

Abstract
Hepatitis C (HCV) protein 3/4A (NS3/4A) is a bifunctional enzyme comprising two separate domains with protease and helicase activities, which are essential for viral propagation. Both domains are stable and have enzymatic activity separately, and the relevance and implications of having protease and helicase together as a single protein remains to be explored. Altered in vitro activities of isolated domains compared with the full-length NS3/4A protein suggest the existence of interdomain communication. The molecular mechanism and extent of this communication was investigated by probing the domain–domain interface observed in HCV NS3/4A crystal structures. We found in molecular dynamics simulations that the two domains of NS3/4A are dynamically coupled through the interface. Interestingly, mutations designed to disrupt this interface did not hinder the catalytic activities of either domain. In contrast, substrate cleavage and DNA unwinding by these mutants were mostly enhanced compared with the wild-type protein. Disrupting the interface did not significantly alter RNA unwinding activity; however, the full-length protein was more efficient in RNA unwinding than the isolated protease domain, suggesting a more direct role in RNA processing independent of the interface. Our findings suggest that HCV NS3/4A adopts an “extended” catalytically active conformation, and interface formation acts as a switch to regulate activity. We propose a unifying model connecting HCV NS3/4A conformational states and protease and helicase function, where interface formation and the dynamic interplay between the two enzymatic domains of HCV NS3/4A potentially modulate the protease and helicase activities in vivo.
Keywords: HCV NS3/4A, protease-helicase interaction, dynamic coupling, catalytic activity, bifunctional enzyme, interdomain communication
Introduction
Hepatitis C virus (HCV) is the causative agent of non-A non-B viral hepatitis that infects 3% of the world population.1 HCV infection, if not cleared by the host, eventually progresses into chronic hepatitis.2 Upon infection of the host, the single-stranded positive sense RNA genome is translated into a 3000 amino acid polyprotein which is then processed by a combination of host and viral proteases to yield structural and proteins.3
HCV NS3/4A is a bifunctional protein that contains both a protease (NS3/4Apro) and a helicase (NS3hel) domain. N-terminal 181 amino acids of the NS3 protein constitute a chymotrypsin-like serine protease formed by N-terminal (N-SD) and C-terminal (C-SD) subdomains with β-barrel folds [Fig. 1(A)].4 The hydrophobic β-strand of the NS4A protein (NS4A cofactor – residues 12–23) is integrated into the N-SD, which contributes to the proper folding of the protease domain.5 NS3/4A protease cleaves the viral polyprotein between NS3 and NS4A in cis and downstream sites between NS4A/NS4B, NS4B/NS5A, and NS5A/NS5B in trans, all of which are necessary for viral maturation. The protease domain also cleaves host mitochondrial antiviral signaling protein (MAVS) and TIR-domain-containing adapter-inducing interferon-β (TRIF), which are required in the innate immune response that host cells mount against HCV infection.4
Figure 1.
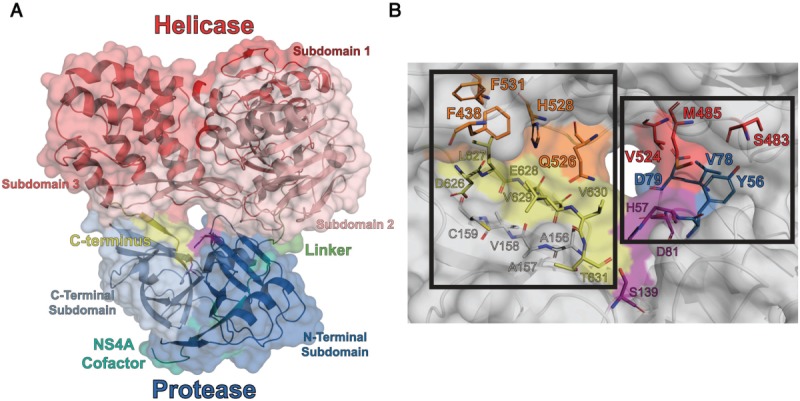
(A) Cartoon representation of HCV scNS3/4A (PDB ID: 1CU1). In this structure, helicase domain covers the otherwise solvent exposed active site of the protease and the C-terminus of the protein occupies the P-side of protease active site. (B) Close-up view of the domain–domain interface. (Left) The substrate interface is formed between the C-terminus (yellow), and residues from the helicase (orange) and the protease. (Right) The direct interface is formed by directly contacting residues between the protease (blue) and the helicase (red). These two interfaces lie on either side of the catalytic triad (magenta).
C-terminal region of NS3 forms a helicase that is classified as a superfamily 2 DExD-box helicase made of two recA-like motor domains (subdomains 1 and 2 – SD1 and SD2) and a third α-helical domain (subdomain 3 – SD3) [Fig. 1(A)].6 NS3 helicase incorporates both polynucleotide stimulated NTPase activity and 3′ to 5′ unwinding activity for duplex RNA and DNA. Exact function of the helicase domain in HCV life cycle is not elucidated yet, however, NS3 helicase likely aids the viral replication machinery by unwinding the RNA duplexes formed during and/or after replication.4,6
Both helicase and protease activities of the HCV NS3/4A are essential for viral propagation, and they have been studied extensively. Many of these studies were performed on isolated protease or helicase domains because such fragments can be more easily expressed and purified as recombinant proteins and are generally more stable in vitro than the full-length NS3/4A. However, catalytic activities in full-length protein differ from those of isolated domains, suggesting bidirectional between the two domains. The influence of helicase domain on NS3 protease function is still controversial. In most studies, helicase domain did not affect protease function,7,8 but Beran and Pyle reported two orders of magnitude enhancement of protease activity in the full-length NS3-4A compared with protease domain alone.9 The effect of protease domain on NS3 helicase function is better established – the RNA to DNA preference and RNA unwinding activity of the full-length protein are both higher compared with the isolated helicase domain.10,11 Similar results for the helicase domain have been reported for Flaviviruses (Dengue and West Nile Virus) NS3/2B, which is an ortholog of HCV NS3/4A complex.12–14 In summary, various studies suggest domain–domain interdependence in NS3/4A protein. However, controversy in the extent and type of influence of the two domains on each other's catalytic activities, and the molecular mechanism underlying this interdependence remains to be addressed.
In crystal structures of HCV NS3/4A, the protease and helicase domains have a proximal arrangement where the two domains make contacts in two discrete interfaces, separated by the protease active site5,15,16 (Fig. 1). In this study, molecular dynamics (MD) simulations further pointed to the importance of the domain–domain interface, as the two domains displayed dynamic coupling through the interface. Hence, we probed the role of this interface in modulating catalytic activities of the protease and helicase domains, to elucidate the molecular mechanism of interdomain communication in NS3/4A. We performed a systematic alanine scanning to disrupt the interface, and determined the effect on substrate cleavage and RNA/DNA unwinding. Most mutations introduced to destabilize the interface enhanced protease and helicase activities. Catalytic activities were also determined for isolated protease and helicase domains wild-type and mutant variants. Our findings suggest that in addition to the conformation observed in the crystal structure, the HCV NS3/4A complex adopts an “extended” conformation,17,18 similar to what is observed in some crystal structures of homologous proteins from Flaviviruses.19,20 This alternate conformation is likely the catalytically active conformation, and interface formation acts as a switch to regulate enzymatic activity.
Results and Discussion
The two domains of HCV NS3/4A are dynamically coupled
In the crystal structures of HCV NS3/4A, the two domains contact each other at two discrete interfaces on either side of the axis passing through the helicase nucleic acid binding groove and the protease catalytic site [Fig. 1(A)]. In the recombinant protein used for crystallization, the NS4A cofactor is covalently tethered to the NS3 N-terminus, forming a single chain NS3/4A protease-helicase construct (scNS3/4A). Six C-terminal residues of the NS3/4A, the P-side product of NS3-NS4A cleavage site, occupy the protease active site. In this “substrate interface”, the product is contacted by residues from both domains [Fig. 1(B) – left]. In the second interface, “direct interface”, residues from both domains form direct contacts [Fig. 1(B) – right].
The role of these two interfaces in facilitating interdomain communication, and the extent of dynamic coupling between the two domains were probed by MD simulations on scNS3/4A and three in silico deletion constructs. Linker deletion (ΔLin) was generated to break the covalent linkage between the two domains, and C-terminus deletion (ΔC-term) to disrupt the association of the protease and helicase domains through the strong product-protease interaction observed in enzymatic studies.21 A double-deletion construct had both linker and C-terminus removed (ΔDouble). Dynamic cross-correlations and interdomain distances during the simulations were calculated to analyze the extent of dynamic interaction between the protease and helicase domains in this bifunctional enzyme.
For all the constructs analyzed, significant interdomain correlations (i.e., absolute values of correlations larger than 0.35 – visualization cutoff) were clustered around the direct interface, with dominant positive correlations compared with negative correlations (i.e., anticorrelation) [Fig. 2(A) – top, Supporting Information Table S1]. Relative to the wild-type, stronger and more numerous positive correlations were observed for the C-terminus deletion constructs (129 correlating atom pairs for wild-type versus 1694 pairs for ΔC-term and 298 pairs for ΔDouble), which were largely around the direct interface. ΔLin exhibited fewer correlations (20 pairs), which were also across the direct interface. For the wild-type and ΔDouble, additional long distance correlations were observed between atoms on the surface of the protein, especially with protease C-SD and helicase SD1. These distal atom pairs exhibit mostly anticorrelated motions, possibly due to the larger scale vibration of the protein. In addition, to quantitate the extent of overall interdomain correlation in all the constructs, the width (w) of correlation distributions were calculated [Fig. 2(A) – bottom, Supporting Information Table S1]. Similar to the trend observed in visualized correlations, protease and helicase domains of the C-terminus deletion constructs (ΔC-term and ΔDouble) were more correlated compared the wild-type (i.e., wider distributions – wΔC-term = 0.26, wΔDouble = 0.30, compared with wwildtype = 0.20) and less correlated for ΔLin (i.e., narrower distributions – wΔLin = 0.15).
Figure 2.
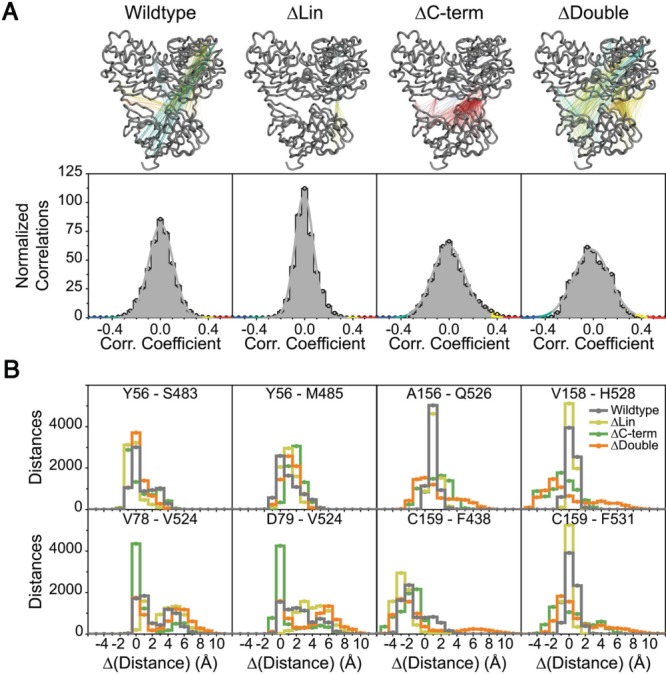
(A) (Top) Visualized cross correlations for wildtype protein and in silico deletion constructs. Correlations between 0.35 and 0.45 were colored yellow, larger than 0.45 red, between −0.35 and −0.45 cyan, lower than −0.45 blue and remaining gray. (Bottom) Distributions of interdomain correlations for each construct. (B) Pairwise distance difference distribution functions for selected residues across interfaces. Each residue pair contains one residue from the protease and one from the helicase. Distance differences are obtained by subtracting crystallographic distances from the simulation distances. Wildtype protein is represented with gray, ΔLin yellow, ΔC-term green, and ΔDouble orange.
The dynamic coupling between the two domains at the interface was further evaluated by Cα-Cα distance distributions during simulation time. From the substrate interface, four residue pairs, A156-Q526, V158-H528, C159-F438, and C159-F531, were selected to probe the relative freedom of movement between the domains [Fig. 2(B)]. When the C-terminus is present (i.e., for wild-type and ΔLin), mobility around the substrate interface is significantly reduced (i.e., sharp peaks around the crystallographic distances). In the absence of the C-terminus, mobility of the domains around the substrate interface increased, as reflected in the broader distance distributions. The C159-F438 pair is an exception, where the distance distributions were similar for all constructs, possibly because F438 is on the outer surface of the protein and solvent exposed. Thus, the C-terminus, which is the P-side cis-cleavage product, holds the two domains in close proximity through substrate-protease contacts and in its absence the substrate interface is largely disrupted.
C-terminus and linker deletions impact the motion around the direct interface differently. Distributions for Y56-S483 and Y56-M485 were similar across various constructs, suggesting the deletions did not affect the mobility around the core Y56. Motion in the Y56-S483-M485 network was also not restricted (i.e., 2–6 Å range), implying that this network, hence the core of the direct interface, is relatively mobile. The pairs V78-V524 and D79-V524 exhibit extensive variation in atomic distances throughout the simulations for all constructs, except ΔC-term. The restriction of motion in this network for the ΔC-term construct reflects strengthening of the network – hence deletion of the C-terminus causes the motion around the direct interface to be more correlated and increases the strength of association. In turn, the weakening of the network around the direct interface for wild-type and ΔLin is accompanied by increased freedom of motion for V78-D79-V524. Thus, the presence of C-terminus weakens correlated motions around the direct interface and increases the mobility of residues comprising the relatively more solvent exposed residues of the direct interface. In ΔDouble, while weaker, the correlation network exhibited similar characteristics to ΔC-term, however the mobility of V78-D79-V524 in this construct was similar to ΔLin. In summary, correlated motions through the core of the direct interface are modulated by the C-terminus, and linker deletion weakens the extent of correlated motion by disrupting the interaction of residues in the outer shell of the direct interface.
Analysis of protein dynamics through MD simulations shows that the two domains in NS3/4A are dynamically coupled, and this coupling is modulated through the substrate and direct interfaces, which work in an anti-correlated manner. Thus, our simulations implicate the interfaces in interdomain communication, which was experimentally investigated through site specific mutations and enzymatic activity assays.
Design of amino acid substitutions to disrupt the protease–helicase interface
To evaluate the functional relevance of the interfaces, a total of 10 residues at scNS3/4A interdomain interface were mutated to alanine to yield two sets of variants. The first set of variants had mutations designed to disrupt the direct interface, and included three variants with single point mutations Y56A, V78A, D79A in the protease N-SD, and one with all three substitutions (Y56A/V78A/D79A i.e., Triple Prot) [Fig. 1(B) – blue]. An additional four variants contained mutations in NS3hel SD2 and SD3, three variants with single point mutations – S483A, M485A, V524A, and one variant with all three substitutions (S483A/M485A/V524A, i.e. Triple Hel) [Fig. 1(B) – red]. The ninth variant in this first set (Hexa) had all six amino acid substitutions in the direct interface. The second set of variants was generated to disrupt the substrate interface, with one of the following amino acid substitutions – F438A, Q526A, H528A, and F531A [Fig. 1(B) – orange]. All mutant proteins were expressed, purified and biochemically characterized for protease, DNA/RNA unwinding, DNA binding and RNA-stimulated ATPase activities; in comparison with wild-type scNS3/4A and the isolated protease or helicase (scNS3/4Apro and, NS3hel).
Disrupting the direct interface enhanced protease cleavage activity
The catalytic efficiencies of scNS3/4A variants were determined using a FRET-based cleavage assay (Fig. 3, Supporting Information Table S2). Variants with single mutations in the direct interface, with the exception of Y56A, had higher catalytic efficiency (between 1.5 and 2.2 fold), compared with scNS3/4A. However, this activation was absent in the Triple Hel when the mutations were combined. Variants containing Y56A were less efficient in cleavage assays; Triple Prot performing the worst (0.3 fold) followed by Hexa (0.4 fold) and Y56A (0.9 fold). Y56 is near the protease active site next to the catalytic H57, so this residue may play a direct role in proteolysis, and is also reported to be conserved as a large hydrophobic side chain even in observed polymorphisms.22 The catalytic efficiencies of the proteins with substitutions in the substrate interface were similar to scNS3/4A, except H528A, which was more active (1.3 fold). The protease domain (i.e. scNS3/4Apro) activity was enhanced relative to full-length scNS3/4A (1.7 fold), similar to the interface mutants. These results signify that disruption of the direct interface enhances protease activity.
Figure 3.
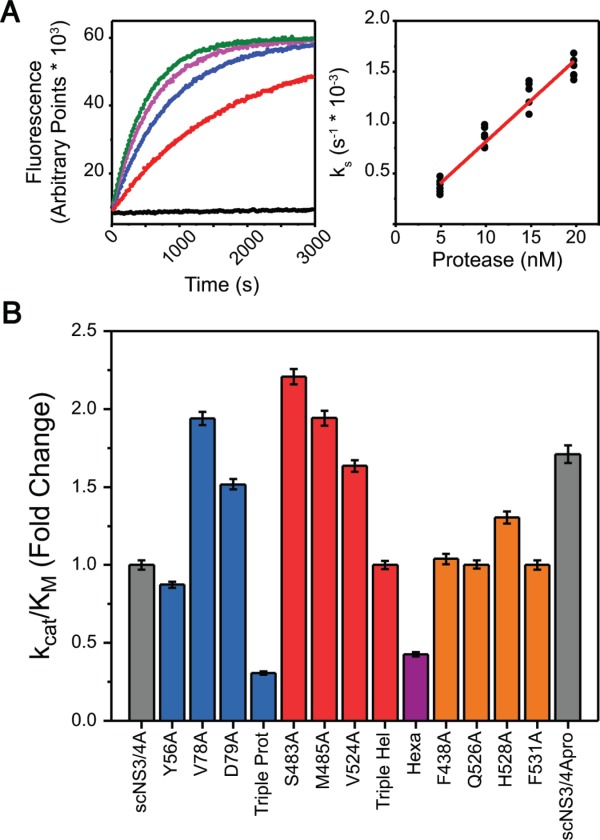
(A) (Left) Upon cleavage of the RET-S1 substrate, the FRET pair is liberated and the increase in fluorescence intensity is recorded kinetically (black – no protein, red to green – increasing enzyme concentration). Progress curves were fit to first order rate equations to obtain specificity constants (ks). (Right) Specificity constants at various enzyme concentrations were subjected to concatenate linear fit to yield catalytic efficiencies. (B) Substrate cleavage catalytic efficiencies of each protease variant, normalized to scNS3/4A. Error bars represent propagated standard errors (n = 6).
Mutations in the protease–helicase interface enhanced DNA unwinding rates
The ability of each variant to unwind DNA was assessed by measuring rates of helicase catalyzed DNA unwinding upon ATP addition [Fig. 4(A)]. To compare the specific activity of each protein, rates were plotted against protein concentration and linear regions of these curves from independent replicates were fitted concatenately [Fig. 4(B)]. These specific activities (V/E) were determined using a DNA molecular beacon based unwinding assay,23 performed both in the presence and absence of an enzyme trap under multiple and single turnover conditions [Fig. 4(C) Supporting Information Table S3]. Under multiple turnover conditions, the same amount of NS3hel unwound DNA faster (5.7 fold) compared with wild-type scNS3/4A. All scNS3/4A proteins with mutations designed to disrupt the helicase-protease interface unwound DNA with rates between those of scNS3/4A and NS3hel. Y56A, V78A, and V524A were each significantly more active (3.2, 5.6, and 4.8 fold, respectively) than scNS3/4A. In addition, Triple Hel performed two fold better compared with NS3hel, unwinding DNA significantly faster than wild-type full-length protein (11.6 fold). Similar to the substitutions that disrupt the direct interface, the unwinding rates catalyzed by proteins harboring substitutions designed to disrupt the substrate interface were also between the full-length protein and the isolated helicase (Q526A–5.5 fold, H528A–2.0 fold, F531A–2.3 fold), with the exception of F438A (0.8 fold).
Figure 4.
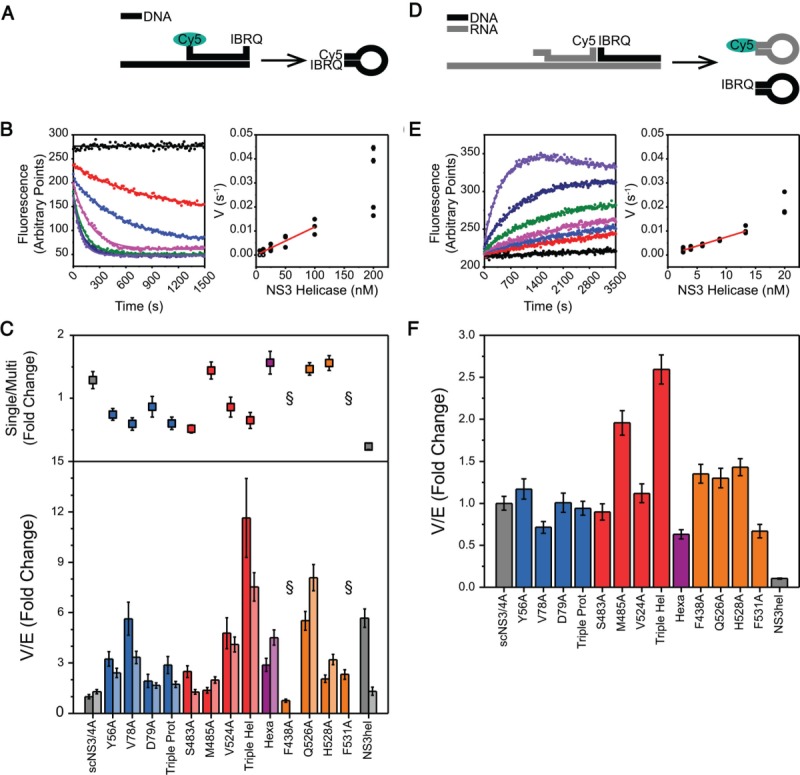
(A) DNA unwinding assay. dsDNA substrate for DNA unwinding assays consists of a top strand labeled with Cy5-IBRQ FRET pair at both ends and a longer bottom strand. Upon strand separation, the top strand self-anneals and Cy5 fluorescence is quenched. (B) (Left) Loss of fluorescence is recorded kinetically (black – no protein, red to purple – increasing protein concentration) and rates were obtained from nonlinear regression. (Right) Rates were plotted against enzyme concentration and linear parts of these curves were fitted concatenately to yield specific rates (V/E) from linear regression. (C) (Top) The difference between multiple and single turnover DNA unwinding assays. For each variant, fold change in activities for the single turnover activities with respect to multiple turnover were calculated. Error bars represent propagated standard errors (n = 3). (Bottom) Specific activities of each helicase variant for multiple turnover (bars) and single turnover (bars) DNA unwinding assays, normalized to scNS3/4A multiple turnover activity. Error bars represent propagated standard errors (n = 3). (D) RNA unwinding assay. dsRNA-DNA hybrid substrate for RNA unwinding assays consists of an RNA Cy5-labeled top strand, a DNA IBRQ-labeled top strand and a long RNA bottom strand. Upon unwinding, both top strands are liberated and self-anneal, resulting in increased Cy5 fluorescence intensity. (E) Gain of fluorescence was recorded kinetically (black – no protein, red to purple – increasing protein concentration) and data analysis were performed similar to DNA unwinding assay. (F) Specific activities of each helicase variant for RNA unwinding assays, normalized to scNS3/4A. Error bars represent propagated standard errors (n = 4).
When the ability of the scNS3/4A variants to unwind DNA in the presence of an ssDNA enzyme trap was compared, rates catalyzed under single turnover conditions were generally similar to those seen under multiple turnover conditions (i.e., in the absence of trap) with two noteworthy exceptions [Fig. 4(C) – light bars, Supporting Information Table S3]. Both F438A and F531A were catalytically inactive in the presence of a trap. Both of these residues are conserved, and F438 was previously identified as a critical residue for unwinding activity in NS3hel.24 When multiple and single turnover results for a given variant are compared, direct interface mutants (except M485A and Hexa) exhibited lower activities under single turnover conditions (between 0.5 and 0.9 fold) and the substrate interface mutants that were active under single turnover conditions exhibited higher activities (between 1.5 and 1.6 fold). In addition, compared with scNS3/4A and interface mutants, NS3hel exhibited significant loss of activity (0.2 fold). Taken together, interface mutations enhance helicase activity in unwinding dsDNA substrates under both multiple and single turnover conditions, to a greater extent than they enhance protease activity. The protease domain increases the processivity of the helicase under single turnover conditions, and the direct interface mutants, whereas increasing the overall activity of the helicase, lowers the processivity.
RNA unwinding was enhanced by the protease domain directly
In addition to DNA unwinding rates, RNA unwinding rates and specific activities (V/E) were determined for all variants using a duplex RNA-DNA hybrid substrate in which the helicase must separate an RNA duplex to cause an increase in substrate fluorescence [Fig. 4(D)] and resulting progress curves were processed similar to DNA unwinding assays [Fig. 4(E)].23 Compared with scNS3/4A, NS3hel unwound RNA significantly slower (0.1 fold), confirming the prior observation that full-length NS3/4A unwinds RNA faster than DNA, whereas the isolated NS3 helicase lacking the protease prefers DNA to RNA [Fig. 4(F), Supporting Information Table S3].10,11 Unlike DNA unwinding, where significant activity alterations were observed between variants, RNA unwinding activities for most of the proteins with direct interface mutations were similar to those seen with scNS3/4A (between 0.7 and 1.1 fold), with Triple Hel being more active (2.6 fold) and Hexa slightly less active (0.6 fold). All proteins harboring substitutions designed to disrupt the substrate interface were slightly more active compared with scNS3/4A (between 1.3 and 1.4 fold), except F531A (0.7 fold).
In contrast with DNA, interface mutations only marginally influenced RNA unwinding rates. Rather, the rate enhancement in the full-length enzyme is due to the presence of protease domain, and not mediated through the domain–domain interface. A potential explanation is a direct interaction between the RNA and the protease domain. Indeed, NS3 protease – RNA interactions were reported to inhibit protease activity, therefore probably involve regions near the protease active site.25,26 Potentially, the protease domain may be acting as a helicase cofactor that stabilizes protein-RNA association and modulates RNA unwinding activity and processivity.
Helicase ssDNA affinity was modulated by interface mutations
To act as a helicase, NS3 must first load its substrate by tightly binding single stranded nucleic acid. Fluorescence anisotropy based binding assays were performed to assess the ability of scNS3/4A, the variants, and NS3hel to load on substrates, which gave dissociation constants (KD) for ssDNA binding [Fig. 5(A), Supporting Information Table S4]. In this assay, scN3/4A and NS3hel both exhibited similar affinities to ssDNA. Differences between the variants were greater than those seen in protease and helicase assays, and dissociation constants were spread over a wide range (100 pM–10 nM). Compared with scNS3/4A; V78A, S483A, M485A, Triple Hel and F531A bound ssDNA more weakly (2.6, 2.9, 21.8, 5.2, and 3.0 fold, respectively) whereas D79A, Q526A, and H528A bound ssDNA more tightly (0.2, 0.4, and 0.1 fold, respectively).
Figure 5.
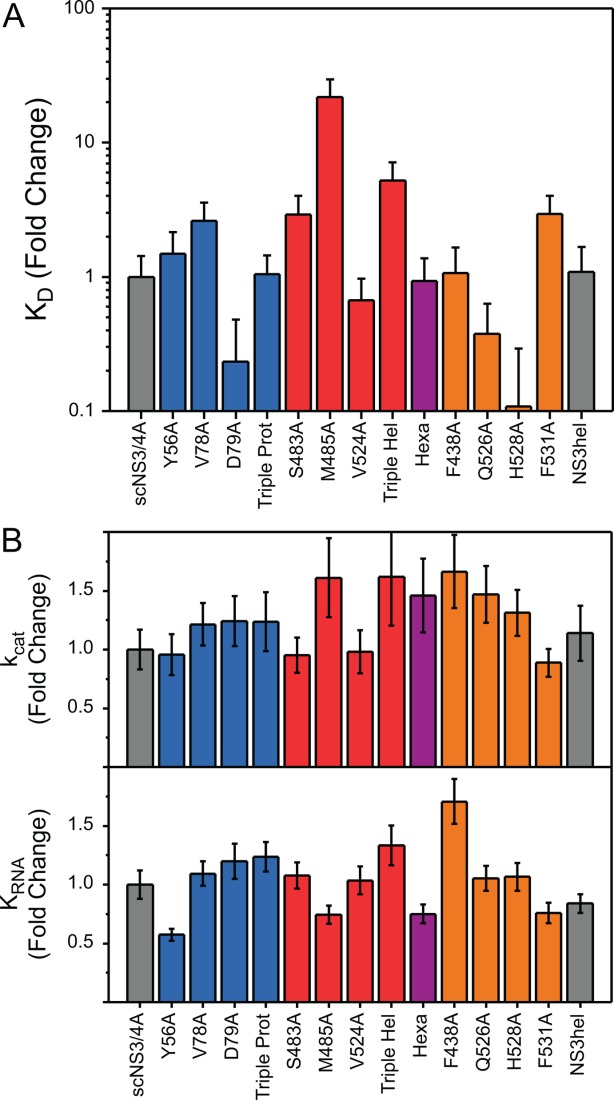
(A) Dissociation constants (KD) for DNA binding, normalized against scNS3/4A. (B) (Top) RNA-dependent catalytic rate constants (kcat) and (Bottom) extent of ATPase stimulation by RNA (KRNA) for all helicase variants, normalized to scNS3/4A. Error bars represent propagated standard errors (n = 3).
RNA-stimulated ATP hydrolysis of the helicase was not influenced by interface mutations
HCV NS3 helicase utilizes ATP to unwind nucleic acids, and in turn, nucleic acid stimulates NS3 catalyzed ATP hydrolysis; thus, in the presence of nucleic acids, NS3 helicase hydrolyzes ATP faster. The extent of ATPase activation by nucleic acids follows apparent Michaelis-Menten kinetics, and kinetic parameters for nucleic acid based activation were obtained. A concentration gradient of poly(U) RNA was used to stimulate NS3 ATPase, and the extent of ATP stimulation by RNA (KRNA), in addition to catalytic rate constants (kcat) were measured [Fig. 5(B), Supporting Information Table S5]. As with RNA unwinding results, KRNA for most of the variants was similar to full-length NS3/4A (between 0.7 and 1.1 fold); with Y56A exhibiting slightly lower KRNA (0.6 fold) and, Triple Hel and Hexa slightly higher (1.3 and 1.7 fold, respectively). kcat values were also similar (between 0.9 and 1.7 fold). Thus, the ATPase activity is largely independent of the presence of the protease domain and the interface.
Interface formation as a molecular mechanism for auto-regulation
Our results suggest a regulatory role for the protease-helicase interface in HCV NS3/4A. Both isolated domains and interface variants exhibit higher peptide cleavage and DNA unwinding activities compared with the wild-type protein. Single turnover DNA unwinding is also modulated by the interface, affecting the processivity of the enzyme. Thus, transient association of two domains through the interface controls the activities of both domains and is potentially the key to auto-regulation of protein activity and function.
The transient and regulatory nature of the interface observed here suggests an alternate functional domain–domain configuration where protease and helicase domains do not associate through the interface. Furthermore, transformation from the crystallographic, “compact” state to an active, “extended” state is possibly a regulatory switch of catalytic activity. A similar mechanism has been proposed for RNA unwinding activity,18 where deletion of the C-terminus lowers the required incubation time for full activity in RNA unwinding. Disrupting the strong C-terminus – protein interactions would lower the strength of the interface, enabling faster transition to the catalytically active “extended” state. Similarly, the interface mutations in the current study destabilize the interfaces, thereby lowering the barrier for transitioning between these states.
Enzymatic studies of the Flavivirus NS3/2B, and membrane dynamics of NS3/4A in cell-based assays are also consistent with an active extended conformation. The protease domain of Flavivirus (Dengue Virus) NS3/2B enhances RNA unwinding activity of the helicase domain, similar to observed here for HCV NS3/4A,12,13 and parallel observations have been reported for the two orthologs.14 However, Flavivirus NS3/2B, which lacks the C-terminus and the terminal alpha-helix, exhibits an extended conformation in crystal and solution structures where the protease is rotated away from the helicase.19,20 In the absence of C-terminus – protease interactions, the conformation Flavivirus NS3/2B adopts may be similar to the active extended conformer proposed here for HCV NS3/4A. In addition, integration of HCV NS3/4A to the host ER membrane is essential for correct function and protein localization,27,28 however the helicase domain clashes with the membrane when modeled in the crystallographic conformation.17
Although these prior studies favor the extended conformation as the major conformation in vivo, a recent study involving allosteric inhibitors of the HCV NS3/4A protein implies existence of the compact conformation in cell-based assays.29 These inhibitors are suggested to bind the cleft between the two interfaces, thereby stabilizing the compact conformer and inhibiting the protein. If the extended conformer were the sole state in vivo, then these inhibitors would have no effect on protein activity in cell-based assays. Thus, the domain–domain interface is likely transiently sampled in vivo, and the protein switches between two the conformations.
A unifying model for the protease and helicase function in the bifunctional HCV NS3/4A
Based on our findings and supported by previous studies, we propose a unifying model for domain–domain arrangement and function in HCV NS3/4A complex (Fig. 6). Cycling between different conformations was proposed before for the protease domain5 and substrate cleavage recently.29 Our model also proposes alternating conformations, and incorporates the regulatory nature of the domain–domain interface for the whole protein [Fig. 6(A)]. In addition to the “compact” and “extended” states, we propose the transition between these two states may proceed through a “semi-extended” state where the substrate interface is disrupted. The N-terminal “P-side” of the substrates have stronger interactions with the protease compared with the C-terminal “P'-side”,21 so the P-side of a substrate can compete out the C-terminus of the helicase, possibly leading to a “semi-extended” conformation where the helicase SD3 and the C-terminus rotates slightly away from the protease active site, which is now occupied by another viral substrate site. The semi-extended conformation is supported by our observations in both MD simulations and mutational analysis: Deletion of the C-terminus led to the destabilization of the substrate interface in silico, increasing the relative freedom of movement across the substrate interface. As the helicase SD3 has relative freedom with respect to SD1 and SD2 from the hinge loop regions,15 a semi-extended conformation that connects these subdomains can be envisioned. In addition, mutations in the substrate interface did not alter protease catalytic efficiency, which is consistent with weak or no contacts between the helicase domain and the substrate (or the product). Further, transition from the “semi-extended” to “extended” conformation can proceed by destabilization of the direct interface by either compact to semi-extended state transition, or by the P'-side of the substrate.
Figure 6.
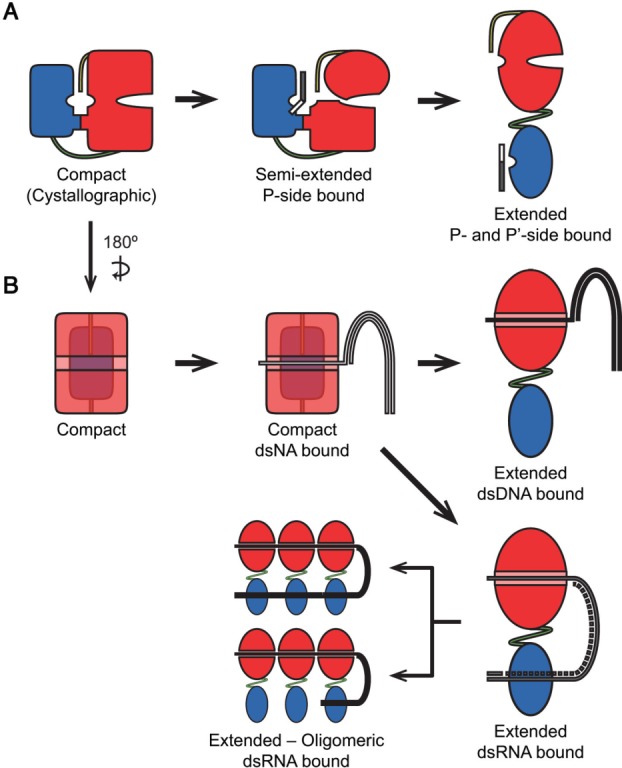
Possible models unifying our experimental results and previous observations on relations between HCV NS3/4A conformational states and protease and helicase function. (A) Binding of the P-side of a substrate induces conformation change from the closed conformation to a semiextended conformation. Conformational switch from the proposed semi-extended conformation to the extended conformation depends on the stability of the direct interface and the protease is fully active in the extended conformation. (B) Binding to the single-stranded region of a nucleic acid (either DNA or RNA) occurs in the closed conformation. For dsDNA, activation of the protein was modulated by interface mutations significantly, possibly due to the alteration of the dynamic interaction between the domains through the protease-helicase interface. For dsRNA, activity is independent of the interface, and protease-RNA association acts a factor modulating the conformational transition. The protease domain potentially associates with the dsRNA as a clamp. When the protein oligomerizes on dsRNA, two possible arrangements for RNA-protease interaction is possible – either all the protease domains or only the domain on the leading monomer associate with dsRNA.
In contrast to protease activity, the unwinding reaction by helicase involves multiple steps (nucleic acid binding, ATP hydrolysis, nucleic acid translocation and oligomerization), and any of these steps may involve a transition between the two different states. In a previous structural study, neither binding a short oligonucleotide nor different states of ATP hydrolysis induced any detectable conformational changes in the protein.15 We expect translocation, and thus unwinding, to happen in the active extended conformation. Binding to a relatively long stretch of nucleic acid may disrupt the interface and cause the conformational switch to the extended form, since maximum activity is observed when the protein is incubated for longer periods of time with dsRNA.18 However, the identity of the bound nucleic acid is relevant in this conformational switch, as we found that DNA unwinding was significantly affected by interface alterations but RNA unwinding was not. Although ssDNA was reported to inhibit protease activity of the isolated domain significantly, the full-length protein was less affected.26 Potentially DNA preferentially associates with the helicase and may induce the conformational switch by altering the inner dynamics of the helicase domain, leading to interface destabilization and liberation of the helicase domain [Fig. 6(B)]. In the case of RNA, a direct interaction between the protease and RNA likely drives the activation of the helicase, as the protease domain has significant binding affinity (low µM – high nM) for both ssRNA and dsRNA25 [Fig 7(B)]. How could the protease domain assist the helicase in unwinding RNA? The ability of the protease to bind both ssRNA and dsRNA, with preference to ssRNA, may be the answer. Association of dsRNA with the protease may disrupt the interface and induce transformation to the extended state, which is subsequently stabilized when the protease domain encounters ssRNA [Fig. 6(B)]. This could explain why the protease domain confers at least an order of magnitude enhancement in RNA unwinding. 3′ to 5′ translocation of the single stranded stretch of the nucleic acid by the helicase forces unzipping of the double stranded region and the equilibrium between unwinding and rewinding is driven forward when the protease binds ssRNA.
Oligomerization may also be a factor driving the compact to extended state transformation. Depending on protein and nucleic acid concentrations, NS3 helicase can adopt monomeric, dimeric or higher oligomeric forms in vitro.30 Oligomerization likely happens in the extended conformation, judged by the DNA-bound dimeric NS3 helicase structure, where the compact conformation clashes with the second helicase domain when juxtaposed.31 However, the nucleic acid – protease association is unclear in the oligomeric state. In this study, we propose two alternative oligomeric helicase – nucleic acid complex models, extending our view of the monomeric NS3-dsRNA complex. The helicase domains associate with ssRNA and only protease domain of the leading monomer (i.e., the monomer in the unwinding fork) is associated with nucleic acid, or the nucleic acid loops back on the leading protease and dsRNA stretch associates with the remaining protease domains [Fig. 6(B)]. Hence, oligomerization and protease – nucleic acid association potentially contribute to the regulation of catalytic activity. In summary, the model we propose here unifies paradigms based on prior biochemical and biophysical observations and our own results to consistently explain the regulatory role of interface formation underlying the modulation of cooperativity, catalytic activity and processivity of the HCV NS3/4A protein.
Materials and Methods
Molecular dynamics simulations
Details of the simulation setup and analytical calculations are explained in Supporting Information. Briefly, the structure of scNS3/4A was obtained from Protein Data Bank (PDB ID: 1CU1)5 and additional in silico deletion constructs ΔC-term (Δ626-631 – C-terminus deletion), ΔLin (Δ184-186 – linker deletion) and ΔDouble (Δ184-186, 626-631 – C-terminus and linker deletion) were created. 20 ns equilibrated MD simulations were performed on quadruplicate on each construct using AMBER10.32 Resulting trajectories were analyzed for cross-correlations between the domains and distance calculations were performed between selected residues across the domains.
Protein expression plasmids
The constructs for the scNS3/4Apro and NS3hel were defined elsewhere.33,34 The single chain NS3/4A (scNS3/4A) protease-helicase construct was generated by ligating the codon optimized genotype 1a helicase construct (H77c) downstream of the Bristol-Myers-Squibb patented scNS3/4A protease (synthesized by GenScript)35 and cloned into a pET28a expression vector (Novagen). Mutant variants Y56A, V78A, D79A, S483A, M485A, V524A, Triple Prot (Y56A-V78A-D79A), Triple Hel (S483A-M485A-V524A), F438A, Q526A, H528A, and F531A were generated using the QuikChange II site-directed mutagenesis kit (Stratagene) with suitable mutagenesis primers generated with PrimerX (http://www.bioinformatics.org/primerx/). The variant Hexa (Y56A-V78A-D79A-S483A-M485A-V524A) was generated by ligating the protease domain of Triple Prot and the helicase domain of Triple Hel. Each mutant variant was sequenced to ensure correctness and full coverage.
Protein expression and purification
The expression and purification of scNS3/4Apro and NS3hel were detailed elsewhere and utilized without modification.33,34 For the scNS3/4A protease-helicase complex, purification scheme is detailed in Supporting Information. Briefly, transformed BL21-CodonPlus (DE3)-RIL E. coli expression cells (Strategene) were grown to an OD600 of 0.6 at 37°C, transferred to 20°C, induced 0.25 mM IPTG, incubated with shaking for 4 h at 20°C, harvested via centrifugation and stored at −80°C. Frozen pellets were resuspended and scNS3/4A was purified in two successive steps using Ni2+ affinity (HisTrap – GE Lifesciences) and cation exchange (Mono S – GE Lifesciences) column chromatography. The final purified protein was judged >90% pure by polacrylamide gel electrophoresis, concentrated, flash frozen in liquid nitrogen and stored at −80°C.
Protease assays
Depsipeptide cleavage assays were performed in a final volume of 60 µL containing protease assay buffer (50 mM Tris, 2.5% glycerol, 0.1% n-octyl-β-D-glucoside (OβG), 5 mM tris(2-carboxyethyl)phosphine (TCEP), 1% DMSO, pH 7.5) and 1–20 nM of NS3/4A protease variants in black 96-well flat bottom nonbinding surface half-area plates (Corning) at room temperature. The protein was pre-incubated in the reaction mixture for 30 min and the reaction was initiated by injecting RET-S1 substrate (Anaspec) with a final concentration of 250 nM in the reaction mixture. The fluorescence output was measured kinetically for 1 h using an EnVision plate reader (Perkin Elmer) with excitation at 340 nm and emission at 492 nm. Dilution series for each protease variant were performed in sextuplicate.
Reaction progress curves were processed using OriginPro 8 (Origin Labs). Progress curves for each variant were fitted globally to Eq. (1) [Fig. 3(A) – left].
| (1) |
In Eq. (1), F is the measured fluorescence, Ff is the final fluorescence, A is the amplitude of the reaction, which is shared globally between different protein dilutions, and ks is the specificity constant, which satisfies Eq. (2) under pseudo-first order conditions when [E]T << [S]T << KM.36 The obtained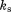 values were plotted against enzyme concentration and fitted linearly using all the data from different experiments (i.e., the concatenate fitting function) using OriginPro 8 (Origin Labs) to Eq. (2) [Fig. 3(A) – right].
values were plotted against enzyme concentration and fitted linearly using all the data from different experiments (i.e., the concatenate fitting function) using OriginPro 8 (Origin Labs) to Eq. (2) [Fig. 3(A) – right].
| (2) |
In Eq. (2), kcat/KM is the catalytic efficiency and [E]T is the total enzyme concentration. Total active enzyme concentrations were accurately measured using an enzymatic inhibition assay with the nanomolar affinity protease inhibitor Danoprevir.37 The enzyme was titrated with an increasing concentration of the inhibitor and the initial velocities from drug dilutions were plotted against drug concentrations respectively. Active enzyme concentrations were calculated from the x-intercept of the intersection of two linear regimens of inhibition curves.38
DNA and RNA unwinding assay
The details of the DNA and RNA unwinding assays were previously described elsewhere,23 and the nucleic acid substrates used in these assays are detailed in Supporting Information. Briefly, DNA unwinding assays were performed in a final volume of 60 µL containing helicase assay buffer (25 mM MOPS, 1.25 mM MgCl2, 0.05% Tween-20, 0.005 mg/mL BSA, 2.5 mM TCEP, pH 6.5), 12.5 nM dsDNA substrate and 2–200 nM of the NS3 helicase variants in white flat bottom 96-well half area plates (Corning) at 25°C. The reaction was initiated by injecting ATP to a final concentration of 1 mM in multiple turnover conditions, or for single turnover reaction, injecting ATP and dT15 ssDNA trap, to final concentrations of 1 mM and 1 µM, respectively. RNA unwinding assays were performed in a final volume of 60 µL containing helicase assay buffer, 25 nM RNA-DNA hybrid substrate and 1–20 nM of the scNS3/4A protease-helicase variants or 20–150 nM of NS3hel in white flat bottom 96-well half area plates (Corning) at 25°C. The reaction was initiated by injecting ATP to a final concentration of 1 mM. Changes in fluorescence as a function of time were monitored using a Varioskan plate reader (Thermo Scientific) for multi turnover DNA and RNA unwinding assays with excitation at 643 nm and emission at 667 nm, and a FLUOstar Omega plate reader (BMG Labtech) for single turnover DNA unwinding assays with excitation at 640 nm and emission at 665 nm. Multiple and single turnover DNA unwinding assays were performed in triplicate and RNA unwinding assays in quadruplicate. Resulting progress curves were analyzed using Prism 5 (GraphPad) and unwinding rates were calculated using the scheme outlined previously.23 Rates from independent repeats for individual variants were plotted together against enzyme concentrations and linear portions of these curves were fitted concatenately using OriginPro 8 (Origin Labs) to calculate specific activities (V/E).
DNA binding assay
The details of fluorescence anisotropy-based DNA binding assays were previously described.39 Briefly, DNA binding assays were performed in a final volume of 60 µL containing helicase assay buffer, 5 nM Cy5-dT15 DNA and 1–100 nM NS3 helicase variants in black flat bottom 96-well half area plates (Corning) at 25°C. The protein-DNA mixture was incubated for 30 min and the fluorescence polarization was measured using an Infinite M1000 plate reader (Tecan) with excitation at 635 nm and emission at 667 nm. DNA dissociation constants (KD) were calculated using Prism 5 (GraphPad). Independent repeats from individual variants were fitted globally using the scheme defined previously,40 sharing KD.
Poly(U)-stimulated ATPase assay
The details of the modified malachite green assay were described previously.33 Briefly, ATPase assays were performed in a final volume of 50 µL containing helicase assay buffer, 0–10 mg/mL poly(U) RNA and 5 nM NS3 helicase variants at 25°C. The reaction was initiated by adding ATP (1 mM final concentration) and incubated for 10 min. 25 µL of this reaction was quenched in 200 µL of malachide green reagent (3 volumes of 0.045% (w/v) malachite green, 1 volume of 4.2% ammonium molybdate in 4 N HCl, 0.01 volume of 10% Tween-20) in clear flat bottom 96-well plates (Corning), immediately followed by the addition of 25 µL of 34% sodium citrate. Color was allowed to develop for 15 min and the absorbance was read using a VarioSkan plate reader (Thermo Scientific) at 630 nm. The absorbance values were converted to inorganic phosphate concentrations using a phosphate standard curve and converted to rates by dividing by incubation times. RNA-stimulated catalytic rate constants (i.e., kcat) and the extent of RNA stimulation (i.e., KRNA) for each helicase variant were calculated using Prism 5 (GraphPad). Independent repeats for individual variants were fitted globally using the scheme defined previously,10 sharing KRNA. kcat values obtained from the global fitting of independent replicates for individual variants were averaged with error-weighing and reported.
Acknowledgments
We thank Hong Cao for valuable assistance in enzymatic assay setup; Akbar Ali for synthesis of ITMN-191; John Hernandez (University of Wisconsin, Milwaukee) for help with preparing RNA substrate; Keith Romano for valuable discussions; and Nese Kurt Yilmaz for editorial help.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
Supplementary Information
References
- 1.Lavanchy D. The global burden of hepatitis C. Liver Int. 2009;29(Suppl 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 2.Liang TJ, Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S62–71. doi: 10.1053/j.gastro.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 3.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 4.Morikawa K, Lange CM, Gouttenoire J, Meylan E, Brass V, Penin F, Moradpour D. Nonstructural protein 3-4A: the Swiss army knife of hepatitis C virus. J Viral Hepat. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 5.Yao N, Reichert P, Taremi SS, Prosise WW, Weber PC. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Structure. 1999;7:1353–1363. doi: 10.1016/s0969-2126(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 6.Frick DN. The hepatitis C virus NS3 protein: a model RNA helicase and potential drug target. Curr Issues Mol Biol. 2007;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 7.Dahl G, Sandstrom A, Akerblom E, Danielson UH. Effects on protease inhibition by modifying of helicase residues in hepatitis C virus nonstructural protein 3. FEBS J. 2007;274:5979–5986. doi: 10.1111/j.1742-4658.2007.06120.x. [DOI] [PubMed] [Google Scholar]
- 8.Thibeault D, Massariol MJ, Zhao S, Welchner E, Goudreau N, Gingras R, Llinas-Brunet M, White PW. Use of the fused NS4A peptide-NS3 protease domain to study the importance of the helicase domain for protease inhibitor binding to hepatitis C virus NS3-NS4A. Biochemistry. 2009;48:744–753. doi: 10.1021/bi801931e. [DOI] [PubMed] [Google Scholar]
- 9.Beran RK, Pyle AM. Hepatitis C viral NS3-4A protease activity is enhanced by the NS3 helicase. J Biol Chem. 2008;283:29929–29937. doi: 10.1074/jbc.M804065200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frick DN, Rypma RS, Lam AM, Gu B. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J Biol Chem. 2004;279:1269–1280. doi: 10.1074/jbc.M310630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beran RK, Serebrov V, Pyle AM. The serine protease domain of hepatitis C viral NS3 activates RNA helicase activity by promoting the binding of RNA substrate. J Biol Chem. 2007;282:34913–34920. doi: 10.1074/jbc.M707165200. [DOI] [PubMed] [Google Scholar]
- 12.Yon C, Teramoto T, Mueller N, Phelan J, Ganesh VK, Murthy KH, Padmanabhan R. Modulation of the nucleoside triphosphatase/RNA helicase and 5'-RNA triphosphatase activities of Dengue virus type 2 nonstructural protein 3 (NS3) by interaction with NS5, the RNA-dependent RNA polymerase. J Biol Chem. 2005;280:27412–27419. doi: 10.1074/jbc.M501393200. [DOI] [PubMed] [Google Scholar]
- 13.Chernov AV, Shiryaev SA, Aleshin AE, Ratnikov BI, Smith JW, Liddington RC, Strongin AY. The two-component NS2B-NS3 proteinase represses DNA unwinding activity of the West Nile virus NS3 helicase. J Biol Chem. 2008;283:17270–17278. doi: 10.1074/jbc.M801719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiryaev SA, Chernov AV, Shiryaeva TN, Aleshin AE, Strongin AY. The acidic sequence of the NS4A cofactor regulates ATP hydrolysis by the HCV NS3 helicase. Arch Virol. 2011;156:313–318. doi: 10.1007/s00705-010-0838-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Appleby TC, Anderson R, Fedorova O, Pyle AM, Wang R, Liu X, Brendza KM, Somoza JR. Visualizing ATP-dependent RNA translocation by the NS3 helicase from HCV. J Mol Biol. 2011;405:1139–1153. doi: 10.1016/j.jmb.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiering N, D'Arcy A, Villard F, Simic O, Kamke M, Monnet G, Hassiepen U, Svergun DI, Pulfer R, Eder J, Raman P, Bodendorf U. A macrocyclic HCV NS3/4A protease inhibitor interacts with protease and helicase residues in the complex with its full-length target. Proc Natl Acad Sci U S A. 2011;108:21052–21056. doi: 10.1073/pnas.1110534108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brass V, Berke JM, Montserret R, Blum HE, Penin F, Moradpour D. Structural determinants for membrane association and dynamic organization of the hepatitis C virus NS3–4A complex. Proc Natl Acad Sci U S A. 2008;105:14545–14550. doi: 10.1073/pnas.0807298105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding SC, Kohlway AS, Pyle AM. Unmasking the active helicase conformation of nonstructural protein 3 from hepatitis C virus. J Virol. 2011;85:4343–4353. doi: 10.1128/JVI.02130-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mastrangelo E, Milani M, Bollati M, Selisko B, Peyrane F, Pandini V, Sorrentino G, Canard B, Konarev PV, Svergun DI, de Lamballerie X, Coutard B, Khromykh AA, Bolognesi M. Crystal structure and activity of Kunjin virus NS3 helicase; protease and helicase domain assembly in the full length NS3 protein. J Mol Biol. 2007;372:444–455. doi: 10.1016/j.jmb.2007.06.055. [DOI] [PubMed] [Google Scholar]
- 20.Luo D, Xu T, Hunke C, Gruber G, Vasudevan SG, Lescar J. Crystal structure of the NS3 protease-helicase from Dengue virus. J Virol. 2008;82:173–183. doi: 10.1128/JVI.01788-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romano KP, Laine JM, Deveau LM, Cao H, Massi F, Schiffer CA. Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J Virol. 2011;85:6106–6116. doi: 10.1128/JVI.00377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallet S, Gouriou S, Nousbaum JB, Legrand-Quillien MC, Goudeau A, Picard B. Genetic heterogeneity of the NS3 protease gene in hepatitis C virus genotype 1 from untreated infected patients. J Med Virol. 2005;75:528–537. doi: 10.1002/jmv.20302. [DOI] [PubMed] [Google Scholar]
- 23.Hanson AM, Hernandez JJ, Shadrick WR, Frick DN. Identification and analysis of inhibitors targeting the hepatitis C virus NS3 helicase. Methods Enzymol. 2012;511:463–483. doi: 10.1016/B978-0-12-396546-2.00021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam AM, Keeney D, Frick DN. Two novel conserved motifs in the hepatitis C virus NS3 protein critical for helicase action. J Biol Chem. 2003;278:44514–44524. doi: 10.1074/jbc.M306444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Umehara T, Fukuda K, Nishikawa F, Sekiya S, Kohara M, Hasegawa T, Nishikawa S. Designing and analysis of a potent bi-functional aptamers that inhibit protease and helicase activities of HCV NS3. Nucleic Acids Symp Ser (Oxf) 2004;48:195–196. doi: 10.1093/nass/48.1.195. [DOI] [PubMed] [Google Scholar]
- 26.Vaughan R, Li Y, Fan B, Ranjith-Kumar CT, Kao CC. RNA binding by the NS3 protease of the hepatitis C virus. Virus Res. 2012;169:80–90. doi: 10.1016/j.virusres.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Y, Weng L, Li R, Li L, Toyoda T, Zhong J. The N-terminal helix alpha(0) of hepatitis C virus NS3 protein dictates the subcellular localization and stability of NS3/NS4A complex. Virology. 2012;422:214–223. doi: 10.1016/j.virol.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 28.Horner SM, Park HS, Gale M., Jr Control of innate immune signaling and membrane targeting by the Hepatitis C virus NS3/4A protease are governed by the NS3 helix alpha0. J Virol. 2012;86:3112–3120. doi: 10.1128/JVI.06727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saalau-Bethell SM, Woodhead AJ, Chessari G, Carr MG, Coyle J, Graham B, Hiscock SD, Murray CW, Pathuri P, Rich SJ, Richardson CJ, Williams PA, Jhoti H. Discovery of an allosteric mechanism for the regulation of HCV NS3 protein function. Nat Chem Biol. 2012;8:920–925. doi: 10.1038/nchembio.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jennings TA, Mackintosh SG, Harrison MK, Sikora D, Sikora B, Dave B, Tackett AJ, Cameron CE, Raney KD. NS3 helicase from the hepatitis C virus can function as a monomer or oligomer depending on enzyme and substrate concentrations. J Biol Chem. 2009;284:4806–4814. doi: 10.1074/jbc.M805540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackintosh SG, Lu JZ, Jordan JB, Harrison MK, Sikora B, Sharma SD, Cameron CE, Raney KD, Sakon J. Structural and biological identification of residues on the surface of NS3 helicase required for optimal replication of the hepatitis C virus. J Biol Chem. 2006;281:3528–3535. doi: 10.1074/jbc.M512100200. [DOI] [PubMed] [Google Scholar]
- 32.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM., Jr Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem. 26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belon CA, Frick DN. Fuel specificity of the hepatitis C virus NS3 helicase. J Mol Biol. 2009;388:851–864. doi: 10.1016/j.jmb.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romano KP, Ali A, Royer WE, Schiffer CA. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc Natl Acad Sci U S A. 2010;107:20986–20991. doi: 10.1073/pnas.1006370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wittekind M, Weinheirner S, Zhang Y, Goldfarb V. Modified forms of hepatitis C NS3 protease for facilitating inhibitor screening and structural studies of protease: inhibitor complexes. United States of America: United States Patent Applications Publication; 2002. p. 26. [Google Scholar]
- 36.Palmier MO, Van Doren SR. Rapid determination of enzyme kinetics from fluorescence: overcoming the inner filter effect. Anal Biochem. 2007;371:43–51. doi: 10.1016/j.ab.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Seiwert SD, Kossen K, Pan L, Liu J, Buckman BO. Antiviral Drugs. John Wiley & Sons, Inc; 2011. Discovery and Development of the HCV NS3/4A Protease Inhibitor Danoprevir (ITMN-191/RG7227) pp. 257–271. [Google Scholar]
- 38.Copeland RA. Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem Anal. 2005;46:1–265. [PubMed] [Google Scholar]
- 39.Mukherjee S, Hanson AM, Shadrick WR, Ndjomou J, Sweeney NL, Hernandez JJ, Bartczak D, Li K, Frankowski KJ, Heck JA, Arnold LA, Schoenen FJ, Frick DN. Identification and analysis of hepatitis C virus NS3 helicase inhibitors using nucleic acid binding assays. Nucleic Acids Res. 2012;40:8607–8621. doi: 10.1093/nar/gks623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lam AM, Rypma RS, Frick DN. Enhanced nucleic acid binding to ATP-bound hepatitis C virus NS3 helicase at low pH activates RNA unwinding. Nucleic Acids Res. 2004;32:4060–4070. doi: 10.1093/nar/gkh743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.