, and 0.4M Na2SO4. The dashed lines represent the ΔG0UI (14.8 kJ/mol for Im7H3M3 and 12.4 kJ/mol for Im7) and the dotted lines represent the ΔG0UN (23.3 kJ/mol for Im7H3M3 and 25.6 kJ/mol for Im7).24 Secondary structure elements are illustrated at the top of the figure, with the extended helix III on the mutant Im7H3M3 displayed in red. The ΔGHX values for Im7 were taken from Gorski et al.17 [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
The ΔGHX values obtained for Im7H3M3 are remarkably similar to those of Im7. Thus, residues in unstructured regions have NH exchange rates that are too fast for their ΔGHX values to be measured. Residues in helices I and IV have ΔGHX values similar to the corresponding ΔG°UN, indicating that their exchange requires global unfolding, while residues in helix II have ΔGHX values that are similar to ΔG°UI determined previously24 using Φ-value analysis. For helix III ΔGHX < ΔG°UI for both Im7 and Im7M3H3 indicating that helix III is not formed in the intermediate ensemble.
NMR characterization of urea-unfolded Im7H3M3
Having characterized the conformational dynamics of the native state of Im7H3M3 and probed the conformation of the intermediate state that is in equilibrium with the native state, we next turned our attention to urea-denatured Im7H3M3. The limited chemical shift dispersion of the 1H-15N HSQC spectrum of Im7H3M3 denatured in 6M urea (Supporting Information Fig. S3), particularly in the 1H dimension, shows the protein to be unfolded, as expected from the fluorescence and CD studies of Knowling et al.24 Despite the poor dispersion almost complete assignments (88 peptide NH groups; 94% completeness excluding Met 1 and the three Pro residues) were obtained from standard triple resonance experiments (CBCANH, CBCA(CO)NH, HNCO) supplemented by a three-dimensional HNN spectrum.
Since the unfolded states of proteins are highly dynamic, the observed NMR parameters are a population-weighted average over all structures in the conformational ensemble. Nevertheless, deviations of chemical shifts from their expected random coil values, secondary chemical shifts (Δδ = δobs - δrc, where the chemical shift δ is referenced to a random coil shift δrc), are a useful measure of transient secondary structure.40 However, determination of secondary chemical shifts is dependent on an appropriate choice of random coil chemical shifts. We have used the latest random coil values reported in the literature,41,42 which take into account a set of sequence corrections to the random coil values for all nuclei (for pH, temperature, and neighbouring residues), following the approach of Schwarzinger et al.43 The widely used method for identification of protein secondary structure elements uses 13C chemical shift data,40 which reflect the relative population of backbone dihedral angles in the α and β regions of conformational space.44 The secondary chemical shifts incorporating sequence corrections (Fig. 6) suggest that though the protein is largely unfolded in 6M urea there are regions that may be involved in transient secondary structure. This is suggested by the predominantly positive secondary shifts for 13Cα and 13CO, particularly for regions of the protein corresponding to native helices, since positive values are indicative of α-helices.45 Though 13Cβ chemical shifts are less sensitive to the presence of α-helices,46 Δδ13Cα − Δδ13Cβ values are a useful tool to reveal secondary structure propensities. For Im7H3M3 in 6M urea, Δδ13Cα − Δδ13Cβ values [Fig. 6(D)] suggest that residues forming helices I, III, and IV of native Im7H3M3 have a preference for φ/Ψ angles close to those required for α-helices, because they are positive. In contrast, the negative Δδ13Cα − Δδ13Cβ values [Fig. 6(D)] for residues that form helix II in native Im7H3M3 indicate that these residues have a preference for φ/Ψ angles in the β region. As Pashley et al.27 note in their NMR study of a Im7 variant that is unfolded in the absence of chaotrophs, though positive 13Cα chemical shift changes indicate formation of helices the magnitude of the shifts are less than the ∼2.6 ppm change observed in folded proteins45 which means that the tendency for some residues in unfolded proteins, and here we include Im7H3M3, to adopt helical structure is weak. Pashley et al.27 found that residues in their unfolded Im7 variant that form the four helices in native Im7 had helical character, with those from the native helices I and IV having most. Given that their sample was unfolded in the absence of urea and had 0.2M Na2SO4 present our findings for Im7H3M3 in the presence of urea and absence of Na2SO4 are in reasonable agreement.
Figure 6.
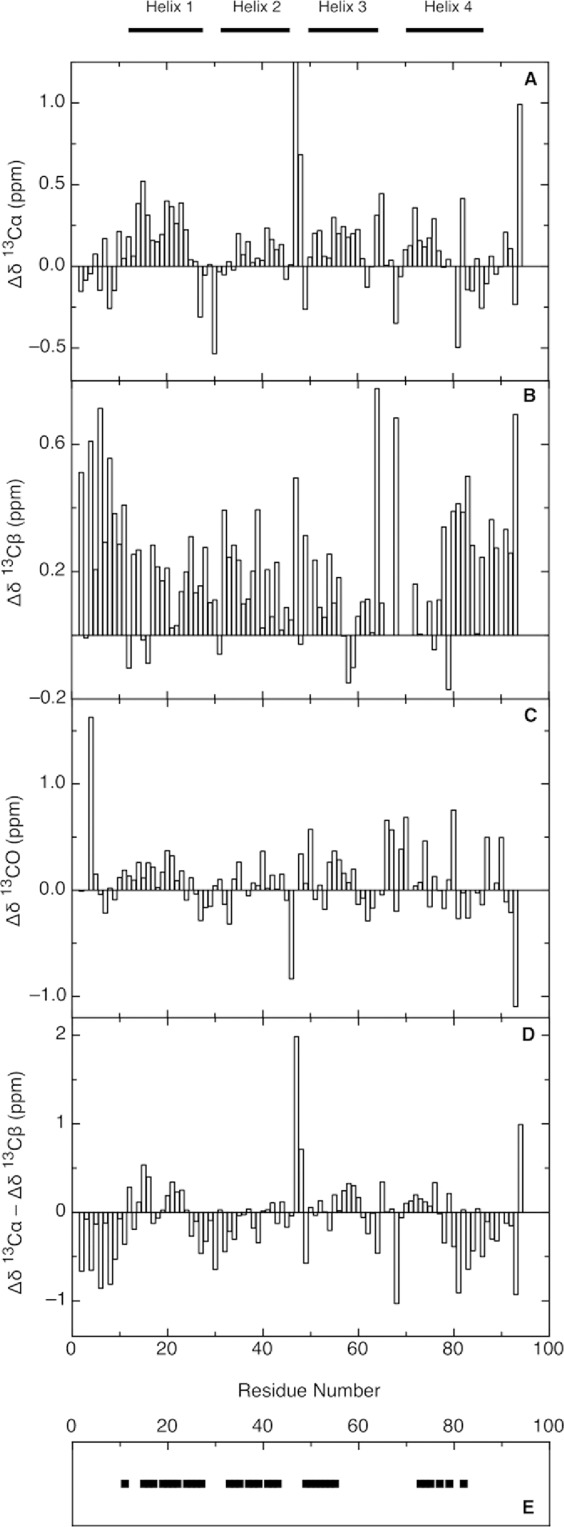
Chemical shift deviations of Im7H3M3 in the urea-unfolded state (6M urea) for 13Cα (A), 13Cβ (B), and 13CO (C), in 50 mM phosphate buffer, pH 7 at 25°C. Intrinsic random coil chemical shifts were used according to reference.41,42 Secondary chemical shift differences Δδ13Cα − Δδ13Cβ (D) provide a valuable probe of secondary structure propensities; positive values indicate α-structure propensity and negative values indicate β-structure propensity. His 47 show a large 13Cα deviation ∼2.5 ppm (A), nevertheless a linear analysis of chemical shifts (LACS) was performed validating all Im7H3M3 chemical shifts. Summary of the assigned cross-peaks in the 1H-1H-15N NOESY-HSQC spectrum of Im7H3M3 (E) showing NOEs between the HN of residue i and the HN of residue i + 1 (black squares). Though some NOE connectivities between NH resonances could be observed in the three-dimensional NOESY-HSQC spectrum, the severe overlap of the amide resonances limited the number of NOE peaks that could be assigned. The horizontal bars on the top of the figure indicate the secondary structure elements present in the native state.
Even with disordered proteins NOEs can provide valuable structural information though they are not readily interpreted quantitatively because of the conformational averaging. However, as Yao et al.47 showed, dNN(i,i + 1) NOEs measured at long mixing times are good indicators of helical content in disordered proteins. For Im7H3M3 in 6M urea dNN(i,i+1) NOEs were observed [Fig. 6(E)] for regions that are helices in the folded structure, correlating well with the regions suggested by the chemical shift analyses to have transient helical content. Long range NOEs indicative of the presence of preferred topologies, were not observed for Im7H3M3 in 6M urea.
Polypeptide chain dynamics of urea-unfolded Im7H3M3
Backbone dynamics of urea-unfolded Im7H3M3 were investigated with 15N R1, 15N R2, and {1H}-15N heteronuclear NOE data recorded at 1H frequencies of 600 and 800 MHz at 10°C. Relaxation parameters were determined for 80 of the 94 backbone amides (Supporting Information Fig. S4) as described in Materials and Methods. For residues 2 to 94, the average R2 values at 600 MHz and 800 MHz are 4.46 (± 0.08) s−1 and 5.03 (± 0.13) s−1, respectively, while the average R1 values at 600 and 800 MHz are 1.76 (± 0.04) s−1 and 1.54 (± 0.04) s−1, respectively. The {1H}-15N NOE values alone indicate considerable flexibility throughout the urea-unfolded Im7H3M3 sequence, below the average value of +0.78 expected for backbone amides of a rigid globular protein tumbling isotropically.48
The slight increase in the R2 values for Im7H3M3 compared with those of urea-unfolded Im7 suggests that a change in the conformational ensemble has occurred. To explore this we determined the hydrodynamic radius (Rh) of Im7H3M3 from NMR diffusion experiments (see Materials and Methods). At 25°C and 10°C, respectively, it was 29.7 ± 0.4 Å and 25.5 ± 0.6 Å compared with the theoretical maximum value of 30.7 Å, calculated as described by Wilkins et al.,49 and the Rh of urea-unfolded Im7* at 10°C, 29.8 ± 1.6 Å.26 Thus, as with the native folded proteins, Im7H3M3 appears to be more compact than Im7* in their urea-unfolded states. Im7H3M3 in 6M urea shows a similar degree of compaction as the Im7 mutant L18A-L19A-L37A unfolded in the absence of urea since the hydrodynamic radius of the latter at 10°C is 26.1 ± 0.6 Å.
Dynamics of a polypeptide chain can be deduced from the backbone NH relaxation parameters R1, R2, and {1H}-15N NOE through the use of the reduced spectral density functions J(0), J(ωN), and J(0.87ωH).50–52 The magnitudes of the spectral density functions are sensitive to motions at the corresponding frequencies, zero, ωN and 0.87ωH. Thus, J(0) reflects slow internal motions on the millisecond to microsecond time scale as well as slow global rotational diffusion while J(0.87ωH) reports on the presence of fast internal motions, on the picosecond timescale.50–52 In the case of urea-unfolded Im7H3M3, J(0) is most informative as it shows that many residues involved in secondary structure elements in the native state have restricted mobility, with J(0) values above the average [Fig. 7(A)]. However, those residues comprising helix III of the native state of Im7H3M3 fall into two distinct groups: the N-terminal segment from residues Gly 50 to Glu 56, whose J(0) values [Fig. 7(A)] indicate restricted motion, and the C-terminal segment from residues Ala 57 to Asn 64 which appears to have largely random fast motions on the picosecond timescale unrestricted by whatever perturbs the motions of the N-terminal segment. Comparing the J(0) values between the urea-unfolded states of Im7H3M3 and wild-type Im7 [compare Fig. 7(A) with Fig. 6 of Ref. 26], both measured at 600 MHz, gives further insight into the motional variations due to the elongation of helix III. While residues forming helices I and II of native Im7 have similar J(0) values for urea-unfolded Im7H3M3 and wild-type Im7, smaller J(0) values were observed for the residues forming the C-terminal region of helix III in native Im7H3M3 and the adjacent loop, indicating that these residues are more flexible than those forming the loop between helices III and IV of native Im7 in urea-unfolded Im7.
Figure 7.
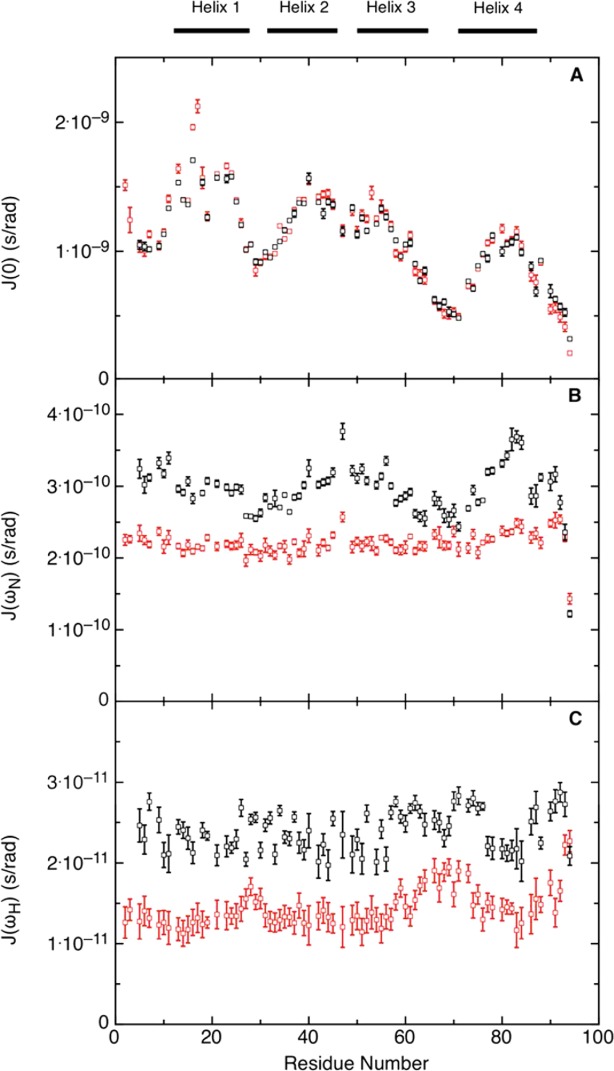
Reduced spectral density mapping of urea-unfolded Im7H3M3. Data recorded at 600 MHz (black) and 800 MHz (red), at pH 7 and 10°C; (A) J(0), (B) J(ωN), and (C) J(ωH) calculated as described in Materials and Methods. The black bars at the top of the figure indicate the location of the α-helices in the native protein. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
As reported for urea-unfolded Im7,26 and shown here to facilitate comparison [Fig. 8(A,C)], the maxima in the sequence profile of J(0) [Fig. 7(A)], indicating motional restrictions on the backbone NH groups, can be accounted for by clusters of side chains coming together to restrict the motions of the polypeptide backbone (Fig. 8). The correlation between the clusters and the average area buried upon folding (AABUF),53 which is proportional to the hydrophobic contribution of a residue to the conformational free energy of a protein, and not the helix propensity as determined by AGADIR54 [Fig. 8(B,D)], confirms that it is the hydrophobicity of the amino acid sequence and not the helix propensity that is the driving force for cluster formation. Nevertheless, as was observed previously with Im7 the clusters are associated with residues forming α-helices in the native structure, which is a consequence of many of the residues that promote cluster formation also promoting helix formation. This is also shown by the correspondence between the location of the α-helices of the native state and hydrophobic clusters identified by HCADraw.55
Figure 8.
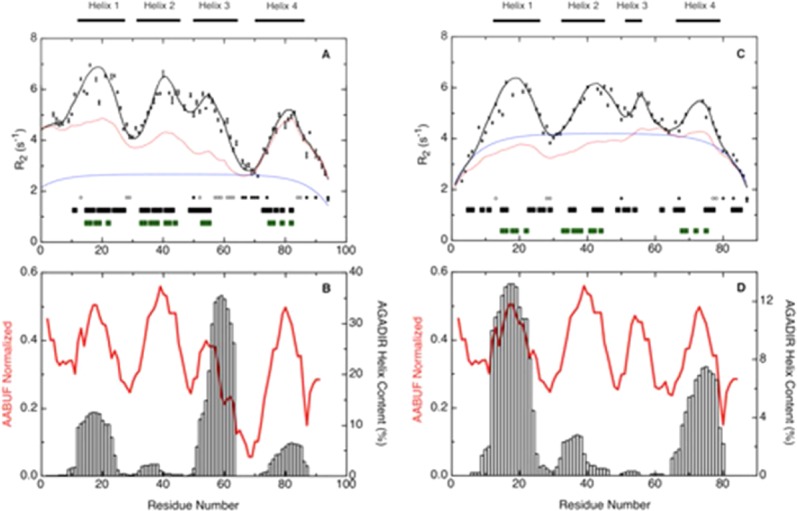
R2 relaxation rates for (A) Im7H3M3 in 6M urea and (C) Im7 in 6M urea, pH 7 and at 10°C, and the average area buried upon folding (AABUF)53 and helix propensity (AGADIR)54 for (B) Im7H3M3 and (D) Im7, plotted as a function of residue number. Measured R2 rates are shown as black squares with error bars; red lines in (A) and (C) shows the R2 values calculated according to the volume-dependent model58 incorporating only the side chain radius of gyration and with persistence length λ0 = 7 except for Gly and Ala for which the value of λ0 was set to 2, and a Gaussian term incorporating four clusters centered at residues Leu 18, Val 42, Gly 56, and Lys 80 (Im7H3M3). The blue lines in (A) and (C) show the R2 values described by the segmental motion model.56,57 Black bars at the bottom of graphs (A) and (C) represent dNN (i, i + 1) NOEs found in the urea-unfolded states; green bars, show hydrophobic clusters predicted by HCA_Draw;55 and the positions of glycine residues (filled circles) and alanine residues (open circles) in the sequence of each protein is indicated. Open vertical bars in (B) and (D), AGADIR54 prediction of helix propensity; the average area buried upon folding (red line) was normalized from 0 to 1 with a window size of seven residues. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Characteristics of the clusters can be obtained from fitting the observed R2 rates to models for polypeptide motion. We have used the segmental motion model56,57 and the volume dependent model,58 as described in Materials and Methods, because there is not clear agreement in the literature on which is most applicable. However, the key features extracted about the clusters (Table III) were the same for both models: clusters I, II, and IV of urea-unfolded Im7H3M3 are the same size as the corresponding clusters of wild-type Im7, which is not surprising since the engineered insert into Im7H3M3 is not in these sequence regions and the clusters are largely non-interacting. Cluster III, the smallest in Im7 is still one of the smallest in Im7H3M3. We return to this observation below.
Table III.
Clusters in Urea-Unfolded Im7H3M3 and Urea-Unfolded Wild-Type Im7 Determined from Fits of 15N R2 Relaxation Data to Models for Polypeptide Motion
| Helical residuesa | Cluster centre | Cluster width | |
|---|---|---|---|
| 12-27 (Helix I) | Leu 18 | 9 | |
| 32-45 (Helix II) | Val 42 | 9 | |
| Im7H3M3 | 50-64 (Helix III) | Glu 56 | 3 |
| 72-86 (Helix IV) | Lys 80 | 6 | |
| 12-24 (Helix I) | Leu 18 | 9 | |
| 32-45 (Helix II) | Val 42 | 9 | |
| Im7b | 51-56 (Helix III) | Tyr 56 | 3 |
| 66-79 (Helix IV) | Lys 73 | 6 |
From the corresponding native structure: Im7H3M3 (2K0D.pdb) and wild-type Im7 (1AYI.pdb), respectively.
From Ref.25.
Implication for the folding mechanism of Im7
In the previous study of Knowling et al.24 the notion that helix III in Im7 is the last to fold because it has the lowest helical propensity was dispelled by engineering the helix to contain an extended poly-Ala sequence in Im7M3H3. Remarkably, the extended helix III contributes to an enhanced network of highly frustrated contacts without hindering formation of the minimally frustrated contacts and without perturbing the folding pathway. The data reported here allow us to see why the extended helix III does not perturb the folding pathway by demonstrating that it is the properties of the unfolded ensemble that favour the folding of Im7 via a three helical intermediate. The observation that the clusters in the urea-denatured state of Im7H3M3 mirror those of urea-denatured Im7 (Table III), as well as Im7 denatured in the absence of chaotrope27 are the critical findings that underpin this conclusion. However, cluster III of Im7H3M3 is the smallest of the four clusters, as it is in Im7 despite this region having the highest helix propensity (Fig. 8). The reason is clear; the high helix propensity has been achieved largely by inserting a polyalanine helix, and because Ala is small it has a low AABUF53 (Fig. 8) and thus does not give rise to a large cluster. Thus, hydrophobic collapse involving the interaction of the largest clusters early in folding creates the three helical intermediate that is common to the folding pathways of both Im7 and Im7H3M3 in which their largest clusters, I, II, and IV, interact. Since such an interaction promotes these clusters adopting their preferred helical conformations the similarity of the collapsed states leads to similar three-helical intermediate states. Furthermore, it is clear that such cluster interactions can be correlated with the network of minimally frustrated contacts observed in the native state. It is noteworthy that the elongation of the sequence with the insert to create Im7H3M3 inevitably means that there is a greater separation in sequence-space between residues in clusters I, II, and IV, however, this greater separation in sequence-space does not materially affect the folding pathway (Fig. 1), consistent with it being the inter-cluster interactions that drive the early stages of folding.
General implication for protein folding
Numerous studies of many small proteins have contributed to the current view that the rates of folding for proteins that do not involve kinetic intermediates are determined by the topology of the native state.59 Following the initial analyses of Baker and his colleagues,60–63 who showed there was a direct correlation between the rate at which such a protein folds and the average sequence separation between contacting residues expressed as an absolute value or relative to the sequence length, which they called the contact order, there have been other analyses confirming that the long-range order of the native state is an important determinant of folding rate.5,64–67 Grantcharova et al.62 discussed some of the implications of the correlation of the folding rate with contact order and pointed out that this correlation implies that the contact order of the native state is correlated with the contact order of the transition state ensemble. The work presented here adds to this view, suggesting that the conformational properties of the folding intermediate of Im7 are determined by the nature of hydrophobic clusters in the denatured state and the network of highly frustrated interactions in the native state. Consistent with this view of the significance of structure in the unfolded state, Nishimura et al.68 and Felitsky et al.69 used NMR measurements to show that transient long-range contacts in unfolded apomyoglobin, some of which are non-native but some native-like, are important for folding, suggesting that the contact order of the native state does indeed start to appear early on the folding pathway. Calculations also support the idea that native-like contacts are formed early in protein folding linked to hydrophobic collapse.70 Overall, therefore, the detailed analyses of Im7H3M3 presented here, combined with previous NMR analyses of wild-type Im7,18,19 urea denatured Im7,26 Im7 denatured in the absence of chaotrope27 and Im919 all point to the collapsed status of the denatured protein playing a role in determining the details of the folding landscape and the topology of the native state. Since sequence determines both collapse in the denatured state and its inherent secondary structure propensity, the correlation of structure in the denatured state, the ruggedness of the folding energy landscape, and the rate of folding to the native state is perhaps not surprising. Finally, our study reveals the importance of considering minimal frustration for protein design, where rational engineering of frustrated regions may provide clues about folded states.
Materials and Methods
Sample preparation
15N labeled and 13C/15N double-labeled samples of Im7H3M3 were produced and purified as described previously.71 For NMR experiments lyophilized samples were resuspended in 50 mM phosphate buffer, pH 7, 10% 2H2O/90% H2O at a concentration of ∼0.5 to 1 mM. For urea-unfolded studies the lyophilized protein was dissolved in 50 mM phosphate buffer, 10% 2H2O/90% H2O containing 6M urea, pH 7.0. The urea concentration was determined using a refractometer, as described by Pace.72
Frustration analysis of Im7 and Im7H3M3
The Im7 crystal structure (1AYI.pdb) and the Im7H3M3 NMR solution structure (2K0D.pdb) were used in the calculation of the residue-based configurational frustration using the web server at http://www.frustratometer.tk/. The algorithm quantifies the degree of frustration manifested in spatially local interatomic interactions.28
NMR spectroscopy
All NMR experiments were performed at 25°C (unless otherwise specified) and acquired with Bruker Avance III 800 MHz, Avance II+ 600 and Avance II+ 400 spectrometers or with Varian Unity Inova spectrometers operating at 500 and 600 MHz proton Larmor frequencies. The Avance II+ 600 MHz spectrometer at Lisbon was additionally equipped with a cryogenic probe. Proton chemical shifts were referenced against external DSS while nitrogen and carbon chemical shifts were referenced indirectly to DSS using absolute frequency ratios. All NMR data were processed using NMRPipe73 or Bruker TopSpin 2.1 software and analyzed with CCPNMR74 or NMRView.75
15N relaxation measurements of native Im7H3M3
15N R1 and R2 relaxation rates and {1H}-15N heteronuclear NOE values for native Im7H3M3 were measured at a 1H frequency of 600 MHz and 400 MHz at 25°C by standard procedures.76,77 The R1 measurements included a recycle delay between scans of 4.0 s and an array of ten different relaxation delays: 0.01 (in duplicate), 0.05, 0.08, 0.2 (in duplicate), 0.5, 0.75, 1, 2 s. The R2 relaxation delays were: 0.01 (in duplicate), 0.03, 0.05 (in duplicate), 0.07, 0.11, 0.15 (in duplicate), 0.25 s. A 3 s saturation delay was applied during d1 in all {1H}-15N steady-state NOE experiments with a total recycle delay of 5 s to allow the longitudinal magnetization to relax back to equilibrium. For both R1 and R2 data, monoexponential two-parameter decay functions were fit to peak intensity versus measured relaxation delay profiles using the CURVEFIT program freely available from Arthur G. Palmer, III.78 Uncertainties in the derived R1 and R2 values were estimated using Monte-Carlo simulations with 1000 random Gaussian noise iterations, taking into account the root mean square noise in the spectra.29 Heteronuclear NOE values were calculated as the ratio of peak volumes in spectra recorded with and without saturation. In the experiment without saturation, a total recycle delay, d1, of 5 s was used in place of the saturation delay to ensure the same recycle delay between scans for both experiments. Errors in the NOE values were calculated from the uncertainties in the peak volume measurements estimated by the root mean square noise in each of the two spectra.
For model-free analysis, an initial estimate of the rotational diffusion tensor was obtained from the R2/R1 ratios of the individual residues and the PDB coordinates of the solution structure of Im7H3M3 (2K0D.pdb) using the programs pdbinertia, r2r1[lowem]diffusion and quadric[lowem]diffusion distributed by Arthur G. Palmer, III.34 After exclusion of residues with hetNOE values lower than 0.65, or with R1 or R2 values exceeding one standard deviation from the mean, according to the criteria proposed by Tjandra,79 15N transverse relaxation data were analyzed by the extended model free approach using FAST-Modelfree80 and automated version of Modelfree 4.2.29 Fitting of the R2/R1 ratios was performed using the combined magnetic field data for different rotational diffusion tensors: isotropic, axial and fully anisotropic with the model selection criteria based on the methods proposed by Palmer,29 including the use of the F-test to judge the statistical significance of invoking any additional parameter. A 15N magnetogyric ratio of −2.71, a CSA of the 15N atom −160 ppm and a NH bond distance of 1.02 Å were used. The following five models were used to describe the spin relaxation data: the first model (model 1) was based simply on fitting the generalized order parameter S2 alone (τf = τs = 0) used to fit the amplitude of internal motions on the picosecond to nanosecond timescale; model 2, incorporated the presence of fast internal motions (τf < 100–200 ps) by fitting both S2 and τe (the effective correlation time for internal motions); models 3 and 4 added an Rex term to the model-free formalism to take into account the loss of transverse magnetization due to chemical and/or conformational exchange for microsecond to millisecond motions and provided fits to S2 and Rex (model 3) and S2, τe, and Rex (model 4), respectively. Finally, the last model (model 5) considered the presence of internal motions slower than τf but faster than the overall rotational correlation time of the protein by fitting S2, τe, and S2f (model 5). Residues were individually fitted to the five dynamic models by hierarchical fitting29 to subsets of the parameters in the extended Lipari-Szabo expression33 for the spectral density. The exchange terms were scaled quadratically with respect to the different magnetic fields. A grid search was used to obtain initial estimates for the values of the remaining model parameters by minimising the χ2 function defined within the program documentation.29,80 Statistical properties of the model-free parameters were obtained from Monte Carlo simulations using 600 randomly distributed synthetic data sets.35 The quality of the fit between the experimental data and theoretical model was assessed for each spin by comparing the optimal value of Γi with the α = 0.05 critical value of the distribution of Γi obtained from the Monte Carlo simulations. Model selection was conducted according to the protocol outlined by Palmer et al.29 and implemented in FAST-Modelfree.80 For comparison purposes with experimental data rotational diffusion tensor was also predicted using HYDRONMR,36 using an atomic element radius of 3.3 Å.
N1H/N2H exchange
15N-labeled samples were used to analyze the decay of amide proton signal intensities due to hydrogen exchange with 2H2O. A 15N sample lyophilized from water was dissolved into 100% 2H2O buffer, 50 mM phosphate buffer, with 0.4M Na2SO4 and containing 0.01% sodium azide, pH* 6.96, (*indicating direct meter reading uncorrected for any isotope effects). Spectra were acquired at a sample temperature of 10°C. To reduce the time required for the sample to reach temperature the buffer solution was pre-equilibrated at 10°C for 40 min before dissolution of the lyophilized protein. The dissolved protein was then immediately placed in the NMR tube and inserted into the NMR spectrometer, previously tuned and shimmed using a sample with the same buffer characteristics. The dead time elapsed between dissolving the sample in 2H2O and recording the first spectrum was approximately 2 min. Consecutive 1H-15N HSQC spectra were recorded on a Varian Unity Inova spectrometer operating at 1H frequency 600 MHz with successive increase in the number of scans, in order to get an acceptable signal/noise ratio maintained along the experiment as peak intensities decreased with amide hydrogen exchange for deuterium. After approximately 6 h the majority of amide protons had exchanged completely. Cross-peak volumes were obtained using NMRPipe73 and normalized over the number of scans of each 1H-15N HSQC spectrum. To calculate the exchange rates, the normalized peak volumes corresponding to each amide peak acquired as a function of the exchange time (defined as the period from the suspension of the lyophilized sample in 2H2O to the successive two-dimensional 1H-15N HSQC spectra) were fitted to the following three-parameter single-exponential decay function using Origin (OriginLab, Northampton, MA).
where C is the baseline noise offset, I0 is the amplitude of the exchange curve at zero time, t is the time in minutes and kex is the exchange rate. Intrinsic exchange rates, kint, were obtained by using the web program SPHERE81 with default activation energies: Eacid = 15 kcal/mol, Ebase = 2.6 kcal/mol. In the transiently open condition, a kinetic competition between exchange and reclosing ensues. If reclosing is faster kcl >> kint, the structural opening reaction appears as a preequilibrium step prior to the rate-limiting chemical exchange, and the observed rate constant (kex) is kex = Kop kint where Kop is the equilibrium constant for structural opening (Kop = kop/kcl) – EX2 mechanism. From the Boltzmann relationship (ΔGHX = -RT ln Kop) one can then calculate the free energy change for the structural opening reaction that exposes the hydrogen to exchange. To guarantee the same exchange mechanism (EX2) as previously reported for wild-type Im717 the same conditions were used (ionic strength, pH, and temperature) to monitor the amide hydrogen exchange for Im7H3M3.
Urea-unfolded Im7H3M3
13C/15N labeled and 15N labeled samples of Im7H3M3 in 6M urea were used for backbone resonance assignment and for relaxation studies, respectively. All NMR measurements were done with freshly prepared samples that were allowed to reach equilibrium before NMR acquisition over a period of 5 h. Standard triple resonance experiments for backbone assignment (CBCANH, CBCA(CO)NH, HNCO, and HNN) were measured at 25°C on Bruker Avance III 800 MHz and Varian INOVA 500 MHz spectrometers, equipped with room temperature triple resonance probes. The spectral widths for the three-dimensional NMR experiments recorded at 500 MHz (CBCA(CO)NH, HNCO, and HNN) were 5629 Hz for 1H, 1320 Hz for 15N, 7535 Hz for 13Cαβ, and 1294 Hz for 13CO; at 800 MHz spectral widths were 11,161 Hz for 1H, 1953 Hz for 15N, and 12,500 Hz for 13Cαβ (CBCANH).
To probe the existence of inter- and/or intra-residue NOEs in urea-unfolded Im7H3M3, a three-dimensional 1H-1H-15N NOESY-HSQC experiment was recorded at 800 MHz with a mixing time of 200 ms. The buffer conditions were the same as used for the backbone assignment but the temperature was lowered to 10°C. To monitor for temperature dependence of the chemical shifts, two-dimensional 1H-15N HSQC spectra were recorded from 10 to 25°C, which allowed us to follow completely the full backbone assignment of the protein.
Residue-specific backbone amide 15N longitudinal (R1) and transverse (R2) relaxation rates and steady-state heteronuclear {1H}-15N NOE were collected on uniformly 15N-enriched Im7H3M3 in 6M urea at two static magnetic fields strengths, 600 and 800 MHz, respectively, and at 10°C using standard procedures described in the literature.76,77 15N R1 data were acquired with the following relaxation delay times: 10 (duplicate, 2×), 50, 80, 200 (2×), 500 (2×), 750, 1000, and 2000 ms. Similarly, 15N R2 values were obtained from a series of 20 experiments recorded in an interleaved manner with (randomly distributed) relaxation delays of 16 (2×), 32 (2×), 48, 64 (2×), 80, 96, 112, 128 (2×), 144, 160 (2×), 192, 208, 240, 320, 400 ms. The interpulse delay for the 180° 15N pulse in the CPMG experiment was 625 μs. The rates were fit with the program CURVEFIT,78 as for the relaxation studies in the native state. Steady-state {1H}-15N NOE values were obtained by recording spectra with and without 1H saturation. {1H}-15N NOE values were calculated as the ratio of peak volumes from spectra recorded with and without saturation. Three repeats of the NOE measurement were performed and the results were averaged together. The errors in the NOE values were calculated from the uncertainties in the peak volume measurements estimated by the root mean square noise in each spectrum.
Following the procedure in Le Duff et al.,26 R2 relaxation rate profiles were fitted to a segmental motion model56,57 and to a segmental motion model incorporating a residue volume dependence.58 The first model predicts a bell-shaped profile distribution for the dynamics of a linear peptide, with increased flexibility at the termini, as shown by the first term of equation:
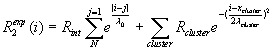 |
It assumes that the influence of the neighbouring residues in a polypeptide chain is independent of side chain volume or hydrophobicity, and decays exponentially as the distance from a given residue increases; Rint is the intrinsic relaxation rate, which depends on temperature and viscosity, λ0 is the persistence length of the polypeptide chain (in terms of number of residues) and N is the total chain length. The second term of the equation accounts for the residue volume dependence. This Gaussian term is characterized by the position of the cluster in the protein (residue number) xcluster, the cluster width λcluster, and a distinct relaxation rate for each cluster, Rcluster. Overall, the first term of the equation characterises the baseline, whereas the second term fits clusters yielding the deviation from the baseline relaxation profile.
The spectral density at zero frequency, J(0), was calculated as described by Lefevre51 using the reduced spectral density. From the relaxation parameters 15N R1, R2, and {1H}-15N NOE, reduced spectral densities were calculated using the jw[lowem]mapping.py python script incorporated in the relax program.82 The spectral density functions were obtained assuming that at higher frequencies J(ωH) ≍ J(ωH + ωN) ≍ J(ωH − ωN) ≍ J(<ωH>)51 and that J(<ωH>) is equivalent to J(0.87ωH) or J(ωH + ωN), where ωH + ωN < ωH since the Larmor frequencies of proton and nitrogen have opposite sign. Thus, J(0) is represented as follows:
where,
The constants c2 and d2 are approximately equal to 1.25 × 109 (rad/s)2 and 1.35 × 109 (rad/s)2, respectively, at 14.1 T (ωH = 600 MHz), and 2.25 × 109 (rad/s)2 and 1.35 × 109 (rad/s)2 at 18.8 T (ωH = 800 MHz). 15N chemical shift anisotropy was considered to be −160 ppm and the NH bond length 1.02 Å. Uncertainties in the spectral density values were estimated from 500 Monte Carlo simulations using the relax program.82
NMR diffusion experiments
Pulsed-field gradient diffusion NMR experiments were carried out with lyophilized Im7H3M3 dissolved in 100% D2O and with 20 μL 1,4-dioxane added as internal molecular radius standard. The PG-SLED pulse sequence49 was used to collect pulsed-field-gradient diffusion experiments on a Bruker Avance III 800MHz at 10°C, respectively. Fifteen gradient experiments were acquired for each data set, with the gradient strengths augmented linearly through the acquisition from 0 to 30 G/cm and all other delays and pulses held constant. Gradient pulses (δ) were applied for 6.3 ms with a recovery time of 0.7 ms, and diffusion delay (Δ) of 100 ms. This was found to be adequate to give a total decay of more than 90%. Thirty-two transients were acquired per gradient experiment. Data were analyzed using the variable gradient fitting routines in Bruker TopSpin 2.1 software and in all cases protein resonances were fit with a single exponential decay function using peak intensities. Theoretical hydrodynamic radii (Rh) values were calculated from the empirical equation for folded and denatured proteins.49 Experimental Rh values for Im7H3M3 were determined as follows: (Dref/Dprotein) × Rh(ref), where Dref and Dprotein are the measured diffusion coefficients of dioxane and the protein, respectively, and Rh(ref) is the effective hydrodynamic radius of dioxane, taken to be 2.12 Å.49
Hydrophobic analysis
The per-residue average area buried upon folding (AABUF) was calculated using the method described by Rose et al.53 using the ExPaSy tool ProtScale (http://us.expasy.org/tools/protscale.html), with a window size of seven residues and normalized from 0 to 1. Hydrophobic cluster analysis was performed using the program HCADraw55 on the ExPaSy tool web server (http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py?form=HCA).
Acknowledgments
The authors thank Nick Cull and Colin Macdonald for technical assistance.
Glossary
- AABUF
average area buried upon folding
- DSS
2,2-(dimethylsilyl)propanesulfonic acid
- fid
free induction decay
- HSQC
heteronuclear single quantum coherence
- Im7
the immunity protein for colicin E7
- Im7*
His-tagged Im7
- Im7H3M3
Im7 variant containing an engineered helix III
- MD
molecular dynamics
- NOE
nuclear Overhauser enhancement
- ppm
parts per million
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Sosnick TR, Jackson S, Wilk RR, Englander SW, DeGrado WF. The role of helix formation in the folding of a fully alpha-helical coiled coil. Proteins. 1996;24:427–432. doi: 10.1002/(SICI)1097-0134(199604)24:4<427::AID-PROT2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 2.Lopez-Hernandez E, Cronet P, Serrano L, Munoz V. Folding kinetics of Che Y mutants with enhanced native alpha-helix propensities. J Mol Biol. 1997;266:610–620. doi: 10.1006/jmbi.1996.0793. [DOI] [PubMed] [Google Scholar]
- 3.Islam SA, Karplus M, Weaver DL. Application of the diffusion-collision model to the folding of three-helix bundle proteins. J Mol Biol. 2002;318:199–215. doi: 10.1016/S0022-2836(02)00029-3. [DOI] [PubMed] [Google Scholar]
- 4.Meisner WK, Sosnick TR. Fast folding of a helical protein initiated by the collision of unstructured chains. Proc Natl Acad Sci USA. 2004;101:13478–13482. doi: 10.1073/pnas.0404057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivankov DN, Finkelstein AV. Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc Natl Acad Sci USA. 2004;101:8942–8944. doi: 10.1073/pnas.0402659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karplus M, Weaver DL. Protein-folding dynamics. Nature. 1976;260:404–406. doi: 10.1038/260404a0. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin RL. How does protein folding get started? Trends Biochem Sci. 1989;14:291–294. doi: 10.1016/0968-0004(89)90067-4. [DOI] [PubMed] [Google Scholar]
- 8.Fernandez A, Kardos JJ, Goto Y, Fernández A. Protein folding: could hydrophobic collapse be coupled with hydrogen-bond formation? FEBS Lett. 2003;536:187–192. doi: 10.1016/s0014-5793(03)00056-5. [DOI] [PubMed] [Google Scholar]
- 9.Daggett V, Fersht AR. Is there a unifying mechanism for protein folding? Trends Biochem Sci. 2003;28:18–25. doi: 10.1016/s0968-0004(02)00012-9. [DOI] [PubMed] [Google Scholar]
- 10.Ferguson N, Capaldi AP, James R, Kleanthous C, Radford SE. Rapid folding with and without populated intermediates in the homologous four-helix proteins Im7 and Im9. J Mol Biol. 1999;286:1597–1608. doi: 10.1006/jmbi.1998.2548. [DOI] [PubMed] [Google Scholar]
- 11.Friel CT, Smith DA, Vendruscolo M, Gsponer J, Radford SE. The mechanism of folding of Im7 reveals competition between functional and kinetic evolutionary constraints. Nat Struct Mol Biol. 2009;16:318–324. doi: 10.1038/nsmb.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.James R, Penfold CN, Moore GR, Kleanthous C. Killing of E. coli cells by E group nuclease colicins. Biochimie. 2002;84:381–389. doi: 10.1016/s0300-9084(02)01450-5. [DOI] [PubMed] [Google Scholar]
- 13.Osborne MJ, Breeze AL, Lian LY, Reilly A, James R, Kleanthous C, Moore GR. Three-dimensional solution structure and 13C nuclear magnetic resonance assignments of the colicin E9 immunity protein Im9. Biochemistry. 1996;35:9505–9512. doi: 10.1021/bi960401k. [DOI] [PubMed] [Google Scholar]
- 14.Dennis CA, Videler H, Pauptit RA, Wallis R, James R, Moore GR, Kleanthous C. A structural comparison of the colicin immunity proteins Im7 and Im9 gives new insights into the molecular determinants of immunity-protein specificity. Biochem J. 1998;333:183–191. doi: 10.1042/bj3330183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capaldi AP, Shastry MC, Kleanthous C, Roder H, Radford SE. Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat Struct Biol. 2001;8:68–72. doi: 10.1038/83074. [DOI] [PubMed] [Google Scholar]
- 16.Capaldi AP, Kleanthous C, Radford SE. Im7 folding mechanism: misfolding on a path to the native state. Nat Struct Biol. 2002;9:209–216. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
- 17.Gorski SA, Le Duff CSCS, Capaldi AP, Kalverda AP, Beddard GS, Moore GR, Radford SE. Equilibrium hydrogen exchange reveals extensive hydrogen bonded secondary structure in the on-pathway intermediate of Im7. J Mol Biol. 2004;337:183–193. doi: 10.1016/j.jmb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 18.Whittaker SB-M, Spence GR, Günter Grossmann J, Radford SE, Moore GR. NMR analysis of the conformational properties of the trapped on-pathway folding intermediate of the bacterial immunity protein Im7. J Mol Biol. 2007;366:1001–1015. doi: 10.1016/j.jmb.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whittaker SB-M, Clayden NJ, Moore GR. NMR characterisation of the relationship between frustration and the excited state of Im7. J Mol Biol. 2011;414:511–529. doi: 10.1016/j.jmb.2011.09.038. [DOI] [PubMed] [Google Scholar]
- 20.Gsponer J, Hopearuoho H, Whittaker SB-M, Spence GR, Moore GR, Paci E, Radford SE, Vendruscolo M. Determination of an ensemble of structures representing the intermediate state of the bacterial immunity protein Im7. Proc Natl Acad Sci USA. 2006;103:99–104. doi: 10.1073/pnas.0508667102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sutto L, Latzer J, Hegler JA, Ferreiro DU, Wolynes PG. Consequences of localized frustration for the folding mechanism of the IM7 protein. Proc Natl Acad Sci USA. 2007;104:19825–19830. doi: 10.1073/pnas.0709922104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueda Y, Taketomi H, Go N. Studies on protein folding, unfolding, and fluctuations by computer simulation. II. A. Three-dimensional lattice model of lysozyme. Biopolymers. 1978;17:1531–1548. [Google Scholar]
- 23.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein-folding - a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 24.Knowling SE, Figueiredo AM, Whittaker SB-M, Moore GR, Radford SE. Amino acid insertion reveals a necessary three-helical intermediate in the folding pathway of the colicin E7 immunity protein Im7. J Mol Biol. 2009;392:1074–1086. doi: 10.1016/j.jmb.2009.07.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maiti R, Van Domselaar GH, Zhang H, Wishart DS. SuperPose: a simple server for sophisticated structural superposition. Nucleic Acids Res. 2004;32:W590–W594. doi: 10.1093/nar/gkh477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Duff CSCS, Whittaker SB-M, Radford SE, Moore GR. Characterisation of the conformational properties of urea-unfolded Im7: implications for the early stages of protein folding. J Mol Biol. 2006;364:824–835. doi: 10.1016/j.jmb.2006.09.037. [DOI] [PubMed] [Google Scholar]
- 27.Pashley CL, Morgan GJ, Kalverda AP, Thompson GS, Kleanthous C, Radford SE. Conformational properties of the unfolded state of Im7 in nondenaturing conditions. J Mol Biol. 2012;416:300–318. doi: 10.1016/j.jmb.2011.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jenik M, Parra RG, Radusky LG, Turjanski A, Wolynes PG, Ferreiro DU. Protein frustratometer: a tool to localize energetic frustration in protein molecules. Nucleic Acids Res. 2012;40:W348–W351. doi: 10.1093/nar/gks447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandel AM, Akke M, Palmer AG. Backbone dynamics of Escherichia-coli ribonuclease HI - correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 30.Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 31.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 32.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 33.Clore GM, Szabo A, Bax A, Kay LE, Driscoll PC, Gronenborn AM. Deviations from the simple two-parameter model-free approach to the interpretation of nitrogen-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 34. Available from: http://www.palmer.hs.columbia.edu/software/diffusion.html. Accessed on August 1 2013.
- 35.Palmer AG, Rance M, Wright PE. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance C-12 heteronuclear NMR-spectroscopy. J Am Chem Soc. 1991;113:4371–4380. [Google Scholar]
- 36.García de la Torre J, Huertas ML, Carrasco B, de la Torre J. HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J Magn Reson. 2000;147:138–146. doi: 10.1006/jmre.2000.2170. [DOI] [PubMed] [Google Scholar]
- 37.Clore GM, Gronenborn AM, Szabo A, Tjandra N. Determining the magnitude of the fully asymmetric diffusion tensor from heteronuclear relaxation data in the absence of structural information. J Am Chem Soc. 1998;120:4889–4890. [Google Scholar]
- 38.Englander SW. Protein folding intermediates and pathways studied by hydrogen exchange. Annu Rev Biophys Biomol Struct. 2000;29:213–238. doi: 10.1146/annurev.biophys.29.1.213. [DOI] [PubMed] [Google Scholar]
- 39.Englander SW, Mayne L, Bai Y, Sosnick TR. Hydrogen exchange: the modern legacy of Linderstrom-Lang. Protein Sci. 1997;6:1101–1109. doi: 10.1002/pro.5560060517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 41.Kjaergaard M, Brander S, Poulsen FM. Random coil chemical shift for intrinsically disordered proteins: effects of temperature and pH. J Biomol NMR. 2011;49:139–149. doi: 10.1007/s10858-011-9472-x. [DOI] [PubMed] [Google Scholar]
- 42.Kjaergaard M, Poulsen FM. Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution. J Biomol NMR. 2011;50:157–165. doi: 10.1007/s10858-011-9508-2. [DOI] [PubMed] [Google Scholar]
- 43.Schwarzinger S, Kroon GJ, Foss TR, Chung J, Wright PE, Dyson HJ. Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc. 2001;123:2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 44.Dyson HJ, Wright PE. Defining solution conformations of small linear peptides. Annu Rev Biophys Biophys Chem. 1991;20:519–538. doi: 10.1146/annurev.bb.20.060191.002511. [DOI] [PubMed] [Google Scholar]
- 45.Spera S, Bax A. Empirical correlation between protein backbone conformation and C.alpha. and C.beta. 13C nuclear magnetic resonance chemical shifts. J Am Chem Soc. 1991;113:5490–5492. [Google Scholar]
- 46.Avbelj F, Kocjan D, Baldwin RL. Protein chemical shifts arising from alpha-helices and beta-sheets depend on solvent exposure. Proc Natl Acad Sci USA. 2004;101:17394–17397. doi: 10.1073/pnas.0407969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yao J, Chung J, Eliezer D, Wright PE, Dyson HJ. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry. 2001;40:3561–3571. doi: 10.1021/bi002776i. [DOI] [PubMed] [Google Scholar]
- 48.Farrow NA, Zhang O, Forman-Kay JD, Kay LE. A heteronuclear correlation experiment for simultaneous determination of 15N longitudinal decay and chemical exchange rates of systems in slow equilibrium. J Biomol NMR. 1994;4:727–734. doi: 10.1007/BF00404280. [DOI] [PubMed] [Google Scholar]
- 49.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 50.Peng JW, Wagner G. Mapping of the spectral densities of N-H bond motions in eglin c using heteronuclear relaxation experiments. Biochemistry. 1992;31:8571–8586. doi: 10.1021/bi00151a027. [DOI] [PubMed] [Google Scholar]
- 51.Lefevre JF, Dayie KT, Peng JW, Wagner G. Internal mobility in the partially folded DNA binding and dimerization domains of GAL4: NMR analysis of the N-H spectral density functions. Biochemistry. 1996;35:2674–2686. doi: 10.1021/bi9526802. [DOI] [PubMed] [Google Scholar]
- 52.Farrow N a, Zhang O, Szabo A, Torchia Da, Kay LE. Spectral density function mapping using 15N relaxation data exclusively. J Biomol NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- 53.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 54.Lacroix E, Viguera AR, Serrano L. Elucidating the folding problem of alpha-helices: local motifs, long-range electrostatics, ionic-strength dependence and prediction of NMR parameters. J Mol Biol. 1998;284:173–191. doi: 10.1006/jmbi.1998.2145. [DOI] [PubMed] [Google Scholar]
- 55.Gaboriaud C, Bissery V, Benchetrit T, Mornon JP. Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett. 1987;224:149–155. doi: 10.1016/0014-5793(87)80439-8. [DOI] [PubMed] [Google Scholar]
- 56.Schwalbe H, Fiebig KM, Buck M, Jones JA, Grimshaw SB, Spencer A, Glaser SJ, Smith LJ, Dobson CM. Structural and dynamical properties of a denatured protein. Heteronuclear 3D NMR experiments and theoretical simulations of lysozyme in 8 M urea. Biochemistry. 1997;36:8977–8991. doi: 10.1021/bi970049q. [DOI] [PubMed] [Google Scholar]
- 57.Klein-Seetharaman J, Oikawa M, Grimshaw SB, Wirmer J, Duchardt E, Ueda T, Imoto T, Smith LJ, Dobson CM, Schwalbe H. Long-range interactions within a nonnative protein. Science. 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]
- 58.Schwarzinger S, Wright PE, Dyson HJ. Molecular hinges in protein folding: the urea-denatured state of apomyoglobin. Biochemistry. 2002;41:12681–12686. doi: 10.1021/bi020381o. [DOI] [PubMed] [Google Scholar]
- 59.Go A, Kim S, Baum J, Hecht MH. Structure and dynamics of de novo proteins from a designed superfamily of 4-helix bundles. Protein Sci. 2008;17:821–832. doi: 10.1110/ps.073377908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 61.Plaxco KW, Simons KT, Ruczinski I, Baker D. Topology, stability, sequence, and length: defining the determinants of two-state protein folding kinetics. Biochemistry. 39:11177–11183. doi: 10.1021/bi000200n. [DOI] [PubMed] [Google Scholar]
- 62.Grantcharova V, Alm EJ, Baker D, Horwich AL. Mechanisms of protein folding. Curr Opin Struct Biol. 2001;11:70–82. doi: 10.1016/s0959-440x(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 63.Ivankov DN, Garbuzynskiy SO, Alm E, Plaxco KW, Baker D, Finkelstein AV. Contact order revisited: influence of protein size on the folding rate. Protein Sci. 2003;12:2057–2062. doi: 10.1110/ps.0302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gromiha MM, Selvaraj S. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: application of long-range order to folding rate prediction. J Mol Biol. 2001;310:27–32. doi: 10.1006/jmbi.2001.4775. [DOI] [PubMed] [Google Scholar]
- 65.Zhou H, Zhou Y. Folding rate prediction using total contact distance. Biophys J. 2002;82:458–463. doi: 10.1016/S0006-3495(02)75410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nölting B, Schälike W, Hampel P, Grundig F, Gantert S, Sips N, Bandlow W, Qi PX. Structural determinants of the rate of protein folding. J Theor Biol. 2003;223:299–307. doi: 10.1016/s0022-5193(03)00091-2. [DOI] [PubMed] [Google Scholar]
- 67.Ouyang Z, Liang J. Predicting protein folding rates from geometric contact and amino acid sequence. Protein Sci. 2008;17:1256–1263. doi: 10.1110/ps.034660.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishimura C, Lietzow MA, Dyson HJ, Wright PE. Sequence determinants of a protein folding pathway. J Mol Biol. 2005;351:383–392. doi: 10.1016/j.jmb.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 69.Felitsky DJ, Lietzow MA, Dyson HJ, Wright PE. Modeling transient collapsed states of an unfolded protein to provide insights into early folding events. Proc Natl Acad Sci USA. 2008;105:6278–6283. doi: 10.1073/pnas.0710641105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camilloni C, Sutto L, Provasi D, Tiana G, Broglia RA. Early events in protein folding: Is there something more than hydrophobic burst? Protein Sci. 2008;17:1424–1433. doi: 10.1110/ps.035105.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gorski SA, Capaldi AP, Kleanthous C, Radford SE. Acidic conditions stabilise intermediates populated during the folding of Im7 and Im9. J Mol Biol. 2001;312:849–863. doi: 10.1006/jmbi.2001.5001. [DOI] [PubMed] [Google Scholar]
- 72.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 73.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPIPE—a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 74.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 75.Johnso BA, Blevins RA. NMR VIEW—a computer-program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 76.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 77.Kay LE, Nicholson LK, Delaglio F, Bax A, Torchia DA. Pulse sequences for removal of the effects of cross-correlation between dipolar and chemical-shift anisotropy relaxation mechanism on the measurement of heteronuclear T1 and T2 values in proteins. J Magn Reson. 1992;97:359–375. [Google Scholar]
- 78. CurveFit. Available from: http://cpmcnet.columbia.edu/dept/gsas/biochem/labs/palmer/software/curvefit.html. Accessed on August 1 2013.
- 79.Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from N-15 NMR relaxation. J Am Chem Soc. 1995;117:12562–12566. [Google Scholar]
- 80.Cole R, Loria JP. FAST-Modelfree: a program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 81.Hydrogen Exchange Prediction. Available from: http://www.fccc.edu/research/labs/roder/sphere/sphere.html. Accessed on August 1 2013.
- 82.d'Auvergne EJ, Gooley PR. Optimisation of NMR dynamic models I. Minimisation algorithms and their performance within the model-free and Brownian rotational diffusion spaces. J Biomol NMR. 2008;40:107–119. doi: 10.1007/s10858-007-9214-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.