

Abstract
Calcitonin gene-related peptide (CGRP) and adrenomedullin (AM) are related peptides that are potent vasodilators. The CGRP and AM receptors are heteromeric protein complexes comprised of a shared calcitonin receptor-like receptor (CLR) subunit and a variable receptor activity modifying protein (RAMP) subunit. RAMP1 enables CGRP binding whereas RAMP2 confers AM specificity. How RAMPs determine peptide selectivity is unclear and the receptor stoichiometries are a topic of debate with evidence for 1:1, 2:2, and 2:1 CLR:RAMP stoichiometries. Here, we describe bacterial production of recombinant tethered RAMP-CLR extracellular domain (ECD) fusion proteins and biochemical characterization of their peptide binding properties. Tethering the two ECDs ensures complex stability and enforces defined stoichiometry. The RAMP1-CLR ECD fusion purified as a monomer, whereas the RAMP2-CLR ECD fusion purified as a dimer. Both proteins selectively bound their respective peptides with affinities in the low micromolar range. Truncated CGRP(27-37) and AM(37-52) fragments were identified as the minimal ECD complex binding regions. The CGRP C-terminal amide group contributed to, but was not required for, ECD binding, whereas the AM C-terminal amide group was essential for ECD binding. Alanine-scan experiments identified CGRP residues T30, V32, and F37 and AM residues P43, K46, I47, and Y52 as critical for ECD binding. Our results identify CGRP and AM determinants for receptor ECD complex binding and suggest that the CGRP receptor functions as a 1:1 heterodimer. In contrast, the AM receptor may function as a 2:2 dimer of heterodimers, although our results cannot rule out 2:1 or 1:1 stoichiometries.
Keywords: class B G protein-coupled receptor, calcitonin family peptide, vasodilator, neuropeptide
Introduction
Calcitonin gene-related peptide (CGRP) and adrenomedullin (AM) are multi-functional calcitonin family peptides.1,2 CGRP is a neuropeptide that acts in the central and peripheral nervous systems as a potent arterial and venous vasodilator. AM is widely distributed in the cardiovascular system where it has vasodilator effects. AM is essential during development for formation of blood and lymph vasculature.3,4 AM also has cardioprotective functions and plays a role in normal pregnancy and pregnancy complications.4,5 In addition, CGRP and AM both exhibit angiogenic activity.6,7 As a consequence of the various actions of CGRP and AM and their involvement in several pathophysiological states, their receptors are of interest as drug targets for a variety of disorders including migraine headache, acute myocardial infarction, pulmonary hypertension, preeclampsia, sepsis, and cancer.4,8–10
The CGRP and AM receptors are heteromeric protein complexes comprised of the calcitonin receptor-like receptor (CLR), a class B/Secretin family G protein-coupled receptor (GPCR), in association with a receptor activity modifying protein (RAMP).11 Three distinct RAMPs associate with CLR to determine peptide selectivity. RAMP1-CLR heteromers are CGRP receptors, whereas CLR heteromers with RAMP2 or -3 are AM receptors. RAMPs also regulate calcitonin family peptide binding to another class B GPCR, the calcitonin receptor (CTR), which is closely related to CLR. CTR alone is a receptor for calcitonin (CT), which regulates calcium homeostasis, whereas CTR heteromers with any of the three RAMPs are receptors for amylin (AMY), which regulates blood glucose levels.1,12–14
CLR and the RAMPs exhibit a similar plasma membrane topology with an N-terminal extracellular domain (ECD) followed by a membrane-embedded portion and an intracellular C-terminal tail. The integral membrane portion of CLR is a 7-transmembrane helix bundle typical of GPCRs whereas the RAMPs have a single transmembrane helix. Structural studies elucidated the folds of the CLR and RAMP1 and -2 ECDs and revealed how they interact in the absence of peptide ligands.15–17 The CLR ECD fold is common to class B GPCR ECDs and consists of an N-terminal α-helix followed by a short consensus repeat fold that is composed of two β-sheets each with two β-strands. Three conserved disulfide bonds hold the secondary structure elements together. The RAMP ECDs form a 3-helix bundle that is held together by three disulfide bonds in RAMP1 and two disulfide bonds in RAMP2. The RAMP2 ECD also has an N-terminal extension of ∼30 residues of unknown structure and function that is absent in RAMP1 and -3. In the CLR-RAMP ECD complex structures, RAMP α-helices 2 and 3 pack against the CLR N-terminal α-helix, adjacent to the putative peptide binding site of the CLR ECD such that the peptide ligands likely contact both proteins.
Peptide binding to class B GPCRs follows a two-domain model in which the C-terminal half of the peptide binds to the receptor ECD, which confers high affinity and specificity, and the N-terminal half of the peptide binds to the receptor helical bundle to activate signaling.18 CGRP and AM binding to their receptors appears to follow this model,19 but peptide-bound structures of CLR-RAMP ECD complexes are not available. CGRP has 37 residues and AM has 52 residues, but the first 12 AM residues are dispensable for function.20 CGRP and AM contain a conserved disulfide bond structure in their N-terminal regions that interact with the receptor helical bundle and variable sequence in their C-terminal regions that interact with the receptor ECDs.21 It is unclear if CGRP and AM bind to their receptor ECDs as continuous α-helices as observed for other class B GPCR ECD-peptide pairs. Chimeric receptor and mutagenesis studies indicated that the RAMP ECDs are sufficient to determine peptide selectivity and identified receptor and CGRP and AM peptide residues that are important for their interactions,22–27 but precisely how RAMPs determine peptide selectivity remains uncertain.
The CGRP and AM receptor stoichiometries are a topic of debate. Crystal structures of the CLR:RAMP1 ECD complex revealed a 1:1 heterodimer.16 In contrast, bimolecular fluorescence complementation and bioluminescence resonance energy transfer experiments with intact receptors in cells indicated a RAMP1 monomer interacting with a CLR homo-oligomer.28 Kusano et al. demonstrated that recombinant CLR:RAMP2 ECD complex was a heterodimer at low concentration and a tetramer at high concentration, and their crystal structure of the complex showed a 1:1 heterodimer in the asymmetric unit and a 2:2 dimer of heterodimers formed by crystal symmetry.17 Watkins et al. reported the production and biochemical characterization of recombinant CLR:RAMP2 ECD complex with evidence for a 2:1 CLR:RAMP2 stoichiometry.27 We previously described novel methodology for bacterial production of the AM receptor ECD complex as a maltose binding protein (MBP)-CLR ECD fusion protein in association with the RAMP2 ECD.29 Fusion of the CLR ECD to MBP was designed to facilitate crystallization as for other class B GPCR ECDs.30–35 The complex appeared to be a dimer of heterodimers, but a 2:1 MBP-CLR ECD:RAMP2 ECD stoichiometry could not be ruled out. Here, we describe bacterial production of recombinant CGRP and AM receptor ECD complexes as tethered MBP-RAMP ECD-CLR ECD fusion proteins, which ensures complex stability and enforces 1:1 CLR:RAMP stoichiometry within the fusion protein. Biochemical characterization of the peptide binding properties of the tethered fusion proteins identified critical CGRP and AM determinants for ECD complex binding.
Results
Bacterial expression and purification of tethered RAMP ECD-CLR ECD fusion proteins
In an effort to obtain stable RAMP ECD-CLR ECD complexes of defined 1:1 stoichiometry we devised a tethered fusion protein approach for producing the CGRP (RAMP1-CLR) and AM (RAMP2-CLR) receptor ECD complexes [Fig. 1(A)]. The protein expression and purification strategy was based on our previously reported methodology for bacterial production of disulfide-bond containing class B GPCR ECDs as soluble MBP fusion proteins engineered for crystallization.29,31 Crystal structures of peptide-free RAMP ECD-CLR ECD complexes16,17 were examined to decide how to connect the two ECDs with minimal chance of altering RAMP ECD-CLR ECD interactions or interfering with peptide binding. We chose a RAMP-CLR connection topology and a flexible ten-residue length (Gly-Ser)5 tether. The tethered fusion protein constructs consisted of MBP followed by the RAMP ECD 3-helix bundle (residues 24-111 for RAMP1 or 55-140 for RAMP2) followed by a ten-residue linker and the CLR ECD (residues 29-144) with a C-terminal H6 tag. The N-terminal signal peptides of the RAMPs and CLR were omitted. In the course of plasmid construction we also obtained an adventitious deletion mutant of the AM receptor ECD fusion construct with a shortened (GS)3 linker.
Figure 1.
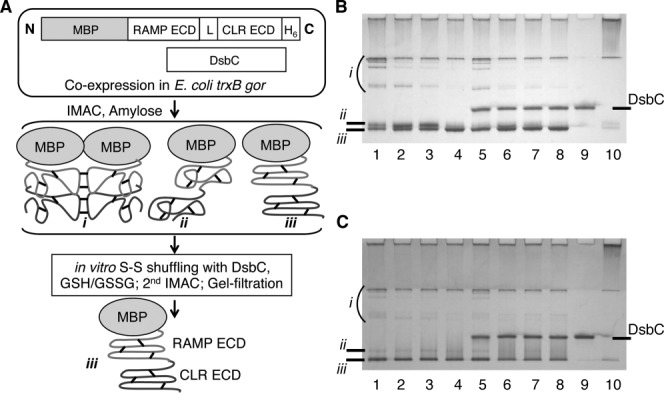
Overview of protein expression and purification methodology and native gel analysis of in vitro disulfide shuffling. A: Schematic outline of the expression and purification methodology for production of tethered MBP-RAMP ECD-CLR ECD fusion proteins. “L” represents the (GS)X linker. Species i, ii, and iii represent intermolecular disulfide-linked multimers, monomeric fusion protein with misfolded ECDs, and monomeric fusion protein with properly folded ECDs, respectively. Short black lines indicate disulfide bonds. B: Native gel analysis of small-scale disulfide shuffling reactions for the MBP-RAMP1 ECD-(GS)5-CLR ECD fusion protein purified by IMAC and Amylose affinity chromatography. The fusion protein was incubated overnight at 20°C under the following conditions: 1, buffer alone; 2, 1 mM GSH, 5 mM GSSG; 3, 1 mM each GSH and GSSG; 4, 5 mM GSH, 1 mM GSSG; 5, DsbC (no redox reagents); 6, DsbC, 1 mM GSH, 5 mM GSSG; 7, DsbC, 1 mM each GSH and GSSG; 8, DsbC, 5 mM GSH, 1 mM GSSG; 9, as in Lane 7, but no fusion protein; 10, 20 mM DTT. Approximately 5 μg of protein was loaded in each lane of a 12% gel and the gel was stained with coomassie brilliant blue. C: Native gel analysis of in vitro disulfide shuffling for the MBP-RAMP2 ECD-(GS)3-CLR ECD fusion protein as in Panel B.
The tethered CGRP and AM receptor ECD fusion proteins were coexpressed with DsbC in the oxidizing cytoplasm of E. coli trxB gor and purified by IMAC and amylose affinity chromatography under native conditions [Fig. 1(A)]. Analysis of the affinity-purified samples by native gel electrophoresis revealed heterogenous populations consisting of disulfide-linked multimers (species i), misfolded monomer (species ii), and correctly folded monomer (species iii) [lane 1 of Fig. 1(B,C)]. Our assignment of the molecular nature of the species is based on our previous experience applying this methodology to several other class B GPCR ECDs including a noncovalent MBP-CLR ECD:RAMP2 ECD complex.29–34 Several in vitro disulfide shuffling conditions with various GSH:GSSG ratios in the absence and presence of purified DsbC were tested for their ability to decrease the presence of species i and ii and increase the presence of species iii [lanes 2-8 of Fig. 1(B,C)]. Overnight incubation of the tethered CGRP receptor ECD fusion protein in 5 mM GSH, 1 mM GSSG in the absence [Fig. 1(B), lane 4] or presence [Fig. 1(B), lane 8] of DsbC significantly reduced species i and ii and increased species iii. Inclusion of DsbC yielded a slightly more defined species iii band on the native gel, so this condition was chosen as optimal. For the tethered AM receptor ECD fusion protein, overnight incubation in 1 mM GSH, 1 mM GSSG in the presence of DsbC was chosen as optimal [Fig. 1(C), lane 7], although the effects were not as dramatic as for the CGRP receptor ECD fusion. The AM receptor ECD fusion protein containing the (GS)3 linker was used for the small-scale disulfide shuffling experiments [Fig. 1(C)]. All experiments hereafter utilized the AM receptor ECD construct with a ten residue-length linker for comparison with the CGRP receptor ECD fusion protein.
For large-scale purifications of the tethered ECD fusion proteins we started with 6L bacterial cultures. Each protein was purified by IMAC and Amylose affinity chromatography and subjected to overnight in vitro disulfide shuffling in the presence of DsbC using the redox buffer conditions identified above. After removal of untagged DsbC by a second IMAC step, the tethered fusion proteins were further purified by gel-filtration chromatography, which resulted in homogeneous, highly purified samples for both the CGRP receptor ECD [Fig. 2(A)] and AM receptor ECD [Fig. 2(B)] fusion proteins. The oligomeric states of the tethered fusion proteins were determined by estimating their molecular masses from their gel-filtration elution volumes [Fig. 2(C)]. The CGRP receptor ECD fusion eluted at a volume corresponding to a molecular mass of ∼69 kDa, consistent with a monomeric fusion protein (calculated monomer MM = 66,293 Da). In contrast, the AM receptor ECD fusion eluted at a volume corresponding to a molecular mass of ∼128 kDa, consistent with a dimeric fusion protein (calculated monomer MM = 66,367 Da). The final yields of the CGRP and AM receptor ECD fusion proteins were 29 mg and 32 mg, respectively.
Figure 2.
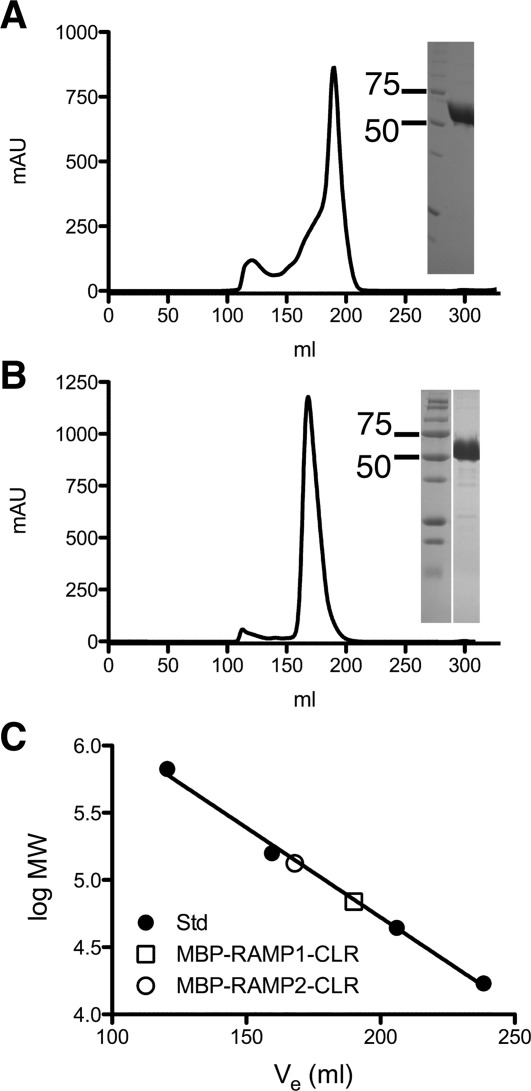
Gel-filtration purification of the MBP-RAMP ECD-CLR ECD fusion proteins after disulfide shuffling. A: Elution profile for the MBP-RAMP1 ECD-(GS)5-CLR ECD fusion protein from a 320 mL bed volume Superdex200 column. The inset shows a coomassie blue-stained, nonreducing SDS-PAGE analysis of the peak fraction compared to molecular weight markers (in kDa). B: As in Panel A, except for the MBP-RAMP2 ECD-(GS)2-RS-(GS)2-CLR ECD fusion protein. C: Molecular mass estimation of the fusion proteins from a standard curve obtained with Bio-Rad gel-filtration molecular mass standards.
Isothermal titration calorimetry characterization of CGRP and AM peptide binding
The peptide binding ability of the tethered CGRP receptor ECD fusion protein was assessed by isothermal titration calorimetry (ITC). The sample cell contained the CGRP receptor ECD fusion protein (44 μM) and synthetic CGRP(8-37)NH2 peptide (450 μM) was injected from the syringe. CGRP bound the tethered CGRP receptor ECD fusion protein with a KD of 57 μM and stoichiometry 0.85 (Fig. 3). More accurate ITC data would be obtained using a protein concentration ∼ 5x higher than the KD, but this was not possible because of CGRP peptide solubility limitations. We also assessed the binding of AM(22-52)NH2 to the tethered AM receptor ECD fusion protein in several ITC experiments using protein at 40–80 μM and AM at 400–800 μM. The data reproducibly indicated binding of the peptide with exothermic heats, but the data were not of sufficient quality to determine the thermodynamic parameters (data not shown). An alternative binding assay format indicated that the tethered AM receptor ECD fusion protein was functional (see below).
Figure 3.
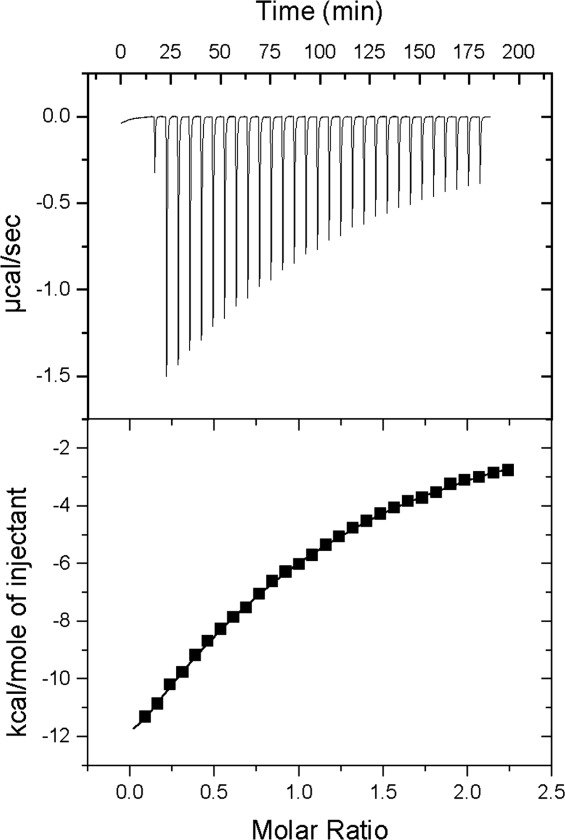
Isothermal titration calorimetry analysis of the binding of CGRP(8–37)NH2 to the MBP-RAMP1 ECD-(GS)5-CLR ECD-H6 fusion protein at 27°C. The sample cell contained 44 μM protein and the syringe contained 450 μM peptide. (Upper) Raw data. (Lower) Integrated heats/mol of peptide vs. the molar ratio of peptide:protein. Nonlinear regression fitting to a single site model yielded a binding constant KA = 1.74e4 M−1 (KD = 57 μM); number of binding sites, n = 0.85; ΔH = −29,830 calories per mol; ΔS = −80.0 calories per mol per degree.
AlphaScreen luminescent proximity assay characterization of peptide binding
The tethered CGRP and AM receptor ECD fusion proteins were further characterized using a luminescent AlphaScreen peptide binding assay. In this bead-based proximity assay excitation of “donor” beads generates singlet oxygen molecules that activate “acceptor” beads to emit light when the beads are brought into proximity by a molecular interaction. An advantage of this assay format over ITC is the small sample requirements, which is beneficial when working with costly synthetic peptides. In addition, multivalency resulting from the two-bead assay format and the amplification properties of the technology enable the detection of low affinity interactions even with low concentrations of protein and peptide. N-terminally biotinylated peptides were attached to the surface of streptavidin-coated donor beads and the tethered CGRP or AM receptor ECD fusion proteins were attached to the surface of Ni-chelate-coated acceptor beads via their C-terminal H6-tags. For the biotinylated peptides, the N-terminal portions containing the disulfide bond structure was omitted because this region is not involved in ECD binding. In a saturation binding assay format, we detected binding of both biotin-CGRP(8-37)NH2 and biotin-AM(19-52)NH2 to the CGRP receptor ECD fusion protein, but CGRP binding was stronger [Fig. 4(A)]. These results indicated that the tethered CGRP receptor ECD protein was selective for CGRP over AM. In contrast, the tethered AM receptor ECD fusion protein exhibited specific AM binding [Fig. 4(B)]. Neither fusion protein exhibited detectable binding of biotin-salmon calcitonin or biotin-rat amylin.
Figure 4.
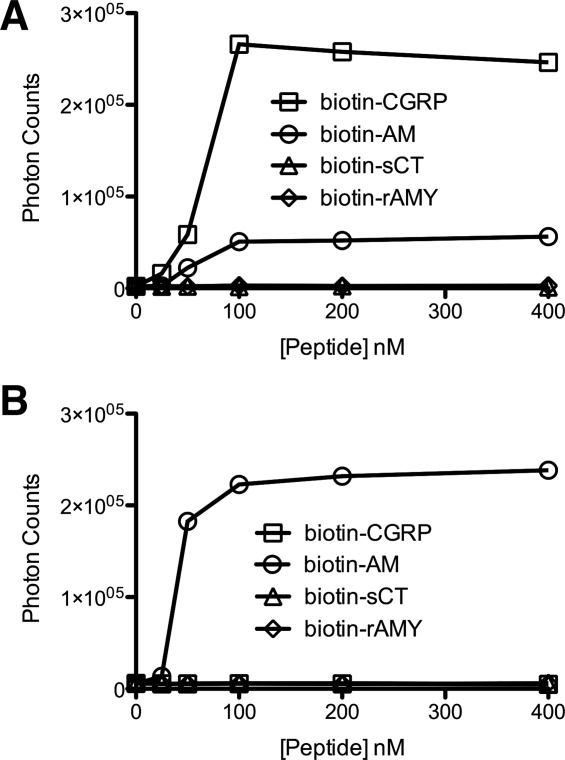
AlphaScreen luminescent peptide binding assay. A: Saturation binding for the MBP-RAMP1 ECD-(GS)5-CLR ECD fusion protein (300 nM) with the indicated biotinylated peptides. B: As in panel A, except for the MBP-RAMP2 ECD-(GS)2-RS-(GS)2-CLR ECD fusion protein. Data shown are the average of duplicate samples. Error bars are smaller than the symbols.
Peptide binding selectivity/specificity was further examined in a competition binding assay format (Fig. 5). Unlabeled CGRP(8-37)NH2 displaced the interaction of biotin-CGRP and the tethered CGRP receptor ECD fusion protein with an IC50 value of 24 μM [Fig. 5(A)], which is close to the KD determined by ITC. The free acid form of CGRP exhibited diminished, but still detectable binding [Fig. 5(A)], which indicated that the C-terminal amide group contributes to, but is not essential for receptor binding. AM(22-52)NH2 also bound the tethered CGRP receptor ECD fusion, but with a diminished IC50 compared to CGRP(8-37)NH2 [Fig. 5(A)], again indicating selectivity of the receptor for CGRP over AM. Unlabeled AM(22-52)NH2 displaced the interaction of biotin-AM and the tethered AM receptor ECD fusion protein with an IC50 value of 24 μM [Fig. 5(B)]. The free acid form of AM did not bind to the tethered AM receptor ECD fusion [Fig. 5(B)], which indicated that the AM C-terminal amide group is essential for receptor binding. CGRP(8-37)NH2 did not bind the tethered AM receptor ECD fusion in the competition assay [Fig. 5(B)], again indicating specificity for AM.
Figure 5.
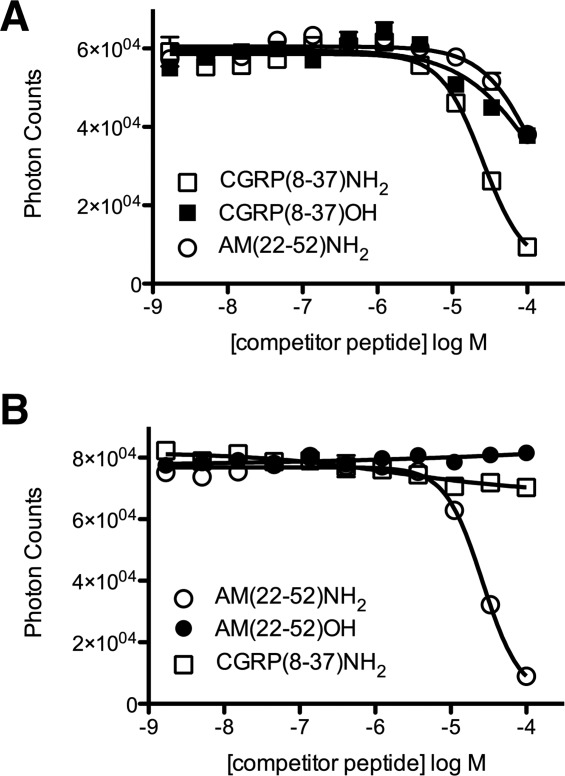
AlphaScreen luminescent competition binding assay to assess peptide affinity and specificity. A: Competition binding with Biotin-CGRP (50 nM) and MBP-RAMP1 ECD-(GS)5-CLR ECD-H6 (50 nM) in the presence of the indicated unlabeled peptides. B: As in planel B, except for Biotin-AM (100 nM) and MBP-RAMP2 ECD-(GS)2-RS-(GS)2-CLR ECD-H6 (100 nM). Data shown are the average of duplicate samples. Error bars are smaller than the symbols for most data points.
Previous studies indicated that the C-terminal (27-37) fragment of CGRP retained the ability to bind the CGRP receptor, albeit with low affinity.22,26,36,37 It was recently demonstrated that the C-terminal eight amino acids of AM, in the context of the 22-52 fragment, constituted the primary AM receptor ECD binding epitope.27 We examined the ability of N-terminally truncated CGRP and AM peptides of varying lengths to bind their respective tethered receptor ECD fusion proteins in the competition AlphaScreen assay. In agreement with previous studies, we observed that CGRP peptides as short as (27-37) retained the ability to bind the tethered CGRP receptor ECD fusion protein and that the affinity of the (27-37) fragment was only slightly reduced compared to the (8-37) fragment (Table I). The AM(37-52) fragment retained the ability to bind the tethered AM receptor ECD fusion with similar affinity as the (22-52) fragment, but the (39-52) fragment exhibited significantly diminished binding and the (41-52) fragment did not exhibit any detectable binding (Table I). These results indicated that the C-terminal fifteen or sixteen residues of AM are minimally required for receptor ECD binding.
Table I.
AlphaScreen Competitiona Assays for Truncated CGRP and AM Peptides
| Competitor peptide | IC50 (μM) | 95% confidence intervals (μM) |
|---|---|---|
| MBP-RAMP1-CLR fusion | ||
| CGRP(8-37)NH2 | 24 | 17–35 |
| CGRP(11-37)NH2 | 14 | 9–21 |
| CGRP(15-37)NH2 | 37 | 22–64 |
| CGRP(23-37)NH2 | 48 | 33–70 |
| CGRP(25-37)NH2 | 49 | 29–81 |
| CGRP(27-37)NH2 | 45 | 28–71 |
| MBP-RAMP2-CLR fusion | ||
| AM(22-52)NH2 | 24 | 17–33 |
| AM(25-52)NH2 | 12 | 9–15 |
| AM(29-52)NH2 | 30 | 25–35 |
| AM(34-52)NH2 | 53 | 45–61 |
| AM(37-52)NH2 | 35 | 31–39 |
| AM(39-52)NH2 | >100b | |
| AM(41-52)NH2 | NBc |
The competitor concentration range was 1.7 nM to 100 μM. Competition curves were fit to data values that were the average of duplicate reactions.
Approximately 30% signal inhibition was observed at 100 μM competitor.
No binding detected.
Binding of alanine-scan CGRP and AM peptides
We sought to identify individual CGRP and AM amino acid side chains that were important for receptor ECD binding by examining the ability of alanine-scan peptides to bind the tethered receptor ECD fusion proteins in the competition AlphaScreen assay. All non-Gly or -Ala residues in CGRP and AM were individually substituted with Ala in the context of the CGRP(23-37)NH2 and AM(37-52)NH2 backbones and the ability of a single high concentration (100 μM) of the peptides to inhibit the binding signal as compared to the wild-type peptides was assessed (Fig. 6). Substitution of CGRP residues T30, V32, or F37 with alanine resulted in essentially a complete loss of CGRP receptor ECD binding and substitution of residues F27, V28, P29, N31, or K35 diminished receptor binding to a lesser, but still significant degree [Fig. 6(A)]. Substitution of AM residues P43, K46, I47, or Y52 with alanine abrogated AM receptor ECD binding, whereas substitution of residues S48, P49, or Q50 resulted in somewhat diminished binding [Fig. 6(B)]. The ALA-scan results are summarized with an alignment of the CGRP and AM peptides [Fig. 6(C)].
Figure 6.
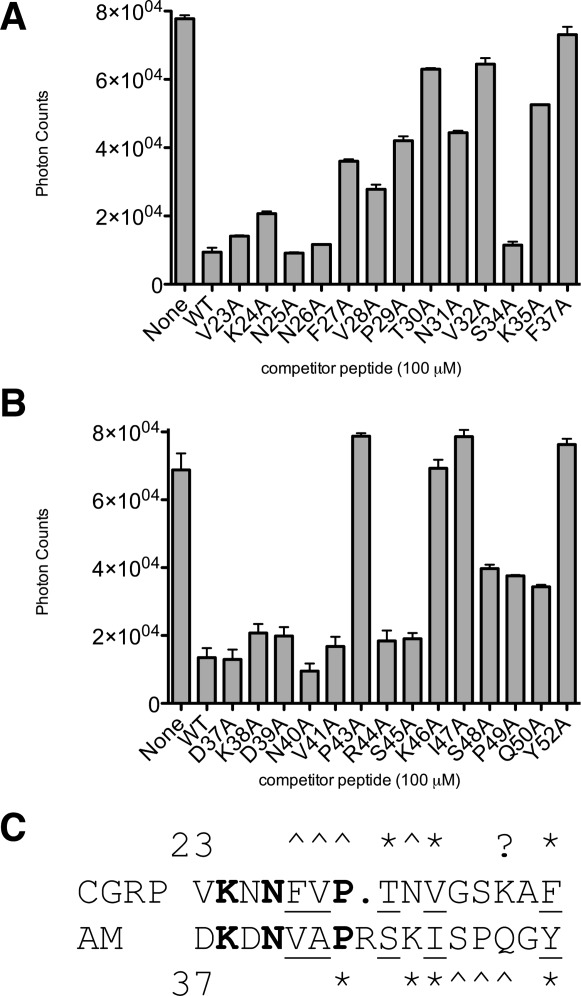
AlphaScreen luminescent competition binding assay with ALA-scan CGRP and AM peptides. A: Competition binding with Biotin-CGRP (50 nM) and MBP-RAMP1 ECD-(GS)5-CLR ECD-H6 (50 nM) in the presence of the indicated unlabeled ALA-scan peptides in the CGRP(23-37)NH2 backbone. B: As in Panel B, except for Biotin-AM (100 nM) and MBP-RAMP2 ECD-(GS)2-RS-(GS)2-CLR ECD-H6 (100 nM) in the presence of the indicated unlabeled ALA-scan peptides in the AM(37-52)NH2 backbone. Data shown are the average of duplicate samples. C: Amino acid sequence alignment of CGRP(23-37) and AM(37-52) summarizing the ALA-scan mutagenesis results. Resides that are identical between CGRP and AM are shown in bold and conservative substitutions are underlined. Residues where mutation to ALA most significantly diminished binding are highlighted with a * and residues where mutation to ALA diminished binding to a lesser, but still significant degree are highlighted with a ∧. The ? denotes that our finding of CGRP K35A diminishing binding is at odds with previous findings.
Discussion
The CGRP and AM receptors are of considerable interest as drug targets. Here, we presented novel methodology for cost- and time-efficient bacterial production of mg quantities of recombinant CGRP and AM receptor ECD complexes as tethered fusion proteins. Our rationale for covalent linkage of the two ECDs was that the tether would ensure stability of the complex in the absence of the membrane portions of the receptors and enforce 1:1 RAMP:CLR stoichiometry within the fusion protein. The tethered fusion proteins may prove useful for structural studies of the receptors and aid screening for small molecule drugs. Our DsbC-assisted disulfide shuffling methodology is distinct from the traditional denaturant-based refolding protocols employed by other groups to produce CLR-RAMP ECD complexes.17,27,37 We previously employed our methodology to produce several other class B GPCR ECDs with 3 disulfide bonds.30–34 Here, the methodology was successful for proteins with up to 6 disulfide bonds, which suggests that it may be more broadly applicable to various disulfide bond-containing proteins.
The tethered fusion proteins were properly folded as evidenced by their selective CGRP and AM binding. The tethered CGRP receptor ECD fusion bound CGRP with a KD of 57 μM by ITC and an IC50 of 24 μM by AlphaScreen competition assay, which agreed well with the KD of 24 μM previously reported for the binding of CGRP to a noncovalent RAMP1:CLR ECD complex as measured by surface plasmon resonance (SPR).37 These results suggested that the MBP tag and covalent tether did not alter CGRP binding. The tethered AM receptor ECD fusion bound AM with an IC50 of 24 μM by AlphaScreen competition assay, which agreed well with our previous determination of a 10 μM IC50 for AM binding to a noncovalent MBP-CLR:RAMP2 ECD complex,29 and with the 5 μM KD reported for the binding of AM to a noncovalent RAMP2:CLR ECD complex as measured by ITC.27 Our results disagreed with the 70 nM KD reported for the binding of AM to a noncovalent RAMP2 ECD:CLR ECD complex as measured by SPR.17 The basis for this discrepancy is unclear, but it seems unlikely that the ECD complex binds AM with nM affinity because several other class B GPCR ECD-peptide pairs exhibit μM binding affinities.30–33,35 Although we were unable to obtain reliable ITC data for the tethered AM receptor ECD fusion, our AlphaScreen results nonetheless suggested that the MBP tag and covalent tether did not significantly alter AM binding.
We used the tethered fusion proteins to identify critical determinants of CGRP and AM binding to their receptor ECD complexes. Consistent with previous studies,22,26,37 we observed that the minimal CGRP(27-37)NH2 fragment retained essentially wild-type binding. Our ALA-scan experiments were in agreement with those of other groups22,26 that CGRP residues T30, V32, and F37 are most critical for ECD binding. One discrepancy is our result that the CGRP K35A mutation significantly reduced binding, which is in contrast to previous findings. The basis for this discrepancy is unclear. We identified the AM(37-52)NH2 fragment as the minimal ECD binding region, even though our ALA-scan experiments indicated that the side chains of residues 37-41 were not important for ECD binding. These residues may indirectly contribute to ECD binding via effects on AM structure and stability. Our AM ALA-scan results indicated that AM residues P43, K46, I47, and Y52 are most critical for ECD binding and were generally in agreement with those of Watkins et al., although they did not test P43.27 One discrepancy is their finding that the AM R44A mutation diminished binding, which we did not observe. However, our single point assay may have missed subtle effects of the R44A mutation. Some of the amino side chains of CGRP and AM that are critical for ECD complex binding are similar between the two peptides, such as CGRP V32, F37 and AM I47, Y52, which probably reflects similar receptor interactions for these residues [Fig. 6(C)]. In contrast, other residues that are critical for ECD complex binding differ between the two peptides, including CGRP T30 and AM K46, suggesting that these residues may contribute to selective binding of their respective receptor ECD complexes [Fig. 6(C)]. Another notable difference between the two peptides is that the AM amide group is essential for ECD binding, whereas the CGRP amide group is not essential. The ALA-scan and C-terminal amide mutation results thus highlight important differences in how CGRP and AM bind to their receptor ECD complexes. Crystal structures of peptide bound complexes will be required to fully understand how selectivity arises.
It is interesting to consider the oligomeric states of the tethered fusion proteins. The tethered CGRP receptor ECD fusion protein was a monomer, which is consistent with the CGRP receptor functioning as 1:1 heterodimer, at least with respect to CGRP binding. Nonetheless, in the context of the full-length receptors in the cell membrane, other stoichiometries are possible.28 The 1:1 RAMP:CLR heterodimer may bind CGRP, but additional CLR subunits could be involved in signaling, especially in light of the many examples of GPCR oligomerization.38 The AM receptor stoichiometry may be more complex. The tethered AM receptor ECD fusion protein was dimeric, which is consistent with the 2:2 dimer of heterodimers observed by Kusano et al.17 Watkins et al. proposed a 2:1 CLR:RAMP2 stoichiometry for their purified ECD complex based on their data that indicated a 1.5:1 molar ratio of the two ECDs.27 We have noticed that under certain chromatographic conditions our noncovalent MBP-CLR ECD:RAMP2 ECD complex29 exhibited instability and appeared to dissociate (Moad and Pioszak, unpublished data). Complex instability could possibly account for the non-integer molar ratio obtained by Watkins et al. Nonetheless, we cannot formally rule out a 2:1 CLR:RAMP2 stoichiometry as the functional form in our dimeric tethered fusion protein because one RAMP2 ECD could be making productive interactions in the dimer while the other RAMP2 ECD is simply “along for the ride”. We also cannot rule out a 1:1 stoichiometry for the AM receptor because under the low protein concentration conditions of our AlphaScreen assays the tethered AM receptor ECD fusion could be monomeric as observed for the noncovalent CLR:RAMP2 ECD complex of Kusano et al.17 On the other hand, immobilization of the tethered fusion protein on the AlphaScreen bead surface might enable dimer formation even at low protein concentration. It is unclear if our inability to obtain reliable ITC data for the tethered AM receptor ECD fusion was due to oligomeric state differences at high protein concentration or an unknown technical artifact. Moreover, it is unclear if higher order CLR:RAMP2 species form with the intact proteins in cells. Ultimately, additional approaches will be required to determine the functional oligomeric state(s) of the AM receptor.
Materials and Methods
Plasmid construction
Construction of E. coli expression plasmids that encode maltose binding protein (MBP)-RAMP ECD-CLR ECD fusion proteins with the ECDs connected by a flexible (Gly-Ser)X linker and containing a C-terminal (His)6 tag utilized standard PCR- and restriction enzyme-based cloning methods. Human RAMP1, RAMP2, and CLR cDNAs were obtained from the Missouri S&T cDNA resource center. The RAMP and CLR ECDs were PCR-amplified with addition of restriction endonuclease sites and linker sequences via the primers. PCR primer sequences are available from the authors upon request. The RAMP1 ECD was amplified as an EcoRI-BamHI fragment including ½ of the GS linker sequence at the downstream end. The RAMP2 ECD was amplified similarly, except that BglII was used instead of BamHI because RAMP2 contains a BamHI site. The CLR ECD was amplified as a BamHI-NotI fragment including ½ the GS linker at the upstream end and an H6-tag and stop codon at the downstream end. The digested RAMP ECD (EcoRI-BamHI or BglII) and CLR ECD (BamHI-NotI) fragments were joined in a 3-piece ligation with an EcoRI-NotI-digested, previously described,31 pETDuet1-based plasmid engineered for cytoplasmic co-expression of MBP fusion proteins with the bacterial disulfide isomerase DsbC. The 1st multiple cloning site of this vector encodes MBP ending with the sequence NAAAEF and the 2nd multiple cloning site encodes untagged DsbC. Joining BglII-BamHI for the RAMP2 construct resulted in an altered GS linker sequence as noted below. The plasmids constructed were (residue numbers used for the ECDs are indicated for RAMPs and CLR): pHH033, MBP-RAMP1.24–111-(GS)5-CLR.29–144-H6; pHH034, MBP-RAMP2.55–140-(GS)2-RS-(GS)2-CLR.29-144-H6. In the course of constructing pHH034, we also obtained an adventitious deletion mutant, pHH041, which lacked the BglII-BamHI junction and encoded expression of MBP-RAMP2.55-140-(GS)3-CLR.29-144-H6. The coding regions of the plasmids were verified by automated DNA sequencing at the OUHSC Microgen core facility.
Protein expression and purification
E. coli Origami B DE3 cells (Novagen), which contain the trxB gor mutations, were transformed with the expression plasmids. Protein expression was performed in baffled shake flasks with a culture volume of 6 L. The cultures were grown in LB lennox broth supplemented with 50 μg/mL ampicillin at 37°C to mid-log phase, the temperature was reduced to 16°C, and 0.4 mM IPTG was added to induce the cultures overnight. Cells were harvested by centrifugation and stored at −80°C. The purification protocols for the MBP-RAMP1 ECD-CLR ECD-H6 and MBP-RAMP2 ECD-CLR ECD-H6 fusion proteins were identical, except where noted below, and they were similar to our previously reported protocol for a noncovalent complex of an MBP-CLR ECD fusion protein and the RAMP2 ECD.29 All steps were performed at 4°C unless otherwise noted and the column chromatography steps utilized an AKTA purifier (GE Healthcare). Buffers for cell resuspension, IMAC and Amylose chromatography, and IMAC and Amylose procedures were as described.29 The harvested cells were resuspended and lysed by sonication, the lysate was clarified by centrifugation, and the soluble fusion protein was purified from the supernatant by IMAC and Amylose affinity chromatography under native conditions and then subjected to in vitro disulfide shuffling overnight at 20°C in a glutathione redox buffer in the presence of purified DsbC. The fusion proteins eluted from Amylose resin in a buffer (50 mM Tris-HCl, pH 8.0, 5% (vol/vol) glycerol, 150 mM NaCl, ∼3 mM Maltose) suitable for in vitro disulfide shuffling. DsbC was purified as previously described31 and added at a 0.5:1 molar ratio (DsbC dimer:fusion protein monomer). Optimal redox buffer conditions were determined in small-scale disulfide shuffling reactions that tested various GSH:GSSG ratios. The redox conditions used for large-scale purifications were 5 mM GSH, 1 mM GSSG for MBP-RAMP1-CLR and 1 mM GSH, 1 mM GSSG for MBP-RAMP2-CLR. After disulfide shuffling, IMAC was used to remove the untagged DsbC and the eluted fusion protein was concentrated and applied to a Superdex200 HR (GE Healthcare) gel-filtration column in a buffer of 50 mM Tris-HCl, pH 7.5, 10% (vol/vol) glycerol, 150 mM NaCl. Peak fractions from gel-filtration were pooled, dialyzed to storage buffer (50 mM Tris-HCl, pH 7.5, 50% (vol/vol) glycerol, 150 mM NaCl), and stored at −80°C. Tethered fusion protein concentrations were determined by Bradford assay with a BSA standard curve and are stated in terms of the monomer.
Native gel electrophoresis
Nondenaturing polyacrylamide gel electrophoresis for monitoring protein folding states was performed as described31 except that the resolving gel was pH 7.5, the stacking gel was pH 6.8, and Tris-Borate pH 7.5 running buffer was used instead of Tris-Glycine pH 9.0.
Peptides
Custom synthesized and HPLC-purified peptides were purchased from RS Synthesis (Louisville, KY). The lyophilized peptides were reconstituted in sterile ultrapure water and their concentrations were determined by UV absorbance at 280 nm using the extinction coefficients calculated based on Tyr residues. A single Tyr was added to the N-terminus of CGRP to facilitate its quantitation. The sequences of the N-terminally biotinylated, C-terminally amidated peptides used for AlphaScreen assays were as follows:
B-αCGRP(8-37)NH2: Biotin-GGYVTHRLAGLLS RSGGVVKNNFVPTNVGSKAF-NH2
B-AM(19-52)NH2[C21A]: Biotin-GTATVQKLAH QIYQFTDKDKDNVAPRSKISPQGY-NH2
B-sCT(1-32)NH2[C1A/C7A]: Biotin-ASNLSTAVL GKLSQELHKLQTYPRTNTGSGTP-NH2
B-rAMY(5-37)NH2[C7A]: Biotin-ATAATQRLAN FLVRSSNNLGPVLPPTNVGSNTY-NH2
The biotinylated peptides were designed to lack the N-terminal disulfide bond structure because this region is unnecessary for ECD binding and because inclusion of the disulfide bond increases the cost of the synthetic peptides and could introduce steric constraints that would affect the accessibility of the peptide at the surface of the streptavidin-coated bead. The sequences of peptides used for ITC and AlphaScreen competition assays were as follows:
αCGRP(8-37)NH2: YVTHRLAGLLSRSGGVKN NFVPTNVGSKAF-NH2
AM(22-52)NH2: TVQKLAHQIYQFTDKDKDN VAPRSKISPQGY-NH2
The free acid forms of CGRP and AM contained a C-terminal carboxylate instead of the amide. Truncated forms of CGRP and AM included the residue numbers indicated. Alanine-scan peptides contained the indicated single Alanine substitution in an otherwise wild-type CGRP(23-37)NH2 or AM(37-52)NH2 backbone.
Isothermal titration calorimetry
ITC experiments were performed with a Microcal VP-ITC calorimeter. Protein and peptide samples were dialyzed overnight at 4°C against 2 L of 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA to ensure identical buffer compositions. Peptide and protein concentrations were determined by UV absorbance at 280 nm. The protein concentrations were also checked by Bradford assay, which gave results nearly identical to the UV absorbance. The samples were degassed and thermostatted to 25°C for 10 min before loading the sample cell with protein, typically at 40–80 μM, and the syringe with peptide at a concentration ∼10-fold higher than the protein concentration. Titrations were carried out at 27°C with ∼30 injections of 10 μL spaced every 350 s and a stirring speed of 300 rpm. Control titrations of peptide injected into buffer provided the heats of dilution, which were subtracted from the peptide into protein data. The data were fit to a single site binding model using nonlinear regression in the Microcal Origin software.
AlphaScreen luminescent proximity peptide binding assay
The reaction mixtures contained 50 mM MOPS pH 7.4, 150 mM NaCl, 7 mg/mL fatty acid-free BSA (PAA Laboratories), 15 μg/mL each (for saturation binding) or 10 μg/mL each (for competition binding) streptavidin-coated donor and nickel-chelate-coated acceptor AlphaScreen beads (Perkin-Elmer), and peptide and protein as indicated. The reactions were prepared in the dark under green light and incubated at room temperature. For saturation binding assays, the MBP-RAMP1-CLR or MBP-RAMP2-CLR fusion proteins (300 nM) were incubated with the indicated concentrations of biotinylated peptides for 5 h before reading. Competition assays were performed in a pre-coupling format. The biotinylated AM or CGRP and the MBP-RAMP1-CLR-H6 or MBP-RAMP2-CLR-H6 fusion proteins were separately mixed with streptavidin donor and nickel-chelate acceptor beads, respectively, and incubated for 1 h to allow attachment of the peptide and protein to their respective beads before initiation of the binding reaction. The pre-coupling reactions were then mixed together and unlabeled competitor peptides were added as indicated and the reactions were incubated an additional 5 h to reach equilibrium. The final concentrations of biotin-CGRP and MBP-RAMP1-CLR-H6 were 50 nM each and biotin-AM and MBP-RAMP2-CLR-H6 were 100 nM each. Photon counts were recorded in 384-well white optiplates (Greiner) with a PolarStar Omega plate reader using filters for AlphaScreen (BMG Labtech). GraphPad Prism 5.0 (GraphPad Software, San Diego) was used for nonlinear regression fitting of the competition data to a 4-parameter logistic equation (variable-slope dose-response inhibition) to determine IC50 values. Control experiments that tested the ability of the unlabeled peptides to inhibit an assay in which a Biotin-(Gly)6-(His)6 peptide was used to bring the beads together showed no nonspecific inhibition at competitor concentrations up to 100 μM, but concentrations higher than 100 μM began to exhibit nonspecific inhibition and were therefore not used (data not shown).
Acknowledgments
The authors thank Alisha Chitrakar for assistance with peptide resuspension and quantitation and Dr. Karla Rodgers for assistance with the Microcal VP-ITC calorimeter.
Glossary
- AM
adrenomedullin
- BSA
bovine serum albumin
- CLR
calcitonin receptor-like receptor
- CTR
calcitonin receptor
- CGRP
calcitonin gene-related peptide
- DsbC
E. coli disulfide bond isomerase
- DTT
dithiothreitol
- ECD
extracellular domain
- GPCR
G protein-coupled receptor
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- IMAC
immobilized metal affinity chromatography
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- ITC
isothermal titration calorimetry MBP, maltose binding protein
- PCR
polymerase chain reaction
- RAMP
receptor activity modifying protein
- rAMY
rat amylin
- sCT
salmon calcitonin
References
- 1.Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, Muff R, Fischer JA, Foord SM. International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev. 2002;54:233–246. doi: 10.1124/pr.54.2.233. [DOI] [PubMed] [Google Scholar]
- 2.Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84:903–934. doi: 10.1152/physrev.00037.2003. [DOI] [PubMed] [Google Scholar]
- 3.Caron KM, Smithies O. Extreme hydrops fetalis and cardiovascular abnormalities in mice lacking a functional Adrenomedullin gene. Proc Natl Acad Sci USA. 2001;98:615–619. doi: 10.1073/pnas.021548898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karpinich NO, Hoopes SL, Kechele DO, Lenhart PM, Caron KM. Adrenomedullin function in vascular endothelial cells: insights from genetic mouse models. Curr Hyperten Rev. 2011;7:228–239. doi: 10.2174/157340211799304761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamid SA, Baxter GF. A critical cytoprotective role of endogenous adrenomedullin in acute myocardial infarction. J Mol Cell Cardiol. 2006;41:360–363. doi: 10.1016/j.yjmcc.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 6.Nikitenko LL, Fox SB, Kehoe S, Rees MC, Bicknell R. Adrenomedullin and tumour angiogenesis. Br J Cancer. 2006;94:1–7. doi: 10.1038/sj.bjc.6602832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toda M, Suzuki T, Hosono K, Hayashi I, Hashiba S, Onuma Y, Amano H, Kurihara Y, Kurihara H, Okamoto H, Hoka S, Majima M. Neuronal system-dependent facilitation of tumor angiogenesis and tumor growth by calcitonin gene-related peptide. Proc Natl Acad Sci USA. 2008;105:13550–13555. doi: 10.1073/pnas.0800767105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gibbons C, Dackor R, Dunworth W, Fritz-Six K, Caron KM. Receptor activity-modifying proteins: RAMPing up adrenomedullin signaling. Mol Endocrinol. 2007;21:783–796. doi: 10.1210/me.2006-0156. [DOI] [PubMed] [Google Scholar]
- 9.Recober A, Russo AF. Calcitonin gene-related peptide: an update on the biology. Curr Opin Neurol. 2009;22:241–246. doi: 10.1097/wco.0b013e32832b2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sexton PM, Poyner DR, Simms J, Christopoulos A, Hay DL. RAMPs as drug targets. Advan Exp Med Biol. 2012;744:61–74. doi: 10.1007/978-1-4614-2364-5_6. [DOI] [PubMed] [Google Scholar]
- 11.McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, Solari R, Lee MG, Foord SM. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature. 1998;393:333–339. doi: 10.1038/30666. [DOI] [PubMed] [Google Scholar]
- 12.Christopoulos G, Perry KJ, Morfis M, Tilakaratne N, Gao Y, Fraser NJ, Main MJ, Foord SM, Sexton PM. Multiple amylin receptors arise from receptor activity-modifying protein interaction with the calcitonin receptor gene product. Mol Pharmacol. 1999;56:235–242. doi: 10.1124/mol.56.1.235. [DOI] [PubMed] [Google Scholar]
- 13.Muff R, Buhlmann N, Fischer JA, Born W. An amylin receptor is revealed following co-transfection of a calcitonin receptor with receptor activity modifying proteins-1 or -3. Endocrinology. 1999;140:2924–2927. doi: 10.1210/endo.140.6.6930. [DOI] [PubMed] [Google Scholar]
- 14.Tilakaratne N, Christopoulos G, Zumpe ET, Foord SM, Sexton PM. Amylin receptor phenotypes derived from human calcitonin receptor/RAMP coexpression exhibit pharmacological differences dependent on receptor isoform and host cell environment. J Pharmacol Exp Ther. 2000;294:61–72. [PubMed] [Google Scholar]
- 15.Kusano S, Kukimoto-Niino M, Akasaka R, Toyama M, Terada T, Shirouzu M, Shindo T, Yokoyama S. Crystal structure of the human receptor activity-modifying protein 1 extracellular domain. Protein Sci. 2008;17:1907–1914. doi: 10.1110/ps.036012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ter Haar E, Koth CM, Abdul-Manan N, Swenson L, Coll JT, Lippke JA, Lepre CA, Garcia-Guzman M, Moore JM. Crystal structure of the ectodomain complex of the CGRP receptor, a class-B GPCR, reveals the site of drug antagonism. Structure. 2010;18:1083–1093. doi: 10.1016/j.str.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Kusano S, Kukimoto-Niino M, Hino N, Ohsawa N, Okuda K, Sakamoto K, Shirouzu M, Shindo T, Yokoyama S. Structural basis for extracellular interactions between calcitonin receptor-like receptor and receptor activity-modifying protein 2 for adrenomedullin-specific binding. Protein Sci. 2012;21:199–210. doi: 10.1002/pro.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- 19.Barwell J, Gingell JJ, Watkins HA, Archbold JK, Poyner DR, Hay DL. Calcitonin and calcitonin receptor-like receptors: common themes with family B GPCRs? Br J Pharmacol. 2012;166:51–65. doi: 10.1111/j.1476-5381.2011.01525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall JM, Siney L, Lippton H, Hyman A, Kang-Chang J, Brain SD. Interaction of human adrenomedullin 13–52 with calcitonin gene-related peptide receptors in the microvasculature of the rat and hamster. Br J Pharmacol. 1995;114:592–597. doi: 10.1111/j.1476-5381.1995.tb17180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watkins HA, Au M, Hay DL. The structure of secretin family GPCR peptide ligands: implications for receptor pharmacology and drug development. Drug Discov Today. 2012;17:1006–1014. doi: 10.1016/j.drudis.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Rist B, Entzeroth M, Beck-Sickinger AG. From micromolar to nanomolar affinity: a systematic approach to identify the binding site of CGRP at the human calcitonin gene-related peptide 1 receptor. J Med Chem. 1998;41:117–123. doi: 10.1021/jm970533r. [DOI] [PubMed] [Google Scholar]
- 23.Fitzsimmons TJ, Zhao X, Wank SA. The extracellular domain of receptor activity-modifying protein 1 is sufficient for calcitonin receptor-like receptor function. J Biol Chem. 2003;278:14313–14320. doi: 10.1074/jbc.M211946200. [DOI] [PubMed] [Google Scholar]
- 24.Qi T, Christopoulos G, Bailey RJ, Christopoulos A, Sexton PM, Hay DL. Identification of N-terminal receptor activity-modifying protein residues important for calcitonin gene-related peptide, adrenomedullin, and amylin receptor function. Mol Pharmacol. 2008;74:1059–1071. doi: 10.1124/mol.108.047142. [DOI] [PubMed] [Google Scholar]
- 25.Qi T, Hay DL. Structure-function relationships of the N-terminus of receptor activity-modifying proteins. Br J Pharmacol. 2010;159:1059–1068. doi: 10.1111/j.1476-5381.2009.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watkins HA, Rathbone DL, Barwell J, Hay DL, Poyner DR. Structure-activity relationships for alpha calcitonin gene-related peptide. Br J Pharmacol. PMID. 2012:23186257. doi: 10.1111/bph.12072. {Medline} [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watkins HA, Au M, Bobby R, Archbold JK, Abdul-Manan N, Moore JM, Middleditch MJ, Williams GM, Brimble MA, Dingley AJ, Hay DL. Identification of key residues involved in adrenomedullin binding to the AM1 receptor. Br J Pharmacol. 2013;169:143–155. doi: 10.1111/bph.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heroux M, Hogue M, Lemieux S, Bouvier M. Functional calcitonin gene-related peptide receptors are formed by the asymmetric assembly of a calcitonin receptor-like receptor homo-oligomer and a monomer of receptor activity-modifying protein-1. J Biol Chem. 2007;282:31610–31620. doi: 10.1074/jbc.M701790200. [DOI] [PubMed] [Google Scholar]
- 29.Hill HE, Pioszak AA. Bacterial expression and purification of a heterodimeric adrenomedullin receptor extracellular domain complex using DsbC-assisted disulfide shuffling. Prot Express Purif. 2013;88:107–113. doi: 10.1016/j.pep.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci USA. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pioszak AA, Parker NR, Gardella TJ, Xu HE. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. J Biol Chem. 2009;284:28382–28391. doi: 10.1074/jbc.M109.022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pal K, Swaminathan K, Xu HE, Pioszak AA. Structural basis for hormone recognition by the Human CRFR2{alpha} G protein-coupled receptor. J Biol Chem. 2010;285:40351–40361. doi: 10.1074/jbc.M110.186072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pioszak AA, Harikumar KG, Parker NR, Miller LJ, Xu HE. Dimeric arrangement of the parathyroid hormone receptor and a structural mechanism for ligand-induced dissociation. J Biol Chem. 2010;285:12435–12444. doi: 10.1074/jbc.M109.093138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar S, Pioszak A, Zhang C, Swaminathan K, Xu HE. Crystal structure of the PAC1R extracellular domain unifies a consensus fold for hormone recognition by class B G-protein coupled receptors. PloS one. 2011;6:e19682. doi: 10.1371/journal.pone.0019682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakder S, Rattan S. [Tyr0]-calcitonin gene-related peptide 28–37 (rat) as a putative antagonist of calcitonin gene-related peptide responses on opossum internal anal sphincter smooth muscle. J Pharmacol Exp Ther. 1990;253:200–206. [PubMed] [Google Scholar]
- 37.Koth CM, Abdul-Manan N, Lepre CA, Connolly PJ, Yoo S, Mohanty AK, Lippke JA, Zwahlen J, Coll JT, Doran JD, Garcia-Guzman M, Moore JM. Refolding and characterization of a soluble ectodomain complex of the calcitonin gene-related peptide receptor. Biochemistry. 2010;49:1862–1872. doi: 10.1021/bi901848m. [DOI] [PubMed] [Google Scholar]
- 38.Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, Lohse MJ, Milligan G, Palczewski K, Parmentier M, Spedding M. International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev. 2007;59:5–13. doi: 10.1124/pr.59.1.5. [DOI] [PubMed] [Google Scholar]