

Abstract
Recombinant human lecithin-cholesterol acyltransferase Fc fusion (huLCAT-Fc) is a chimeric protein produced by fusing human Fc to the C-terminus of the human enzyme via a linker sequence. The huLCAT-Fc homodimer contains five N-linked glycosylation sites per monomer. The heterogeneity and site-specific distribution of the various glycans were examined using enzymatic digestion and LC-MS/MS, followed by automatic processing. Almost all of the N-linked glycans in human LCAT are fucosylated and sialylated. The predominant LCAT N-linked glycoforms are biantennary glycans, followed by triantennary sugars, whereas the level of tetraantennary glycans is much lower. Glycans at the Fc N-linked site exclusively contain typical asialobiantennary structures. HuLCAT-Fc was also confirmed to have mucin-type glycans attached at T407 and S409. When LCAT-Fc fusions were constructed using a G-S-G-G-G-G linker, an unexpected +632 Da xylose-based glycosaminoglycan (GAG) tetrasaccharide core of Xyl-Gal-Gal-GlcA was attached to S418. Several minor intermediate species including Xyl, Xyl-Gal, Xyl-Gal-Gal, and a phosphorylated GAG core were also present. The mucin-type O-linked glycans can be effectively released by sialidase and O-glycanase; however, the GAG could only be removed and localized using chemical alkaline β-elimination and targeted LC-MS/MS. E416 (the C-terminus of LCAT) combined with the linker sequence is likely serving as a substrate for peptide O-xylosyltransferase. HuLCAT-Fc shares some homology with the proposed consensus site near the linker sequence, in particular, the residues underlined PPPE416GS418GGGGDK. GAG incorporation can be eliminated through engineering by shifting the linker Ser residue downstream in the linker sequence.
Keywords: recombinant human LCAT-Fc, N- and O-linked glycans, site-specific N-linked glycan profile, xylose-based glycosaminoglycan
Introduction
Lecithin-cholesterol acyltransferase (LCAT; phosphatidylcholine-sterol acyltransferase, EC 2.3.1.43) is a key enzyme that catalyzes the esterification of free cholesterol by transfer of a syn-2 fatty acid from phosphatidylcholine to the 3-hydroxyl group of cholesterol to form a cholesterol ester in blood plasma.1,2 LCAT plays a critical role in regulating the composition and concentration of circulating high-density lipoprotein (HDL).3,4 In addition, LCAT is believed to be an important driving force behind the reverse cholesterol transport (RCT) pathway and therefore has been a subject of great interest in cardiovascular research since its discovery in 1962.3–6 However, after half a century of research, the role of LCAT in atherosclerosis is still debated given that patients with LCAT deficiency do not suffer significantly increased levels of cardiovascular disease.7 Conflicting or negative data on the role of LCAT in atherosclerosis have been reported.8–10 In studying transgenic or knockout mouse models, the effect of LCAT expression on HDL regulation and atherosclerosis has also been inconsistent.11–13 In contrast, studies conducted in rabbits and monkeys, which more closely resemble humans in their lipoprotein metabolism, suggest that increased LCAT functionality is likely beneficial for lipid metabolism and atherosclerosis.14,15 It appears that the influence of LCAT on atherosclerosis mostly depends on its effects on proatherogenic apoB-containing lipoproteins and to a lesser extent, if any, on its effects on HDL levels. Furthermore, the observed effects are also highly dependent on the presence of additional key proteins involved in RCT, such as cholesteryl ester transfer protein and the low-density lipoprotein receptor.3
There has been great interest to develop therapeutic agents capable of enhancing LCAT activity to assess its therapeutic benefits on cardiovascular diseases in humans. In an effort to identify therapeutically relevant strategies to stimulate LCAT activity, recent reports have described a class of small-molecule LCAT activators that modulate LCAT functionality.16–18 An enzyme replacement therapeutic approach, such as infusion of recombinant LCAT for treating dyslipidemia and atherosclerosis, has also gained attention. For example, infusion of recombinant LCAT in mice rapidly restored the normal lipoprotein phenotype in LCAT-KO mice and increased cholesterol efflux19; and administration of engineered recombinant LCAT in rabbits effectively raises HDL, promotes RCT, and attenuates atherosclerosis.20 In a small Phase I clinical trial in humans, ACP-501 (a recombinant human LCAT) has been reported to be safe and to “rapidly and substantially elevate HDL cholesterol” due to an increased formation of cholesterol esters.21
The human LCAT gene encodes a 416-amino acid polypeptide chain22–24 with four complex N-linked glycans attached to N20, N84, N272, and N384, and with two O-linked glycans at T407 and S409.25 Therapeutic proteins or peptides, such as human LCAT, often exhibit poor PK properties, including a short serum half-life, in humans and/or research animals.19,20 To extend the serum half-life and in vivo efficacy for preclinical and clinical studies, chimeric molecules with the Fc moiety fused to a protein or peptide can be engineered and produced.26 The fusion of an antibody Fc domain to a therapeutic protein or peptide to create a dimeric fusion molecule has proven to be highly successful for marketed products including protein-Fc chimeras such as TNFR2-Fc (Etanercept)27 and CTLA4-Fc (Abatacept),28 as well as the thrombopoietin mimetic peptide-Fc peptibody (Nplate).26 A construct consisting of human LCAT fused to Fc via a linker (human lecithin-cholesterol acyltransferase Fc fusion [huLCAT-Fc]) has been engineered and produced in our laboratory.29
Each huLCAT-Fc monomer is expected to contain four N-linked and two O-linked carbohydrates in the LCAT portion of the molecule and an N-linked sugar in the Fc domain. The complex N-linked glycans at selected sites are differentially associated with LCAT conformational stability, lipid binding capability, and catalytic activity.30–35 Similar to other glycoprotein therapeutics, the N-linked oligosaccharides of huLCAT-Fc should be considered as a significant quality attributed for therapeutic use as N-linked glycans have been known to affect in vivo efficacy, as well as the pharmacodynamic and pharmacokinetic profile in animals.36–38 Therefore, characterization and quality assessment of the glycans are important even at the early development stage. Here, we report the preliminary analysis of the glycans at the five N-linked sites of huLCAT-Fc by mass spectrometry. In addition, we found that an unusual O-linked glycosaminoglycan (GAG) tetrasaccharide core incorporated into a linker Ser residue, which previously has not been reported in fusion molecules engineered with glycine-rich, serine-containing linkers. Glycans attached at the linker Ser were confirmed to be a xylose-based GAG tetrasaccharide and other intermediate glycoforms of the GAG biosynthetic pathway.39 Redesign of the linker sequence eliminated the consensus sequence for GAG incorporation and was able to successfully produce huLCAT-Fc free of GAG glycans.
Results
Identification and N-linked glycan analysis of tryptic glycopeptides
Figure 1(A) shows a typical full-scan MS base peak chromatogram of tryptic peptides from digest of a Chinese hamster ovary (CHO)-derived sample (see Supporting Information). Figure 1(B) illustrates the extracted ion chromatograms (XICs) at m/z [203.5–204.5] from the surface-induced dissociation (SID) scan. The SID scan permits fragmentation of common carbohydrate marker ions, including N-acetyl hexosamine (HexNAc) at m/z = 204, whose presence directly correlates with the elution position of the different glycopeptides. Six broad regions exhibit intense carbohydrate marker ions at discrete retention times. Because of glycan heterogeneity, related glycopeptides appear to elute as broad peaks (usually 2–3 min or longer). Following collision-induced dissociation (CID) fragmentation of peptides, the five regions labeled with N20, N84, N272, N384, and N499 were confirmed to contain N-linked glycopeptides. The region labeled with Peptide A (T407/S409/GAG) was confirmed to contain both regular and unusual O-linked glycans, and their characterization will be described in the section “Identification of O-linked glycans and the attachment sites.” Two regions around retention time of 31.5 and 63.7 min also exhibit SID signal at m/z = 204 Da. However, MS/MS analysis confirmed that they are not glycopeptides, but rather tryptic peptides with a G-K sequence at the C-terminus that generates a similar SID signal at m/z = 204 Da (data not shown).
Figure 1.
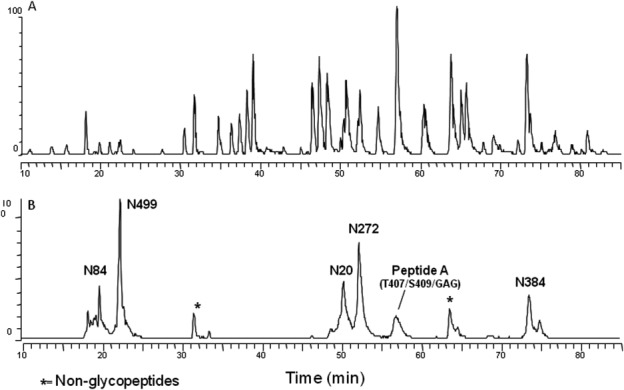
Base peak chromatogram and extracted ion chromatogram for tryptic digest of huLCAT-Fc CHO-A sample. Panel A: Typical full MS base peak chromatogram for all modified and unmodified peptides. Panel B: Extracted ion chromatogram for glycopeptides of huLCAT-Fc detected with diagnostic oxonium ion HexHAc at m/z = 204 Da. Peaks are labeled according to the sites of glycosylation. Peptide A contains multiple O-linked glycosylation indicated in parenthesis.
Supporting Information Figure S2 shows full MS signals averaged from retention time 49.5 to 51.5 min, which corresponds to the elution position of the N20 glycopeptide [see Fig. 1(B)]. Using MassAnalyzer processing and zoom scan data, the detected ions were determined to range from the 3+ to 4+ charge states. Confirmation of a glycan structure in the attached peptide was obtained by MS/MS analysis of the designated glycopeptides; the glycan “types” can then be assigned to the respective glycopeptide ions. Supporting Information Figure S3 shows the CID spectrum of m/z = 1335.4 (4+ charge state), a minor glycopeptide ion that was detected at 50.05 min (see Supporting Information Fig. S2). Significant sequential fragmentation of monosaccharide or oligosaccharide sugar units up to the core structure (composed of two N-acetyl glucosamine and a fucose) was observed. The CID fragmentation of the oligosaccharide is clearly dominant over fragmentation of the peptide bonds, typical of the fragmentation pattern of an N-linked glycopeptide. The result therefore confirmed that the 4+ charge state ion at m/z = 1335.4 and the 3+ charge state ion at m/z = 1780.3 (Supporting Information Fig. S2) belong to the N20 glycopeptide with an average mass of 5337.7 Da. The glycan structure of this glycopeptide is thus assigned to be a fucosylated and disialylated triantennary glycan, A3S2G1F. This 2715-Da glycan, which has a composition of Fuc1Hex6HexNAc5Neu5Ac2, is one of the minor glycans attached at N20 in the A16-K39 tryptic peptide. Using mass data obtained from full scan, zoom scan, and MS/MS fragmentation of glycopeptides, the MassAnalyzer program was able to identify the A16-K39 glycopeptide and to confirm that at least eight different glycans are present at the N20 N-linked site.
Using the above-mentioned strategy, all glycans at the five N-linked sites of huLCAT-Fc were assigned, and the relative distribution amongst the various glycans within the same N-linked site can be determined. The results listed in Supporting Information Table S1 represent the site-specific glycan heterogeneity and distribution of the different glycans. Glycans that were detected below 1.0% level were excluded in the list. For comparison, the glycans were categorized into biantennary, triantennary, and tetraantennary structures. These data indicated that relatively high amount of the glycans at all four N-linked sites in huLCAT are sialylated and fucosylated. All N-linked sites in huLCAT contain predominantly biantennary structure. The disialylated biantennary structure, A2S2F, is the dominant form over the monosialylated form, A2S1G1F, at N20, N84, and N272, except at N384 where the monosialylated biantennary forms are the major species. Sialylated triantennary glycans at all four sites are the second most abundant form. Sialylated tetraantennary glycans represent the least abundant species. Both N20 and N84 contain significant amount of tetraantennary glycans. N272 only contains low level of A4S3G4F, whereas N384 does not have any tetraantennary glycans. N384 has low levels of nonsialylated biantennary glycans such as A2G1F and A2G2F, which were not detected at the other N-linked sites. Glycans detected at N499 of the Fc moiety are all biantennary. Three nonsialylated biantennary glycans, A2G0F, A2G1F, and A2G2F, constitute the major glycoforms. A sialylated biantennary glycan, A2S1G1F, is present but at low levels.
During LC-MS/MS peptide mapping analysis, we tried to identify if any peptides containing consensus N-linked sites were not glycosylated. The results indicated that peptides with no glycan attachment can be identified; however, the MS signal intensity is very weak relative to the total intensity of all respective glycopeptide species. This indicates that all N-linked sites in huLCAT-Fc are almost fully glycosylated.
Intact mass determination of deglycosylated huLCAT-Fc by LC-MS
Because of the heterogeneity and complexity of the glycans described above, intact mass analysis of the parent huLCAT-Fc molecule using LC-MS was not possible. To ascertain the precise mass of the molecule with reduced complexity, huLCAT-Fc was subjected to removal of the terminal sialic acids, followed by the enzymatic removal of N-and O-linked glycans. This was accomplished by digestion of the reduced and alkylated samples with various specific glycosidases, including sialidase, PNGase F, and O-glycanase. Figure 2(A) shows ESI-TOF MS analysis of a sample following sialidase and PNGase F digestion. Three major masses of 74,365.3 Da, 74,998.2 Da, and 75,363.8 Da were observed. Sialidase removes terminal sialic acid present in both the N- and O-linked sugar structures. After sialidase treatment, only the typical mucin O-linked HexNAc-Hex disaccharide remains at each O-linked site. This was confirmed by the 74,365.3-Da mass, which corresponds to deglycosylated huLCAT-Fc with two O-linked glycan cores (GalNAc-Gal of 366 amu). An observed 74,998.2-Da mass was also observed, which is ∼632 Da higher than expected for huLCAT-Fc with two O-linked sugar cores. A lower abundance mass of 75,363.8 Da suggests an additional mass of 366 Da when compared with 74,998.3 Da, indicating the possible presence of a third GalNAc-Gal O-linked core attached to an additional O-linked site (see below for more analysis).
Figure 2.
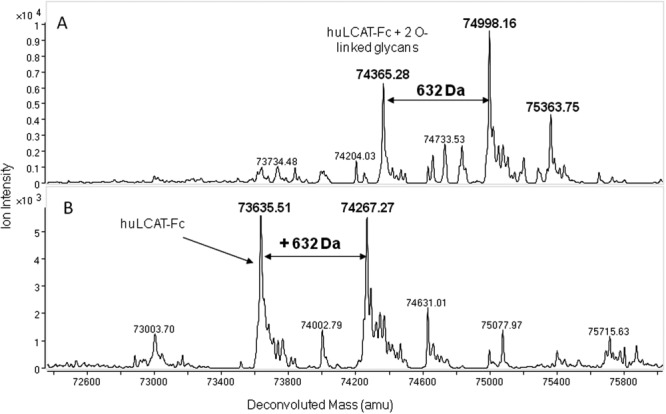
ESI-TOF MS analysis of reduced, alkylated, and deglycosylated huLCAT-Fc. Panel A: Sample treated with sialidase and PNGase F. Panel B: Sample treated with sialidase, PNGase F, and O-glycanase.
For complete removal of the regular N- and O-linked glycans, huLCAT-Fc was digested with sialidase, PNGase F, and O-glycanase to yield deglycosylated huLCAT-Fc. As seen in Figure 2(B), the LC-MS analysis of the deglycosylated molecule identified two major deconvoluted masses: 73,635.5 and 74,267.3 Da. The 73,635.5-Da mass corresponds to fully deglycosylated huLCAT-Fc (theoretical mass of 73,634.6 Da), whereas the 74,267.3-Da mass is ∼632 Da higher. This extra mass is about equal intensity relative to the fully deglycosylated molecule; however, its structure and site of incorporation are not known. The following experiments were thus performed to elucidate the attachment site and its possible molecular structure.
Identification of O-linked glycans and the attachment sites
Characterization and site identification
As shown in Figure 1(B), the glycan peak fraction at retention time of 56.5–58.5 min was collected from multiple HPLC runs and pooled. The major peptide in this fraction contained residues [400–450] of huLCAT-Fc and was designated as “Peptide A” (sequence: Q400GPPASPTASPEPPPPEGSGGGGDKTHTCPPCPAPELLGGPSVFLFPPKPK450). Another hydrophobic tryptic peptide, designated as Peptide B (sequence: T324YIYDHGFPYTDPVGVLYEDGDDTVATR351), was also found to coelute with Peptide A. The pooled fraction from 56.5 to 58.5 min was redigested with Wako endoproteinase Lys-C (Richmond, VA) to yield a pure, early eluting, and more hydrophilic peptide, which was designated as Peptide C (sequence: Q400GPPASPTASPEPPPPEGSGGGGDK424). This shorter peptide composed of residues [400–424] is easily separated from both the C-terminal portion of Peptide A [425–450] and hydrophobic Peptide B [324–351]. In addition to the expected polypeptide mass, Peptide C was again confirmed to contain an additional 632 mass units. These data verify that this additional mass is localized to the C-terminus of LCAT, including sequence at positions [400–416], the linker sequence at positions [417–422], and the first few residues of the Fc moiety at positions [423–424].
To assign T407 and S409 as the expected O-linked glycosylation sites, N-terminal sequencing using Edman degradation was performed on purified Peptide C. A major sequence corresponding to QGPPAS405PX407AX409PEPPPPEGX418GGGGDK was assigned. PTH-Ser was recovered at high yield for residue 405. PTH-Thr and PTH-Ser were totally absent (designated as “X”) from the appropriate cycles for residues 407, 409, and 418, suggesting potential modification at those sites. T407 and S409 have been reported to contain typical mucin O-linked glycans.25 We performed enzymatic deglycosylation followed by chemical β-elimination steps on Peptide C as described in the following section to confirm modification of S418.
Enzymatic deglycosylation and assignment of tetrasaccharide glycan structure
Figure 3(A) shows the deconvoluted monoisotopic [M+H]+ masses between 2000 and 5000 Da averaged across 6.5 min (17–23.5 min) of the LC-MS/MS run for Peptide C. These masses are derived from different glycans attached to the two O-linked sites at T407 and S409, as well as masses that also contain additional 632 Da. Table 1-(A) compares the observed masses with the theoretical glycan masses calculated from peptide mass (sequence position [400–424]) with the addition of both O-linked HexNAc-Hex (i.e., GalNAc-Gal) sugar core and sialic acid. Two major deconvoluted masses of 3583.5 and 3874.6 Da (shown in red) correspond to peptide [400–424] plus two O-linked disaccharide cores with two and three sialic acids, respectively. The attachment of multiple sialic acids distributed between the two O-linked HexNAc-Hex cores generates different combinations of monosialylated and disialylated glycoforms species. Major deconvoluted masses of 4215.7 and 4506.8 Da (shown in blue) were also observed, which are 632 Da greater in mass than their 3583.5 and 3874.61 Da counterparts. Two minor deconvoluted masses of 3715.6 and 4006.7 Da were also found that are about 132 Da greater in mass than the respective counterparts at 3583.5 and 3874.6 Da. Another minor mass of 3565.5 Da is 17 Da less than 3583.5 Da. This 17-Da mass reduction is due to cyclization of the N-terminal glutamine to pyroglutamic acid, which is a common chemical conversion in solution for peptides with an N-terminal Gln.
Figure 3.
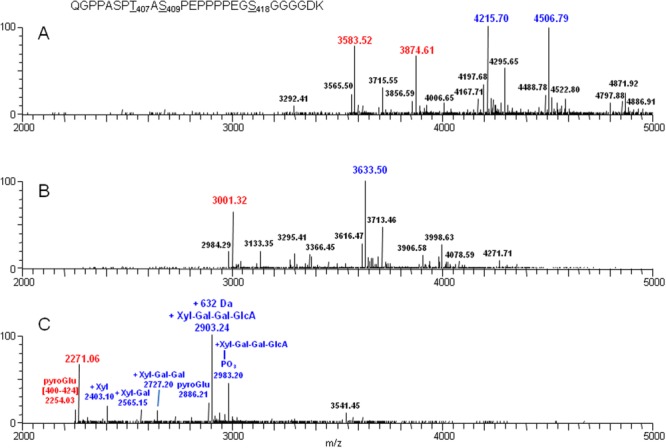
LC-MS analysis of deglycosylated Peptide C obtained from endoproteinase Lys C digestion of Peptide A. Panel A: Peptide C without treatment; Panel B: Peptide C treated with sialidase; and Panel C: Peptide C treated with sialidase and O-glycanase. These masses are derived from different glycans attached to the two O-linked sites at T407 and S409 (shown in red), as well as masses that also contain the +632 Da mass increase or components thereof (shown in blue).
Table 1.
Detected Peptide Masses Related to Mucin-Type O-Linked Sugars, Glycosaminoglycan Glycans, and Other Modifications
| Observed mass | Mass differencea | Calculated glycan massb | HexNAc-Hexc | Sialic acidc | Additional mass |
|---|---|---|---|---|---|
| (A) Untreated, intact peptide | |||||
| 3292.4 | 1021.3 | 1021.9 | 2 | 1 | 0 |
| 3565.5 | 1295.4 | 1313.2 | 2 | 2 | −17.8 (pyroGlu) |
| 3583.5d | 1313.1 | 1313.2 | 2 | 2 | 0 |
| 3715.6 | 1445.5 | 1445.2 | 2 | 2 | 132.1 |
| 3874.6 | 1604.5 | 1604.5 | 2 | 3 | No |
| 4006.7 | 1737.6 | 1604.5 | 2 | 3 | 133.1 |
| 4167.7 | 1896.6 | 1895.7 | 2 | 4 | 0 |
| 4215.7 | 1944.6 | 1313.2 | 2 | 2 | 632.4 |
| 4295.6 | 2024.5 | 1313.2 | 2 | 2 | 711.3 |
| 4506.8 | 2236.7 | 1604.5 | 2 | 3 | 632.2 |
| 4797.9 | 2526.8 | 1895.7 | 2 | 4 | 632.1 |
| 4871.9 | 2600.8 | 1969.5 | 3 | 3 | 631.3 |
| (B) Sialidase-treated peptide | |||||
| 2984.3 | 713.2 | 730.7 | 2 | 0 | −17.5 (pyroGlu) |
| 3001.3 | 730.2 | 730.7 | 2 | 0 | 0 |
| 3133.4 | 862.3 | 730.7 | 2 | 0 | 131.6 |
| 3295.4 | 1024.3 | 730.7 | 2 | 0 | 292.6 |
| 3366.5 | 1095.4 | 1096.0 | 3 | 0 | 0 |
| 3616.5 | 1345.4 | 730.7 | 2 | 0 | 614.2 (pyroGlu) |
| 3633.5 | 1362.4 | 730.7 | 2 | 0 | 631.7 |
| 3713.5 | 1442.4 | 730.7 | 2 | 0 | 711.7 |
| 3998.6 | 1727.5 | 1096.0 | 3 | 0 | 631.5 |
| (C) Sialidase- and O-glycanase-treated peptide | |||||
| Observed mass | Mass differencea | Modificatione | |||
| 2254.0 | −17.1 | N-terminal pyroGlu | |||
| 2271.1 | 0 | Unmodified peptide | |||
| 2403.1 | 132 | Xly | |||
| 2565.2 | 294.1 | Xly-Gal | |||
| 2727.2 | 456.1 | Xyl-Gal-Gal | |||
| 2886.2 | 615.1 | Xyl-Gal-Gal-GlcA (N-terminal pyroGlu) | |||
| 2903.2 | 632.1 | Xyl-Gal-Gal-GlcA | |||
| 2983.2 | 712.1 | Xyl-Gal-Gal-GlcA (+ phosphate on Xyl) | |||
Data were obtained from LC-MS/MS analysis of intact and deglycosylated Peptide C after sialidase and O-glycanase treatment.
Mass difference is obtained by subtracting the theoretical mass of the polypeptide ([M+H]+ = 2271.1 Da) from the observed, monoisotopic glycopeptide mass.
Calculated mass from number of O-linked glycan disaccharide cores (N-acetyl hexosamine-hexose) and sialic acids is listed in Table I.
Estimated HexNAc-Hex cores and sialic acids in the glycopeptides.
Mass in boldface represents major ion intensity signal in LC-MS analysis.
See Figure 4(B) for assignment.
Peptide C was first treated with sialidase to remove terminal sialic acids from the O-linked glycans and then analyzed by LC-MS/MS. Figure 3(B) shows the deconvoluted full MS for the digested sample. Desialylated Peptide C is expected to contain O-linked disaccharide cores as described. Table 1-(B) lists the observed mass, the mass difference after subtraction of the peptide mass ([M+H]+ = 2271.1 Da), and the increase or decrease of other observed additional masses. Major deconvoluted masses of 3001.32 Da (in red; peptide + 2 disaccharide cores) and 3633.50 Da (in blue; peptide + 2 disaccharide cores + 632 Da) were observed at a ratio of roughly 4:6, respectively. Four other minor deconvoluted masses of 2984.3, 3133.4, 3295.4, and 3713.5 Da were observed, which differ from 3001.3 Da (peptide + disaccharide cores) by approximately −17, +132, +294, and 712 Da, respectively. When data in Table 1-(B) were compared with those in Table 1-(A), MS analysis of desialylated Peptide C detected two extra species with additional mass (294 and 711 Da), which were not detected in analysis of the heterogenous parent peptide.
The desialylated peptide was further treated with O-glycanase to specifically remove the Gal-(β1-3)-GalNAc-(α1) glycan attached to the O-linked sites and then reanalyzed by LC-MS/MS. Figure 3(C) displays the deconvoluted full MS of this region, and there are two major and several minor masses between 2000 and 3000 amu. As listed in Table 1-(C), two major masses at m/z 2271.06 (red) and 2903.24 (blue) correspond to the unmodified peptide mass and unmodified peptide mass +632 Da, respectively. The observed 632-Da modification present in Peptide C is resistant to both sialidase and O-glycanase treatment. Several masses detected as minor species appear to show mass increase of 132, 294.1, 456.1, and 712.1 Da [Table 1-(C)]. These species are all related to each other and were later confirmed to be intermediate products of GAG biosynthesis. The 2254.03- and 2886.21-Da masses [see Fig. 3(C) and Table 1-(C)] correspond to pyroGlu versions of the unmodified peptide (m/z=2271.06) and the modified peptide +632 Da, respectively.
MS/MS using CID was performed on the doubly charged ions of both unmodified Peptide C (m/z = 1136.53), as well as the species still retaining the +632 Da modification (m/z = 1452.62) [Fig. 4(A,B)]. These ions would be the nondeconvoluted 2+ ions observed in Figure 3(C). The spectra are similar; however, a few extra ions are observed in the spectra containing the modification. A series of doubly charged fragment ions of m/z = 1136.33 (unmodified peptide), 1202.40, 1283.49, and 1364.54 were observed. The doubly charged 1136.33 fragment ion corresponds to the unmodified Peptide C, suggesting that the 632 Da mass attached to the peptide is labile and is lost during CID fragmentation. The other three fragment ions mentioned above represent the unmodified peptide ions with mass increments of 132, 162, and 162 Da, respectively. The mass difference between m/z = 1452.62 (the modified precursor ion) and m/z = 1364.54 yield a component of +176 Da. This total mass difference is 632 Da. These data indicate that the 632-Da mass is an unusual GAG tetrasaccharide glycan composed of xylose (Xyl; 132 Da), galactose (Gal; 162 Da), galactose, and then glucuronic acid (GlcA; 176 Da). This tetrasaccharide glycan is not a substrate for PNGase F, sialidase, or O-glycanase and was therefore not removed using standard PNGase F or sialidase/O-glycanase treatment. Based on these MS/MS data, the following deconvoluted masses labeled in Figure 3(C) and listed in Table 1-(C) were then assigned (peptide mass with different glycan subunits): 2403.10 (+Xyl), 2565.15 (+Xyl-Gal), 2727.20 (+Xyl-Gal-Gal), and 2903.24 (+Xyl-Gal-Gal-GlcA). In addition to the GAG moiety (Xyl-Gal-Gal-GlcA), the deconvoluted mass of 2983.20 Da also suggests the presence of an additional 80-Da mass. As determined by MS/MS of the doubly charged ion (m/z =1492.60) shown in Figure 4(C), the 80-Da moiety is directly linked to the xylose. This was confirmed to be a phosphate and not sulfate (both 80 Da) by alkaline phosphatase treatment (data not shown).
Figure 4.

MS/MS ion chromatograms of Peptide C after deglycosylation with both sialidase and O-glycanase. Spectra were selected and analyzed for three doubly charged ions. Panel A: m/z = 1136.53, representing parent peptide with no modification; Panel B: m/z = 1452.62, representing peptide with addition of 632 Da; and Panel C: m/z = 1492.60, representing peptide with addition of 712 Da, respectively. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Characterization of additional O-linked site containing GalNAc-Gal core
As shown in Figure 2(A), the detection of a 75,363.8-Da mass suggests that a low abundance 366-Da GalNAc-Gal glycan exists in addition to the three glycosylation sites mentioned above. This additional mass was also found in Peptide C at low abundance at 4871.9 Da in the untreated peptide [Fig. 3(A) and Table 1-(A)] and at 3366.5 and 3998.6 Da in the nonsialylated peptide [Fig. 3(B) and Table 1-(B)]. The mass corresponding to the additional O-linked glycan disappeared after the nonsialylated peptide was treated with O-glycanase [Fig. 3(C) and Table 1-(C)]. The most plausible location for this low level glycan is at S405, although simple extension of the glycan at T407 or S409 cannot be ruled out. Similar results were previously obtained by Schindler et al.25
Identification of the tetrasaccharide attachment site by alkaline β-elimination and LC-MS/MS
The β-elimination reaction is usually inefficient, which leads to an incomplete reaction. If used to remove all three O-linked glycans in Peptide C, a mixture of heterogenous products would result, making it difficult to localize the tetrasaccharide attachment site. To avoid this potential pitfall, the following strategy was used. Purified Peptide C was enzymatically treated with sialidase and O-glycanase to remove all of the regular O-linked glycans and sialic acids attached at T407 and S409. The remaining tetrasaccharide was then chemically deglycosylated by alkaline β-elimination as described in the Materials and Methods section. Alkaline β-elimination resulted in removal of the glycan and an 18-Da mass loss from the previous site of glycan occupancy due to a dehydration process (serine → dehydroalanine or threonine → β-methyldehydroalanine). This mass loss/tag can be monitored and targeted by LC-MS/MS as described below.
After sialidase and O-glycanase treatment of Peptide C but prior to β-elimination, approximately equal amounts of unmodified Peptide C and modified Peptide C +632 Da (extracted m/z [1136.0–1137.0] and [1452.0–1453.0]) exist at retention times T19.5 and T18.6 min, respectively [see Supporting Information Fig. S4(A), Panels 2 and 3]. These extracted mass signals correspond to the doubly charged ions of both unmodified Peptide C and modified Peptide C +632 Da. Pyroglutamate versions of both peptides also exist at 21.6 and 20.6 min at m/z [1127–1128] and [1443–1444] [Supporting Information Fig. S4(A), Panels 4 and 5] due to spontaneous cyclization of the N-terminal glutamine in the peptide to pyroglutamate. As seen in Panels 2 and 3 of Supporting Information Figure S4(B), the unmodified peptide at m/z = [1136–1137] has dramatically diminished, and the modified peptide at m/z = [1452–1453] has completely disappeared after β-elimination. The doubly charged, β-eliminated version of the modified peptide exists at T19.9 min when scanned at m/z = [1127–1128] [Supporting Information Fig. S4(B), Scan 4]. In the same scan, another doubly charged ion at m/z = [1127–1128] was also detected at 21.9 min, which was confirmed to be due to increased pyroglutamate cyclization (−17 Da) of the unmodified peptide (no GAG modification) due to the strong conditions of alkaline β-elimination. Figure 5 shows that CID fragmentation of the 1127.2 Da ion at T19.9 min [Supporting Information Fig. S4(B), Panel 4] yields the [y7] fragment ion of 559.24 Da, suggesting the loss of 18 Da from a Ser residue due to β-elimination. This mass contains the C-terminal fragment of the β-eliminated peptide, S418GGGGDK. If S418 was not the site of tetrasaccharide modification, a fragment ion of 577.25 Da would be expected even after β-elimination. More evidence proving S418 as the GAG glycosylation site was obtained by the detection of other larger C-terminal fragment ions including [y10] (842.36 Da), [y13] (1136.51 Da), [y15] (1359.61 Da), and [y23] (2068 Da), all showing an 18-Da loss in mass. N-terminal fragment ions such as [b17] (1637.77 Da) and [b18] (1694.79 Da) containing both T407 and S409 were also detected. These masses are as expected as the regular O-linked glycans in T407 and S409 were previously removed by sialidase and O-glycanase. Based on the lack of PTH-Ser in this cycle during Edman sequencing as well as the specificity of β-elimination to chemically remove the glycan linkage between the amino acid side chain and glycan chain, the attachment of the unusual GAG tetrasaccharide can be confidently assigned to S418 and not to T407 or S409. As previously confirmed in this study and other studies, the mucin-type O-linked glycosylation is present on both T407 and S409.
Figure 5.
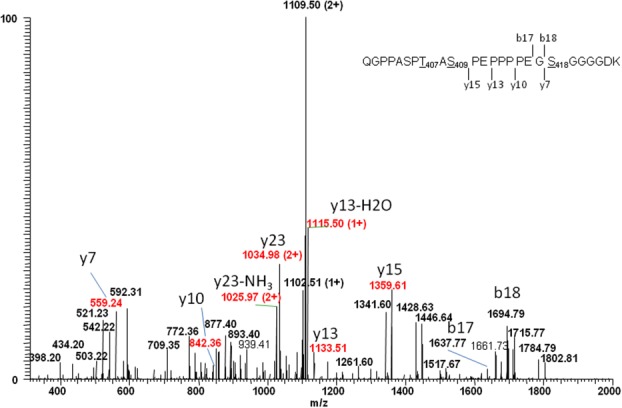
MS/MS spectrum of a doubly charged ion, m/z = 1127.53, for Peptide C (peptide sequence: QGPPASPT407AS409PEPPPPEGS418GGGGDK) treated with sialidase and O-glycanase followed by alkaline β-elimination. Y-series ions including [y7], [y10], [y13], and [y15] have a loss of 18 Da due to GAG glycan elimination at Ser418. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Analysis of GAG incorporation in huLCAT-Fc containing modified linker sequences
Four huLCAT-Fc linker mutants (linker sequences of G4SG, G4S, and G6 together with the original linker sequence, GSG4) have been produced (see Materials and Methods section). The mutant made with a G6 linker contains only Gly residues in the sequence and therefore does not have a Ser residue for GAG incorporation (our unpublished data). Both G4SG and G4S linker sequences still contain a Ser residue; only the G4SG linker still retains the G-S-G sequence. Following reduction and alkylation, these two linker mutants together with a huLCAT-Fc control (GSG4 linker) were digested with trypsin and then further digested with Lys-C to promote cleavage to obtain Peptide C from the huLCAT-Fc control and the equivalent peptides from the two mutants. The tryptic peptide sequences of the two mutants, equivalent to Peptide C from huLCAT-Fc control, are shown below: (a) G4SG linker mutant, Q400GPPASPT407AS409PEPPPPE416G417GGGS421GDK, and (b) G4S linker mutant, Q400GPPASPT407AS409PEPPPPE416G417GGG S421DK. LC-MS/MS results of these three linker-containing tryptic peptides are shown in Figure 6. Regular O-linked glycans are still present in Peptide C and its equivalent peptides as seen by detection of the 3+ ions at m/z =1196.0 and 1293.1 for both the huLCAT-Fc control and the G4SG mutant [Fig. 6(A,B)] and at m/z = 1176.9 and 1274.0 for the G4S mutant [Fig. 6(C)]. The transiently expressed control sample contains significant amounts of GAG incorporation (the 3+ ions at m/z = 1406.4 and 1503.7) at approximately an equal level to the unmodified peptides (the 3+ ions at m/z = 1196.0 and 1293.1). In contrast, the triply charged, precursor ions corresponding to GAG incorporation are expected to be detected at m/z=1406.4 and 1503.7 for the G4SG linker mutant and at m/z = 1387.3 and 1484.6 for G4S linker mutant, yet these ions are all absent. The GAG glycan together with other GAG-related intermediate products were not detected at all in either linker mutant. These data indicate that shifting the Ser position downstream in the linker sequence has eliminated GAG incorporation into the huLCAT-Fc molecule.
Figure 6.
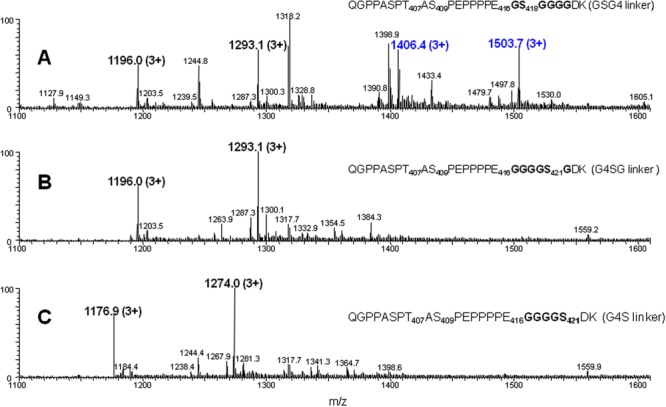
LC-MS/MS analysis of Peptide C and the equivalent peptides after sialidase and O-glycanase treatment. These peptides were derived from both trypsin and endoprotinase Lys C digestion of the reduced/alkylated huLCAT-Fc control and two other linker mutants. Panel A: Peptide from control with GSG4 linker; Panel B: Peptide from G4SG linker mutant; and Panel C: Peptide from G4S linker mutant. The triply charged ions at m/z 1196.0, 1293.1, 1176.9, and 1274.0 represent the glycopeptides containing 2 and 3 sialic acids, respectively. The two triply charged ions at m/z 1406.4 and 1503.7 represent the glycopeptides containing 2 and 3 sialic acids with additional 632 Da, respectively. Individual peptide sequences are indicated. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Discussion
Traditionally, glycan analysis of glycoproteins involves multiple processing steps including enzymatic removal of the glycans, enrichment procedures, and identification and quantification of the released glycans after chemical labeling. The latter steps usually require orthogonal analytical strategies and methods.40–42 After various attempts, we failed when using the above-mentioned procedures for glycan analysis of huLCAT-Fc because of the incomplete removal of LCAT N-linked glycans by glycosidases such as PNGase F (our unpublished observation). In this study, we were able to use in-house software, MassAnalyzer,43 in conjunction with detailed manual verification to perform identification and glycan profiling of the glycans attached at specific N-linked sites of huLCAT-Fc after LC-MS/MS peptide mapping. We found the method to be useful for comparing site-specific glycosylation amongst different huLCAT-Fc preparations in early stage development. Complete peptide bond cleavage during proteolytic digestion step is critical for releasing glycopeptides and for obtaining good N-linked glycan relative distribution data. When compared with traditional glycan mapping methods, MassAnalyzer can provide comparable glycan distribution data for noncharged glycans in different glycoproteins and monoclonal antibodies, and because of decreased ionization efficiency, highly sialylated glycans are consistently underestimated (Z. Zhang, unpublished data). However, comparative glycan profiling data obtained by the LC-MS/MS analysis of glycopeptides are usually reliable and can be used as a quick screening method for clone selection of a desired cell line expressing therapeutic glycoproteins with complex N-linked glycans.
Different mammalian expression systems can produce different types of glycans. For example, the glycan distribution of huLCAT-Fc described here is slightly different from those reported for recombinant human LCAT produced in baby hamster kidney cells44 or human lung cells.45 As confirmed by fast-atom bombardment MS or MALDI-MS, the glycans enzymatically released from recombinant human LCAT in these cells are primarily sialylated tetraantennary followed by triantennary and biantennary glycans. However, recombinant human LCAT produced in a hepatoma-based cell line contains primarily biantennary glycans.46 Likewise, when we analyzed huLCAT-Fc transiently produced in 293 6E cells, levels of triantennary and tetraantennary glycans (both fucosylated and sialylated) were observed to be much higher than those produced in CHO cells (our unpublished data). CHO cell-produced huLCAT-Fc appears to contain glycans similar to those present in human plasma LCAT,25 despite that the latter has triantennary and afucosylated glycans as major glycoforms. These functional molecules may be selected as therapeutic candidates for preclinical and human clinical studies to evaluate their in vivo activity and clinical efficacy.
Following isolation of the O-linked glycopeptide, Edman sequencing, and LC-MS/MS analysis after deglycosylation, we confirmed that huLCAT-Fc also contains similar O-linked glycans at T407 and S409. Each monomer of dimeric huLCAT-Fc contains two GalNAc-Gal cores plus 1 to 4 sialic acids. This suggests that the major O-linked glycans contain the core attached with 0, 1, and 2 terminal sialic acids at one site (T407 or S409), with the second site containing a core with 1 and 2 sialic acids attached. LC-MS/MS analysis of the O-linked glycopeptide indicated that a low-abundance O-linked GalNAc-Gal disaccharide core exists, possibly at S405 or as an extension to that on T407 or S409.
Approximately half of the purified huLCAT-Fc was found to have an unexpected GAG incorporation at S418 of the linker sequence. This is the first observation that we are aware of in which an engineered recombinant fusion molecule is modified by a GAG to yield an O-linked glycosylation site. This observation appears to be unique to huLCAT-Fc as no reports have documented GAG incorporation in other fusion molecules. Most of the linker sequences in these Fc fusion molecules are also rich in Ser and Gly.26 GAGs are one of the most common types of oligosaccharides found in animal glycoconjugates.47–49 A consensus site of a-a-a-a-G-S-G-a-a/G-a (where “a” represents D or E) has been compiled from various natural proteins containing GAGs.39 When a single or repeating acidic amino acid(s) is present at the consensus acidic site, the sequence proved to be a relatively low Km substrate for peptide O-xylosyltransferase and served as a good acceptor for GAG attachment.50 In the case of huLCAT-Fc, the C-terminus (E416) is connected to Fc by a GSG4 hexapeptide linker. The resulting construct creates a sequence having some homology with the proposed xylosyltransferase consensus site, in particular, the residues highlighted in bold: PPPE416G*S418GGGGDK. As GAG incorporation occurs at Ser residues with a specific consensus sequence in the linker, elimination of GAG incorporation can be successfully achieved by engineering out the site. This has been demonstrated by two of the linker mutants we prepared and analyzed. The G4SG linker mutant retains the LCAT C-terminus E416 and a G-S-G motif in the linker sequence, but Ser has been shifted three residues downstream, whereas G4S linker mutant lacks the G-S-G motif, and Ser shifts three residues downstream as well.
Other GAG-related sugars and smaller polysaccharide units such as Xyl, Xyl-Gal, and Xyl-Gal-Gal can also be detected as minor species at the peptide level. Furthermore, the phosphorylated GAG core was also observed as a minor species, whereby the phosphate moiety is directly attached to the xylose moiety. These data thus indicate that incorporation of GAG sugars into huLCAT-Fc is heterogeneous and probably not highly efficient. The GAG core appears to be a key intermediate molecule for further modifications and synthesis of either heparan sulfate or chondroitin sulfate.39 No trace of heparan sulfate or chondroitin sulfate has been detected in huLCAT-Fc. Sulfated and sialylated GAG tetrasaccharide have been found in human urinary-soluble thrombomodulin51; however, such glycans were not detected in huLCAT-Fc.
During preparation of the manuscript, low to medium levels of Ser xylosylation in engineered molecules with (GGGGS)n > 2 linkers was reported by Wen et al.52 The xylosylation was concluded to result from the GSG motif at the linker's sequence repeat. When compared with those found in huLCAT-Fc, there are a few differences in GAG glycans. First, depending on the engineered molecules and the selected clones examined, the levels of O-xylosylation varied considerably,52 whereas huLCAT-Fc with the GSGGGG linker consistently generated high levels of the GAG core. Second, Wen et al. reported that the most abundant O-glycosidic substituent was xylose alone, whereas the major GAG glycan found in huLCAT-Fc was the tetrasaccharide core. Many minor GAG-related glycans were present. Among these, sialylated Xyl-Gal, Xyl-Gal-Gal-Glc-HexNAc, and sulfated Xyl-Gal-Gal-Glc-HexNAc were not found in huLCAT-Fc GAG glycans. Xylose phosphorylation found in huLCAT-Fc GAGs was not reported by Wen et al.52
Materials and Methods
Cloning and expression of huLCAT-Fc in mammalian cells
The huLCAT gene encodes a leader sequence and the 416-amino acid coding sequence of huLCAT23,24 as described below: MGPPGSPWQWVTLLLGLLLPPAAPF1WLLNVLFPPHTTPKAELSNHTRPVILVPGYLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKLKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAYRQGPPASPTASPEPPPPE, where the leader sequence is underlined, the N-terminal amino acid, F1, is labeled, and the different glycosylation sites are in bold. Three huLCAT cDNAs were constructed with amino acid mutations as detailed in our previous report.29 The above-mentioned cDNAs were engineered to connect the huLCAT C-terminus to the N-terminus of human IgG1 Fc sequence (with C-terminal lysine deletion) by using a G-S-G-G-G-G (abbreviated as GSG4) linker sequence. The engineered huLCAT-Fc fusion construct was cloned into a vector for expression in CHO/CS-9 cells under serum-free growth conditions. After evaluation at an early stage, all media were harvested after 10 days of cell culture of clones selected for high expression. The final fusion protein is referred to as huLCAT-Fc. At a later development stage, engineered CHO/CS-9 cells carrying the DHFR− gene were amplified with methotrexate for cloning of stable cell line with high expression of huLCAT-Fc.53
In addition to the G-S-G-G-G-G (GSG4) linker sequence between LCAT and Fc, the following linker sequences, G-G-G-G-S-G (G4SG), G-G-G-G-S (G4S), and G-G-G-G-G-G (G6), were also used to make LCAT-Fc fusion constructs. These constructs together with the construct containing the original GSG4 linker were cloned into a pTT5 plasmid expression vector and transiently transfected into CHO 3E7 cells (NRCC) in Amgen proprietary media. Media were harvested 10 days post-transfection and pooled for the subsequent purification.
Purification of huLCAT-Fc
The harvested condition media containing CHO cell-produced huLCAT-Fc were captured by affinity chromatography using a MabSelect SuRe Protein A column equilibrated with Tris-buffered saline solution (25 mM Tris-HCl, pH 7.9, 150 mM NaCl). Nonspecific binding was washed off with 10 column volumes of 60 mM Tris-HCl, pH 7.9. HuLCAT-Fc was eluted with 87 mM sodium acetate and 150 mM NaCl at pH 3.5, and the eluted fraction was neutralized to neutral pH with 1M Tris-HCl at pH 8.0. The MabSelect SuRe pool was buffer-exchanged into 100 mM sodium acetate and 100 mM NaCl at pH 5.5 and passed through a Fractogel SO3 cationic exchange column. Purified huLCAT-Fc was eluted in the flow through fractions and was collected. The protein was then buffer-exchanged using a 30K-molecular-weight cutoff membrane and formulated in 10 mM potassium phosphate buffer containing 9% sucrose at pH 6.7.
Human LCAT-Fc preparations with various changes in the linker sequence were all produced in a CHO 3E7 transient expression system (see “Cloning and expression of huLCAT-Fc in mammalian cells” section). HuLCAT-Fc in the harvested media was captured on a MabSelect SuRe Protein A column in TBS buffer as described above, and nonspecific binding was washed off by 10 column volumes of TBS. HuLCAT-Fc was eluted by 100 mM acetate, pH 3.5, and neutralized to neutral pH with 1M Tris-HCl, pH 8.0. These samples achieved high purity after affinity capture and were used without further purification to study the effect of linker sequence changes on GAG glycosylation.
HuLCAT-Fc activity assay
Proteoliposome substrate containing [3H]-cholesterol was incubated with aliquots of huLCAT-Fc for a specified period of time. The reaction was terminated by the addition of isopropanol, and free and esterified cholesterol were separated by thin-layer chromatography followed by isolation of the separated lipid zones for scintillation counting.54 Labeled cholesterol and cholesterol esters were recovered from thin-layer chromatograms.55 The enzymatic activity of huLCAT-Fc was confirmed to be similar to that measured for both plasma-derived human LCAT and recombinant human LCAT produced in CHO cells (our unpublished data).
Digestion of huLCAT-Fc for analysis of N-linked glycopeptides
Fifty micrograms of CHO huLCAT-Fc was dried and then resuspended in 12.5 µL of 120 mM Tris, pH 7.5/8M urea/20 mM hydroxylamine/10 mM dithiothreitol (DTT). The sample was reduced for 45 min at 37°C. Iodoacetamide (20 mM) was added, and then the sample was incubated at room temperature for 30 min in the dark. Sample was diluted to 50 µL with water and 1.5 µg of Sigma (St. Louis, MO) proteomics-grade trypsin (3 µL of 0.5 µg/µL) and then incubated overnight at 37°C. About 15 µg of digest was analyzed by LC-MS/MS. For peptide separations, an Agilent 1100 capillary HPLC (Agilent, Santa Clara, CA) was equipped with a Restek (Bellefonte, PA) Viva C18 1.0 mm × 250 mm column (5 µm particle size) and trifluoroacetic acid (TFA)-based solvents (mobile phase A = 0.1% TFA/H2O; mobile phase B = 0.1% TFA/99.9% acetonitrile). Using a flow rate of 60 µL/min, the gradient consisted of initial conditions at 2% B, up to 55% B over 95 min, up to 97% B over 5 min, isocratic at 97% B for 5 min, and then back to 2% B over 5 min. The column effluent was sprayed into a Thermo Scientific LTQ XL ion trap mass spectrometer (San Jose, CA). The LTQ MS method used the following scan events: full MS of m/z [400–2000], zoom scan, CID, pulsed-Q dissociation (PQD), and SID of m/z [150–450]. CID and PQD scans use the following values: collision energy (CE) = 35%, isolation width (IsoW) = 3.0 Da, and minimum signal required = 1000. Other values include: Activation Q = 0.250 (CID) with an activation time of 30 ms, Activation Q = 0.700 (PQD) with an activation time of 0.1 ms. SID fragmentation was set at a relative value of 95%.
Distribution of glycosylation for N-linked glycans
Mass spectrometry-based identification and quantification of glycopeptide glycoforms was performed by computer analysis of LC-MS/MS tryptic peptide maps using the MassAnalyzer program.43 The locations of the various glycopeptides were localized by performing an XIC of m/z [203.5–204.5] from the SID scan following data acquisition.56 The relative abundance of the proposed glycopeptide structures was automatically calculated from the full MS (survey scan) data. Ions corresponding to the various structures were also manually evaluated to insure the accuracy of the assignment.
A simplified N-linked glycan nomenclature system43 shown in Supporting Information Figure S1 was used for assignment of various glycans. Glycan composition is included in parenthesis for each glycan, and the respective branching structures are indicated.
Deglycosylation of huLCAT-Fc
HuLCAT-Fc (2 mg/mL) was denatured and reduced in 4M guanidine hydrochloride (GuHCl)/50 mM Tris, pH 8.3/10 mM DTT for 30 min at 55°C. For alkylation, 8 µL of 300 mM iodoacetic acid (Sigma, MO) was added to the denatured and reduced sample and incubated at room temperature for 15 min in the dark. For PNGase F and sialidase digestion, reduced and alkylated huLCAT-Fc was neutralized to pH 7.5, and then 1 µL of PNGase F (New England Biolab, MA) was added, and the sample was incubated at 37°C for 2 h. Then, 2 µL of sialidase (ProZyme, CA) was added and incubated at 37°C for 30 min. For O-glycanase, sialidase, and PNGase F digestion, 4 µL O-glycanase (ProZyme) and 2 µL sialidase were added to 50 µg of reduced and alkylated huLCAT-Fc followed by incubation at 37°C for 3 h, which was then followed by the addition of 2 µL PNGase F and incubation at 37°C for 1 h. Intact masses of various deglycosylated huLCAT-Fc preparations were determined by LC-MS analysis using an Agilent Technologies 6210 ESI time-of-flight mass analyzer in conjunction with a Waters UPLC system (Bedford, MA). UPLC separation was performed using a Waters Acquity BEH 200SEC column (4.6 mm × 150 mm, 1.7 µm particle size) using buffers of 0.1% TFA, 0.25% formic acid, 15% acetonitrile, and 85% water at a flow rate of 0.4 mL/min. Sample amounts of 20–100 μg were injected.
Sialidase, O-glycosidase, and alkaline β-elimination of isolated O-linked glycan-containing peptide
The O-linked glycopeptide was resuspended in 14 µL deionized water, 4 µL 250 mM sodium phosphate, pH 6.0, and 1 µL recombinant sialidase (QA Bio, Palm Desert, CA) and then incubated for 1 h at 37°C. Further deglycosylation was obtained by the addition of 2 µL of QA Bio O-glycanase and incubation for 1 h at 37°C. To remove the tetrasaccharide by alkaline β-elimination, the sialidase- and O-glycanase-treated peptide was desalted using a Waters Oasis HLB cartridge (Milford, MA), dried, resuspended in 0.3M NaOH, and incubated overnight at 4°C. The deglycosylated and β-eliminated peptide was desalted again prior to LC-MS/MS.
Analysis of O-linked tryptic peptide and the deglycosylated peptide
N-terminal sequencing using Edman degradation was performed to confirm the potential Ser and Thr O-linked glycosylation sites according to a previously reported procedure.57 For LC-MS/MS analysis, a Waters NanoAcquity UPLC (Milford, MA) was utilized which used formic acid buffers (mobile phase A = 0.1% formic acid/H2O; mobile phase B = 0.1% formic acid/99.9% acetonitrile). For peptide separation, the UPLC used a 180 µm × 20 mm Symmetry C18 (5 µm particle size) trap column which was linked to an analytical column (Agilent Zorbax 300SB-C18 column with 0.5 mm × 250 mm; 5 µm particle size). The column effluent was sprayed into a Thermo Scientific LTQ Orbitrap Velos (Bremen, Germany) mass spectrometer using the standard HESI-II probe inside an Ion Max ionization source. A source voltage of 3.0 kV and a capillary temperature of 275°C were used throughout.
The UPLC method consists of 5 min of loading onto the trap column at 15 µL/min (A = 97%, B = 3%). Using a flow rate of 15 µL/min, the gradient consists of initial conditions of 3% B, up to 45% B over 85 min, up to 97% B over 1 min, isocratic at 97% B for 6 min, down to 3% B over 3 min, and then isocratic at 3% B for 20 min.
The MS method used the following scan events: (1) 30K-resolution FT-MS full MS scan of m/z [300–2000], 7.5K-resolution targeted CID (MS/MS) scans in the FT-MS of (2) m/z = 1136.53, (3) m/z = 1452.60, and (4) m/z = 1127.53, and 7.5K-resolution targeted CID (MS3) in the FT-MS of (5) m/z = 1127.53 → 559.39 and (6) m/z = 1127.53 → 1359.74 (CE = 35%, Activation Q = 0.250, time = 20 ms, IsoW = 2.5 Da). Other MS methods consist of the following scan events: (1) 30K-resolution FT-MS full MS scan of m/z [400–2000], and (2) CID (MS/MS) in the IT-MS of the top six most abundant precursors (CE = 35%, Activation Q = 0.250, time = 10 ms, IsoW = 2.0 Da, minimum signal required = 2000).
Acknowledgments
The authors would like to thank Drs. Margaret Karow, Kevin Moore, Jason O'Neill, Janet Cheng, Alison Wallace, and Tom Boone for managerial support, as well as the entire huLCAT Team from the Department of Metabolic Disorders, Amgen Inc.
Glossary
- CHO
Chinese hamster ovary
- CID
collision-induced dissociation
- GAG
glycosaminoglycan
- HDL
high-density lipoprotein
- HuLCAT-Fc
human LCAT Fc fusion
- LCAT
lecithin-cholesterol acyltransferase
- RCT
reverse cholesterol transport
- SID
surface-induced dissociation
- UPLC
ultra-high pressure liquid chromatography
Supplementary material
Additional Supporting Information may be found in the online version of this article.
Supplementary Material Table 1. Glycan assignment and distribution at each individual Asn-linked site (For glycan nomenclature see Supplementary Material Figure 1)
Supplementary Material Figure 1. Selected glycan forms showing antennary branching structures. The nomenclature was adopted from ref. 7. Glycan composition is indicated in parenthesis after each glycan name. Abbreviations for the respective monosaccharides are Hex (mannose or galactose), HexNAc (N-acetyl glucosamine), Neu5Ac (N-acetyl neuraminic acid), and Fuc (fucose).
Supplementary Material Figure 2. Full MS spectra of the N20 glycopeptide averaged across retention time 49.5–51.5 min. (derived from Figure 1 in the main text). The spectra was obtained from tryptic digestion of huLCAT-Fc. Masses assigned to the respective glycan species are indicated. Two co-eluting ions at m/z 1401.4 and 1423.7 Da are from the non-glycosylated huLCAT-Fc tryptic peptides W619-K641 and I245-R256. Several other unassigned ions were also identified to be non-glycopeptides.
Supplementary Material Figure 3. CID spectra of an ion at m/z 1335.12 at 50.05 min selected for fragmentation (see Figure 1 in the main text and Supplementary Material Figure 1). This 4+ charged ion was determined to be the N20 glycopeptide with a sialylated triantennary glycan, A3S2G1F. The charge states, fragmented glycan forms attached to the peptide, and the released sugar units are indicated in the respective fragment ions. SA represents sialic acid (Neu5Ac).
Supplementary Material Figure 4. Targeted LC-MS/MS of sialidase and O-glycanase treated Peptide C before and after β-elimination treatment to remove the O-linked GAG glycan. Column A: Peptide C without alkaline βelimination. Column B: Peptide C with alkaline βelimination. Panel 1: base peak Full MS; Panel 2: Full MS, extracting m/z [1136–1137] (2+ of unmodified peptide); Panel 3: Full MS, extracting m/z [1452–1453] (2+ of modified peptide + 632 Da); Panel 4: Full MS, extracting m/z [1127–1128] (2+ of the pyroGlu unmodified peptide); and Panel 5: Full MS, extracting m/z [1443–1444] (2+ of modified peptide + 632 Da + pyroGlu). The βeliminated peptide was labeled below with a blue arrow. The identity of all extracted masses was confirmed by targeted CID (7.5K resolution). Supplementary Material Figure 1. Selected glycan forms showing antennary branching structures. The nomenclature was adopted from ref. 7. Glycan composition is indicated in parenthesis after each glycan name. Abbreviations for the respective monosaccharides are Hex (mannose or galactose), HexNAc (N-acetyl glucosamine), Neu5Ac (N-acetyl neuraminic acid), and Fuc (fucose).
Supplementary Information
REFERENCES
- 1.Glomset JA. The mechanism of the plasma cholesterol esterification reaction: plasma fatty acid transferase. Biochim Biophys Acta. 1962;65:128–135. doi: 10.1016/0006-3002(62)90156-7. [DOI] [PubMed] [Google Scholar]
- 2.Glomset JA. The plasma lecithins-cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 3.Kunnen SK, Eck MV. 2012) Lecithin:cholesterol acyltransferase: old friend or foe in atherosclerosis? J Lipid Res. 53:1783–1799. doi: 10.1194/jlr.R024513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaysen GA. New insights into lipid metabolism in chronic kidney disease. J Renal Nutr. 2011;21:120–123. doi: 10.1053/j.jrn.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura Y, Kotite L, Gan Y, Spencer TA, Fielding CJ, Fielding PE. Molecular mechanism of reverse cholesterol transport: reaction of pre-beta-migrating high-density lipoprotein with plasma LCAT. Biochemistry. 2004;43:14811–14820. doi: 10.1021/bi0485629. [DOI] [PubMed] [Google Scholar]
- 6.Ohashi R, Mu H, Wang MX, Yao Q, Chen C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. Q J Med. 2005;98:845–856. doi: 10.1093/qjmed/hci136. [DOI] [PubMed] [Google Scholar]
- 7.Kuivenhoven JA, Pritchard H, Hill J, Frohlich J, Assmann G, Kastelein J. The molecular pathology of lecithin-cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38:191–205. [PubMed] [Google Scholar]
- 8.Lee RG, Kelley KL, Sawyer JK, Farese RV, Jr, Parks JS, Rudel LL. Plasma cholesteryl esters provided by lecithin-cholesterol acyltransferase and acyl-coenzyme A:cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circ Res. 2004;95:998–1004. doi: 10.1161/01.RES.0000147558.15554.67. [DOI] [PubMed] [Google Scholar]
- 9.Hovingh GK, Hutten BA, Holleboom AG, Petersen W, Rol P, Stalenhoef A, Zwinderman AH, de Groot E, Kastelein JJ, Kuivenhoven JA. Compromised LCAT function is associated with increased atherosclerosis. Circluation. 2005;112:879–884. doi: 10.1161/CIRCULATIONAHA.105.540427. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi L, Baldassarre D, Castelnuovo S, Conca P, Bocchi L, Candini C, Frigerio B, Amato M, Sirtori CR, Alessandrini P, Arca M, Boscutti G, Cattin L, Gesualdo L, Sampietro T, Vaudo G, Veglia F, Calandra S, Franceschini G. Functional LCAT is not required for efficient atherosclerosis in humans. Circulation. 2009;120:628–635. doi: 10.1161/CIRCULATIONAHA.108.818143. [DOI] [PubMed] [Google Scholar]
- 11.Lambert G, Sakai N, Vaisman BL, Neufeld EB, Marteyn B, Chan CC, Paigen B, Lupia E, Thomas A, Striker LJ, Blanchette-Mackie J, Csako G, Brady JN, Costello R, Striker GE, Remaley AT, Brewer HBJr, Santamarina-Fono S. Analysis of glomerulosclerosis and atherosclerosis in lecithin-cholesterol acyltransferase deficient mice. J Biol Chem. 2001;276:15090–15098. doi: 10.1074/jbc.M008466200. [DOI] [PubMed] [Google Scholar]
- 12.Berard AM, Foger B, Remaley A, Shamburek R, Vaisman BL, Talley G, Hoyt RF, Jr, Marcovina S, Brewer HB, Jr, Santamarina-Fojo S. High plasma HDL concentrations associated with enhanced atherosclerosis in transgenic mice overexpressing lecithin-cholesteryl acyltransferase. Nat Med. 1997;3:744–749. doi: 10.1038/nm0797-744. [DOI] [PubMed] [Google Scholar]
- 13.Foger B, Chase M, Amar MJA, Vaisman BL, Shamburek RD, Paigen B, Fruchart-Najib J, Paiz JA, Koch CA, Hoyt RF, Brewer HB, Jr, Santamarina-Fojo S. Cholesteryl ester transfer protein corrects dysfunctional high density lipoproteins and reduces aortic atherosclerosis in lecithin-cholesterol acyltransferase transgenic mice. J Biol Chem. 1999;274:36912–36920. doi: 10.1074/jbc.274.52.36912. [DOI] [PubMed] [Google Scholar]
- 14.Hoeg JM, Santamarina-Fojo S, Bérard AM, Cornhill JF, Herderick EE, Feldman SH, Haudenschild CC, Vaisman BL, Hoyt RF, Jr, Demosky SJ, Jr, Kauffman RD, Hazel CM, Marcovina SM, Brewer HB., Jr Overexpression of lecithin-cholesterol acyltransferase in transgenic rabbits prevents diet-induced atherosclerosis. Proc Natl Acad Sci USA. 1996;93:11448–11453. doi: 10.1073/pnas.93.21.11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amar MJA, Shamburek RD, Vaisman B, Knapper CL, Foger B, Hoyt RF, Jr, Santamarina-Fojo S, Brewer HB, Jr, Remaley AT. Adenoviral expression of human LCAT in nonhuman primates leads to an antiatherogenic lipoprotein phenotype by increasing high-density lipoprotein and lowering low-density lipoprotein. Metabolism. 2009;58:568–575. doi: 10.1016/j.metabol.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kayser F, Labelle M, Shan B, Zhang J, Zhou M. 2011. Methods for treating atherosclerosis. U.S. Patent US20110206652-A1, 2011.
- 17.Zhou M, Fordstrom P, Zhang J, Meininger D, Schwarz M, Kayser F, Shan B. Novel small molecule LCAT activators raise HDL levels in rodent models (Oral presentation) Arterioscler Thromb Vasc Biol. 2008;28:E65–E66. [Google Scholar]
- 18.Chen Z, Wang SP, Krsmanovic ML, Castro-Perez J, Gagan K, Mendoza V, Rosa R, Shah V, He T, Stout SJ, Geoghagen NS, Lee SH, McLaren DF, Wang L, Roddy TP, Plump AS, Hubbard BK, Sinz CJ, Johns DG. Small molecule activation of lecithin-cholesterol acyltransferase modulates lipoprotein metabolism in mice and hamsters. Metabolism. 2012;61:470–481. doi: 10.1016/j.metabol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Rousset X, Vaisman B, Auerbach B, Krause BR, Homan R, Stonik J, Csako G, Shamburek R, Remaley AT. Effect of recombinant human lecithin cholesterol acyltransferase infusion on lipoprotein metabolism in mice. J Pharmacol Exp Ther. 2010;335:140–148. doi: 10.1124/jpet.110.169540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou M, Sawyer J, Kelley K, Fordstrom P, Chan J, Tonn G, Carlson T, Retter M, Meininger D, Cheng J, Gates A, Woodward A, Delaney J, Zhang R, Tang J, Liu Q, Cao P, Luchoomun J, Voogt J, Turner S, Shan B, Boone T, Rudel L, Schwarz M. Abstract 5920: lecithin cholesterol acyltransferase promotes reverse cholesterol transport and attenuates atherosclerosis progression in New Zealand white rabbits. Circulation. 2009;120:S1175. [Google Scholar]
- 21.Alphacore News. AlphaCore reports positive phase 1 results for ACP-501 (rhLCAT) in patients with stable atherosclerosis. October 9, 2012. Available at: http://www.alphacorepharma.com. Accessed October 16, 2013.
- 22.Yang CY, Manoogian D, Pao Q, Lee FS, Knapp RD, Gotto AM, Jr, Pownall HJ. Lecithin:cholesterol acyltransferase. Functional regions and a structural model of the enzyme. J Biol Chem. 1987;262:3086–3091. [PubMed] [Google Scholar]
- 23.McLean J, Fielding C, Drayna D, Dieplinger H, Baer B, Kohr W, Henzel W, Lawn R. Cloning and expression of human lecithin-cholesterol acyltransferase cDNA. Proc Natl Acad Sci USA. 1985;83:2335–2339. doi: 10.1073/pnas.83.8.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLean J, Wion K, Drayna D, Fielding C, Lawn R. Human lecithin-cholesterol acyltransferase gene: complete gene sequence and sites of expression. Nucleic Acids Res. 1986;14:9397–9406. doi: 10.1093/nar/14.23.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schindler PA, Settineri CA, Collet X, Fielding CL, Burlingame A. Site-specific detection and structural characterization of the glycosylation of human plasma proteins lecithin:cholesterol acyltransferase and apolipoprotein D using HPLC/electrospray mass spectrometry and sequential glycosidase digestion. Protein Sci. 1995;4:791–803. doi: 10.1002/pro.5560040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimamoto G, Gegg C, Boone T, Queva C. Peptibodies: a flexible alternative format to antibodies. MAbs. 2012;4:586–591. doi: 10.4161/mabs.21024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med. 1991;174:1483–1489. doi: 10.1084/jem.174.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreland L, Bate G, Kirkpatrick P. Abatacept. Nat Rev Drug Discov. 2006;5:185–186. doi: 10.1038/nrd1989. [DOI] [PubMed] [Google Scholar]
- 29.Boone T, Meininger DP, Schwarz M, Shan B, Shen W, Zhou M. 2012. Modified lecithin-cholesterol acyltransferase enzymes. U.S. Patent US008168416-B2.
- 30.Collet X, Fielding CJ. Effects of inhibitors of N-linked oligosaccharide processing on the secretion, stability, and activity of lecithin:cholesterol acyltransferase. Biochemistry. 1991;30:3228–3234. doi: 10.1021/bi00227a010. [DOI] [PubMed] [Google Scholar]
- 31.Doi Y, Nishida T. Microheterogeneity and physical properties of human LCAT. J Biol Chem. 1983;258:5840–5846. [PubMed] [Google Scholar]
- 32.Qu SJ, Fan HZ, Blanco-Vaca F, Pownall H. Effects of site-directed mutagenesis on the N-glycosylation sites of human LCAT. Biochemistry. 1993;32:8732–8736. doi: 10.1021/bi00085a002. [DOI] [PubMed] [Google Scholar]
- 33.Kosman J, Jonas A. Deletion of specific glycan chains affects differentially the stability, local structures, and activity of LCAT. J Biol Chem. 2001;276:37230–37236. doi: 10.1074/jbc.M104326200. [DOI] [PubMed] [Google Scholar]
- 34.Karmin O, Hill JS, Wang X, McLeod R, Pritchard PH. Lecithin:cholesterol acyltransferase: role of N-linked glycosylation in enzyme function. Biochem J. 1993;294:879–884. doi: 10.1042/bj2940879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karmin O, Hill JS, Pritchard PH. Role of N-linked glycosylation of LCAT in lipoprotein substrate specificity. Biochim Biophys Acta. 1995;1254:193–197. doi: 10.1016/0005-2760(94)00183-y. [DOI] [PubMed] [Google Scholar]
- 36.Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22:1383–1391. doi: 10.1038/nbt1030. [DOI] [PubMed] [Google Scholar]
- 37.Walsh G, Jefferis R. Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol. 2006;24:1241–1252. doi: 10.1038/nbt1252. [DOI] [PubMed] [Google Scholar]
- 38.Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol. 2009;20:678–684. doi: 10.1016/j.copbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 39.Wilson IB. The never-ending story of peptide O-xylosyltransferase. Cell Mol Life Sci. 2004;61:794–809. doi: 10.1007/s00018-003-3278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schiel JE. Glycoprotein analysis using mass spectrometry: unraveling the layers of complexity. Anal Bioanal Chem. 2012;404:1141–1149. doi: 10.1007/s00216-012-6185-2. [DOI] [PubMed] [Google Scholar]
- 41.An HJ, Froehlich JW, Lebrilla CB. Determination of glycosylation sites and site-specific heterogeneity in glycoproteins. Curr Opin Chem Biol. 2009;13:421–426. doi: 10.1016/j.cbpa.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamada K, Kakehi K. Recent advances in the analysis of carbohydrates for biomedical use. J Pharm Biomed Anal. 2011;55:702–727. doi: 10.1016/j.jpba.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Z. Large-scale identification and quantification of covalent modifications in therapeutic proteins. Anal Chem. 2009;81:8354–8364. doi: 10.1021/ac901193n. [DOI] [PubMed] [Google Scholar]
- 44.Lacko AG, Reason AJ, Nuckolls C, Kudchodkar BJ, Nair MP, Sundarrajan G, Pritchard PH, Morris HR, Dell A. Characterization of recombinant human plasma lecithin: cholesterol acyltransferase (LCAT): N-linked carbohydrate structures and catalytic properties. J Lipid Res. 1998;39:807–820. [PubMed] [Google Scholar]
- 45.Lane SB, Tchedre T, Nair MP, Thigpen AE, Lacko AG. Characterization of lecithin:cholesterol acyltransferase expressed in a human lung cell line. Protein Expr Purif. 2004;36:157–164. doi: 10.1016/j.pep.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 46.Ayyobi AF, Lacko AG, Murray K, Nair M, Li M, Mohuizen HO, Pritchard PH. Biochemical and compositional analyses of recombinant lecithin:cholesterol acyltransferase (LCAT) obtained from a hepatic source. Biochim Biophys Acta. 2000;1484:1–13. doi: 10.1016/s1388-1981(99)00199-7. [DOI] [PubMed] [Google Scholar]
- 47.Varki A, Freeze HH, Manzi AE. Overview of glycoconjugate analysis. Curr Protoc Protein Sci Chapter 12:Unit. 1995;12:1.1–12.1.8. doi: 10.1002/0471140864.ps1201s57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van den Steen PV, Rudd PM, Dwek RA, Opdenakker G. Concepts and principles of O-linked glycosylation. Crit Rev Biochem Mol Biol. 1998;33:151–208. doi: 10.1080/10409239891204198. [DOI] [PubMed] [Google Scholar]
- 49.Lowe JB, Marth JD. Structures common to different types of glycans. In: Varki A, RD Cummings, JD Esko, HH Freeze, P Stanley, CR Bertozzi, GW Hart, ME Etzler, editors. Essentials of glycobiology. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. Chapter 16. [Google Scholar]
- 50.Roch C, Kuhn J, Kleesiek K, Gotting C. Differences in gene expression of human xylosyltransferases and determination of acceptor specificities for various proteoglycans. Biochem Biophys Res Commun. 2010;391:685–691. doi: 10.1016/j.bbrc.2009.11.121. [DOI] [PubMed] [Google Scholar]
- 51.Hiroyuki W, Natsuka S, Mega T, Ostuki N, Isaji M, Naotsuka M, Koyama S, Kanamori T, Sakai K, Hase S. Novel proteoglycan linkage tetrasaccharides of human urinary soluble thrombomodulin, SO4-3GlcAβ1–3Galβ1–3(+/−Siaα2–6) Galβ1–4Xyl. J Biol Chem. 1993;274:5436–5442. doi: 10.1074/jbc.274.9.5436. [DOI] [PubMed] [Google Scholar]
- 52.Wen D, Foley SF, Hronowski XL, Gu S, Meier W. Discovery and investigation of O-xylosylation in engineered proteins containing (GGGGS)n linker. Anal Chem. 2013;85:4805–4812. doi: 10.1021/ac400596g. [DOI] [PubMed] [Google Scholar]
- 53.Rasmussen B, Davis R, Thomas J, Reddy P. Isolation, characterization and recombinant protein expression in veggie-CHO: a serum-free CHO host cell line. Cytotechnology. 1998;28:31–42. doi: 10.1023/A:1008052908496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Batzri S, Korn ED. Single bilayer liposomes prepared without sonication. Biochim Biophys Acta. 1973;298:1015–1019. doi: 10.1016/0005-2736(73)90408-2. [DOI] [PubMed] [Google Scholar]
- 55.Lacko AG, Rutenberg HL, Soloff LA. Recovery of labeled cholesterol and cholesterol esters from thin-layer chromatograms. Clin Chim Acta. 1972;39:506–510. doi: 10.1016/0009-8981(72)90079-4. [DOI] [PubMed] [Google Scholar]
- 56.Carr SA, Huddleston MJ, Bean MF. Selective identification and differentiation of N- and O-linked oligosaccharides in glycoproteins by liquid chromatography-mass spectrometry. Protein Sci. 1993;2:183–196. doi: 10.1002/pro.5560020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haniu M, Horan T, Hui JO, Spahr C, Wang F, Chen C, Richards B, Lu HS. Human Dickkopf-1 (huDKK1) protein: characterization of glycosylation and determination of disulfide linkages in the two cysteine-rich domains. Protein Sci. 2011;20:1802–1813. doi: 10.1002/pro.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.