

Abstract
Structure determination of integral membrane proteins by solution NMR represents one of the most important challenges of structural biology. A Residual-Dipolar-Coupling-based refinement approach can be used to solve the structure of membrane proteins up to 40 kDa in size, however, a weak-alignment medium that is detergent-resistant is required. Previously, availability of media suitable for weak alignment of membrane proteins was severely limited. We describe here a protocol for robust, large-scale synthesis of detergent-resistant DNA nanotubes that can be assembled into dilute liquid crystals for application as weak-alignment media in solution NMR structure determination of membrane proteins in detergent micelles. The DNA nanotubes are heterodimers of 400nm-long six-helix bundles each self-assembled from a M13-based p7308 scaffold strand and >170 short oligonucleotide staple strands. Compatibility with proteins bearing considerable positive charge as well as modulation of molecular alignment, towards collection of linearly independent restraints, can be introduced by reducing the negative charge of DNA nanotubes via counter ions and small DNA binding molecules. This detergent-resistant liquid-crystal media offers a number of properties conducive for membrane protein alignment, including high-yield production, thermal stability, buffer compatibility, and structural programmability. Production of sufficient nanotubes for 4–5 NMR experiments can be completed in one week by a single individual.
Keywords: NMR, DNA nanotube, membrane protein, PALES, DNA origami, Residual Dipolar Coupling, RDC
Introduction
The last three decades have witnessed the exponential development of DNA as a molecular engineering material to create nanostructures with controlled geometries, topologies, and periodicities of increasing complexity1,2. The method of DNA origami, in which a long “scaffold” strand is mixed with hundreds of short “staple” strands to form a parallel bundle of double helices of custom shape, has proven particularly well suited for the robust self-assembly of two- and three-dimensional nanostructures up to ∼10 MDa in size3–5.
One of the most remarkable applications for this technology is in NMR-based membrane-protein structure determination6,7.
Analysis of integral membrane proteins by NMR
In spite of the importance of integral membrane proteins (IMPs)8-12, the structural biology of this class of proteins remains underdeveloped, with only a few hundred high-resolution structural models of membrane proteins determined by crystallography and NMR deposited into the RCSB Protein Data Bank as of summer 2012. However, advances in solution-state NMR spectroscopy are leading to its increased importance in the study of the structure and dynamics of IMPs13–18. There has recently been significant progress in sample-preparation protocols for membrane proteins, and these techniques19 now enable studies of much larger structures through the application of the principles of transverse-relaxation-optimized spectroscopy (TROSY)20.
The classical paradigm for protein structure determination by solution-state NMR spectroscopy is to extract and assign a dense network of 1H-1H NOEs in order to define the three-dimensional fold of a protein. This still presents great challenges for liquid-state NMR-based structural investigations of membrane proteins due to significant peak overlap in the spectra caused by large line widths, limited chemical-shift dispersion, and poor diversity of the amino acids in transmembrane regions of alpha-helical proteins.
An alternative to 1H-1H NOEs as a route to high-resolution structural restraints is found in the controlled re-introduction of anisotropic RDCs21–23. RDCs constitute an excellent source of structural and dynamic information. The dipolar coupling between two atoms, j and k, or Djk, is related to the internuclear distance rjk typically known in advance (e.g. bond length for covalently linked nuclei), and to the angle between the vector connecting the interacting nuclei and the static magnetic field by the relation <3cos2Θ−1>, where the brackets indicate time-averaged sampling. These couplings can be a valuable source of angular structural data for NMR studies of macromolecules. This is because direct information on the orientations of the corresponding bond vectors relative to the protein's steric alignment vector is provided. However, molecular tumbling averages these interactions to zero in conventional isotropic solutions24. It has been shown that RDCs can be measured by utilizing some type of anisotropic media to allow for partial alignment and, therefore, non-vanishing dipole-dipole interactions22. Such incomplete directional averaging of macromolecules in liquid crystalline media would allow routine measurement of residual dipolar couplings, while retaining conditions essential for high-resolution solution-state NMR (i.e. rapid tumbling).
A highly effective method to induce weak-alignment of proteins is through mixing them with a dilute liquid-crystalline medium, such that the interaction between the protein and the medium is weak and highly transient (< ns lifetime). A number of liquid-crystal alignment media have been developed to measure accurate RDCs, including filamentous phage25, DMPC/DHPC bicelles22, C12E5 polyethylene glycol26, ternary mixtures of cetylpyridinium Cl/Br, hexanol, and sodium Cl/Br26,27, cellulose crystallites28, and a highly hydrated anisotropically compressed polyacrylamide gel29.
Except the polyacrylamide gel in some cases30–33 and the fd bacteriophage in one case34, most of these media have been demonstrated to be incompatible with the detergents and lipids needed to solubilize membrane proteins. With the compressed gel, it has been generally difficult to soak membrane proteins to higher than 0.1–0.2 mM because of the inhomogeneous pore size of randomly cross-linked gel matrices, limiting accuracy and signal-to-noise for NMR measurements. Because of this, we have never succeeded to measure accurate RDCs for a single-chain protein longer than 150 residues; thus far our only success has been with homomultimeric proteins31. In addition, for large systems the polyacrylamide gel may not be practical due to strong interactions with the acrylamide mesh, which reduce the molecular tumbling rate. This can be tuned by the gel concentration, but acrylamide gels can't be used at lower concentration than about 4 %, due to mechanical instability.
More recently, two new detergent-compatible liquid crystals have been reported, one based on collagen35, and the other based on nucleic-acid G-tetrad structures36. For these two new media, the reduction in molecular tumbling rates for large systems is much less problematic and they are easy to produce and non-expensive. However, both methods still have issues with signal-to-noise and/or general detergent and buffer compatibility. The alignment induced by collagen gels is quite small when compared to other alignment media. The d(GpG)-based G-tetrad stacks require an excess of potassium which enables effective stacking of pyrene moieties on the exposed guanine tetrads, but some specific detergents can be incompatible with the presence of potassium. Thus measuring RDCs for membrane proteins has remained a difficult challenge.
DNA nanotubes and NMR of Integral Membrane Proteins
Inspired by the architecture of the established phage-based alignment method22,37 and facilitated by the magnetic susceptibility anisotropy of DNA, we designed DNA-nanotube liquid crystals6 as the first detergent-compatible liquid crystal generally suitable for high-resolution NMR study of membrane proteins (Fig. 1). Our six-helix DNA-nanotube technique represents a simple, versatile and stable method to align macromolecules for the measurement of dipolar-coupling interactions. The ability to tune the alignment of molecules over a large selection of detergents, buffers, pH (stable from pH 4 to 9) and large range of temperatures (stable until 60 °C) makes the use of DNA-nanotubes especially attractive for studies of membrane proteins as compared with other alignment media which align membrane proteins in a more limited range of conditions.
Figure 1.
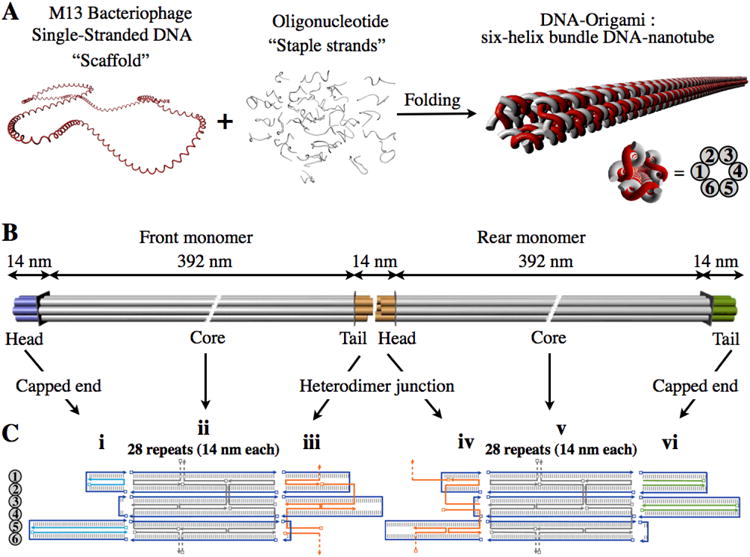
DNA-nanotubes design.
a, Schematic illustration of six-helix bundle DNA-nanotube folding. In red, single-stranded M13-based vector p7308 “scaffold”. In grey, single-stranded “staple” oligonucleotides of length 42 bases, programmed with complementarity of the scaffold. Right, scaffold-plus-staples schematic view of the six-helix bundle DNA-nanotube folded and cross-sectional view of the DNA-nanotube. b, 3D cartoon view of 800 nm-long six-helix bundle heterodimer, (not-to-scale). Left, six-helix bundle front monomer with core module in grey, capped head module in blue and connector tail module for heterodimerization in orange. Right, six-helix bundle rear monomer with core module in grey, connector head module for heterodimerization in orange and capped tail module in green. c, Scaffold-plus-staples schematic view of the heterodimer junction of front and rear monomer. One strand of each double helix is contributed by the scaffold shown in blue, and the other strand is contributed by a staple. Base pairs are depicted as short vertical lines. Helices 1–6 are labeled on the left. In the orientation displayed, the outside surface of the nanotube is facing the viewer. (i) Front monomer head module. Three staple strands serve to cap the front monomer head (shown in cyan). (ii, v) Core module. There are 28 repeats of 42 bp modules for each monomer. A scaffold crossover connecting helix 2 to 3 occurs in the fifteenth repeat and one connecting helix 4 to 5 occurs in the fourteenth repeat (not shown). (iii) Front monomer tail module. Three staple strands with a total of 26 unpaired bases decorate the tail. The scaffold strand is unpaired for 36 bases. (iv) Rear monomer head module. Three staple strands with a total of 36 unpaired bases decorate the head. These unpaired regions are complementary to the corresponding 36 unpaired bases of the front monomer tail scaffold strand. The 26 unpaired bases in the rear monomer head scaffold strand are complementary to the 26 unpaired bases of the three staple strands that decorate the front monomer tail. In the DNA-nanotube heterodimer, these unpaired regions match up to form the complete intermonomer junction. (vi) Rear monomer tail module. Four staple strands serve to cap the rear monomer tail (shown in green).
DNA nanotubes maintain a liquid crystalline phase in the presence of zwitterionic or negatively charged detergents typically used to solubilize membrane proteins for structural study. In our hands, DNA-nanotube liquid crystals have yielded successful weak alignment of a number of membrane or membrane-associated proteins in the presence of different types of detergents. Negatively charged detergents have been used to enable measurement of RDCs for a 40 kDa tetrameric BM2 channel, which includes a 20-residue membrane anchor and a soluble coiled-coil tetramerization domain, reconstituted in 1-myristoyl-2-hydroxy-sn-glycero-3-[phospho-rao-(1-glycerol)] (LMPG) detergent micelles15. Negatively charged detergents have also been used in mixed detergent micelles to enable measurement of RDCs for the human ζ-ζ transmembrane domain of the T-cell receptor6 embedded in a 5:1 molar ratio of dodecylphosphorylcholine (DPC) and dodecylsulfate (SDS) mixed detergent micelles, for the human immunoreceptor DAP12TM dimer fragment, and for a covalently linked NKG2C-DAP12 trimeric intetradecylphosphorylcholine (TDPC)-SDS mixed detergent micelles38. DNA-nanotube liquid crystals are also stable in the presence of zwitterionic detergents and have been used to align a tetrameric M2 channel6 reconstituted with 1,2-diheptanoyl-sn-glycero-3-phosphorylcholine (DHPC) and several mitochondrial carriers reconstituted in DPC or in 1-myristoyl-2-hydroxy-sn-glycero-3- phosphocholine (LMPC) (data not published). However, we never attempted the use of DNA-nanotube to align a membrane protein reconstituted with a positively charged detergent. DNA nanotubes have a net negative surface charge and positively charged detergents might interact too strongly with the nanotube.
More recently, we described a new solution-NMR method, heavily relying on weak alignment by our DNA-nanotube media, for structural characterization of mitochondrial uncoupling protein 2 (UCP2)7, a 33 kDa protein with six transmembrane domains reconstituted in mixed detergents (150 mM DPC, 1 mM cardiolipin and 2 mM DMPC). This method combined orientation restraints derived from NMR residual dipolar couplings and semiquantitative distance restraints from paramagnetic-relaxation-enhancement measurements. The local and secondary structures of the protein were determined by piecing together molecular fragments from the Protein Data Bank that best fit experimental RDCs from samples weakly aligned in a DNA-nanotube liquid crystal.
DNA-nanotube design
While a detailed understanding of DNA-nanotube design is not required for success in the weak-alignment application, we have included a complete description for users who may wish to modify the design. Our DNA-nanotube design involves the self-assembly of a parallel array of six double helices, for which every set of three adjacent helices frames an angle of 120° (Fig. 1a). Adjacent double helices are held together by Holliday-junction crossovers that occur every 42 base-pairs. In order to build nanotubes of 0.8μm uniform length, an assembly strategy was conceived to link two unique 0.4μm monomers (“front” and “rear”) first assembled separately using the DNA-origami method4,5 (Fig. 1b). For each monomer a 7,308-nucleotide (nt) M13-derived single stranded circle of DNA is used as a “scaffold” and 168 single strands of DNA of length 42 nt programmed with complementarity to three separate 14-nt regions of the scaffold are used as “staples” (Fig. 1c). The staples self-assemble with the scaffold into the shape of six parallel double helices curled into a tube. Each DNA-nanotube monomer was divided into three structural modules: core, head and tail (Fig. 1b,c). The inclusion of these three modules in the design allows for the linkage of monomers in a head-to-tail fashion. Three extra staple strands block the head of the front monomer, and four extra staple strands block the tail of the rear monomer (Fig. 1c). To facilitate heterodimerization, three extra staple strands with unpaired bases decorate the tail of the front monomer, and three extra staple strands with unpaired bases decorate the head of the rear monomer (Fig. 1c). After folding, purification and heterodimerization, the aqueous suspension of DNA-nanotube dimers should be concentrated to approximately 25 mg/mL, well above the nematic threshold where they spontaneously align to form a stable liquid crystal as indicated by strong birefringence observed through crossed polarizers (Fig. 2a).
Figure 2.
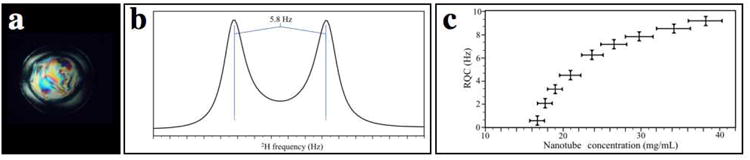
Characterization of DNA-nanotubes liquid crystal.
a, 1 μL drop solution birefringence exhibited between crossed polarizers by DNA-nanotube heterodimers at 25 mg/mL. b, 1D NMR spectrum of 2H2O at 2H frequency of the six helix bundle sample 25 mg/mL in 2 mM MgCl2, 20 mM Tris-HCl pH 7.5, 100 mM DPC, 90%/10% H2O/D2O. The 1D spectrum was recorded at 2H frequency of 600 MHz at 25 °C on a Bruker 600 MHz spectrometer. c, Concentration dependence of the 2H2O Residual Quadrupolar Coupling. Three nanotube preparations were tested, and the vertical error bars represent the standard deviation of the splitting measurement. Horizontal error bars represent the standard deviation in nanotube concentration between the three samples.
When the liquid crystal is aligned in an 11.4 T magnetic field in the presence of 100 mM DPC and 10% D2O, the weakly oriented HDO yields 2H quadrupolar splitting ranging from 4–8 Hz (Fig. 2b). This variation in alignment performance depends on the concentration and the quality of the DNA-nanotube preparation. Figure 2c shows the quadrupolar splitting of the deuterium line which is proportional to the extent of alignment. A nanotube concentration on the lower end of this spectrum (∼4 Hz) has proven sufficient for most of our membrane-protein applications. The 2H quadrupolar splitting can be determined on a 10% D2O DNA-nanotube sample via a simple 1D experiment. The concentration of ordering nanotube medium needed for optimum alignment may be different for positively charged proteins, as DNA nanotubes have a net negative surface charge. If the protein of interest carries a net positive charge, the alignment could be stronger than for a neutral or negatively charged protein. The magnetic-susceptibility anisotropy of the DNA nano-rod is dominated by the diamagnetic purine bases, which have the lowest energy when the external magnetic field is parallel to the plane of the base. Thus the DNA nanotubes align with their long axes orthogonal to the field.
This updated protocol aims to generalize the use of DNA nanostructures as a detergent-resistant liquid crystal for membrane-protein NMR study by offering a user-friendly method for the measurement of membrane-protein RDCs with a high level of accuracy.
Overview of the procedure
The workflow for building DNA-origami nanotubes is illustrated in Figure 3. A single-stranded M13-based p7308 “scaffold” DNA molecule is self-assembled into a nanotube shape using a set of mostly 42 nt “staple” oligodeoxyribonucleotides. The workflow starts with pooling equal amounts of 168 concentration-normalized staple strands for each monomer (“front monomer” and “rear monomer”)(Fig. 3, Box. 1) and, in parallel, producing the p7308 scaffold (Fig. 3, Box. 2). Once “scaffold” DNA is prepared and “staple” oligonucleotide pooling is performed, the self-assembly can be accomplished. Front and rear monomers can be assembled in separate reaction vessels by mixing six-fold excess of pooled staples (“front monomer staples” or “rear monomer staples”) with p7308 ssDNA scaffold in magnesium-containing aqueous buffer (Step 1). After the mixing step, front and rear monomers can be folded separately by heat denaturation, followed by cooling for renaturation (Steps 2–5). Each folded monomer sample can be purified separately from excess staple strands via gravity-flow ion-exchange chromatography (Steps 6–14). Finally, nanotube heterodimers can be self-assembled by combining purified front and rear monomer mixtures together (Steps 15–19). The nanotube heterodimer mixture can then be PEG-precipitated and concentrated. (Steps 20–34). At this point, the sample is ready for mixing with the target protein of interest, further concentration, and subsequent NMR characterization.
Figure 3.
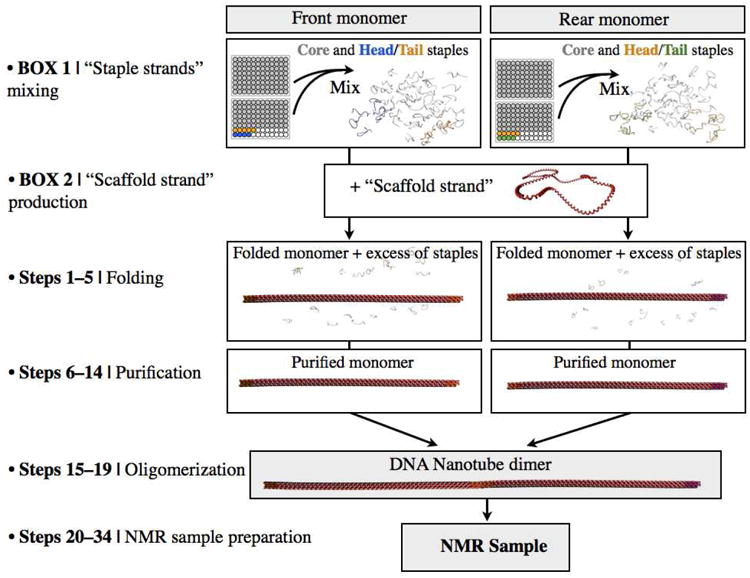
A flowchart diagram summarizing the steps involved in and time required for setting up a large-scale synthesis of detergent-resistant DNA nanotubes.
Step-by-step guide through molecular self-assembly of scaffolded DNA origami nanotube for NMR structure determination of membrane proteins. Box 1 and 2 represent starting materials preparation. DNA nanotubes are illustrated as simplified, not-to-scale. Box 1: involves pooling staple oligonucleotides according to structural modules, Rear and Front monomers (from 96 well plates). Box 2: involves producing the single-stranded M13-based vector p7308 “scaffold”. Step 1-5: folding. Self-assembly reactions are prepared. Front and rear monomers are folded in separate chambers by heat denaturation, followed by cooling for renaturation. Step 6-14: purification. Each folded monomer sample are purified separately from excess staple strands via gravity-flow ion-exchange column. Step 15-19: oligomerization. Nanotube heterodimers are self-assembled by combining purified front and rear monomer mixtures together. Step 20-34: NMR sample preparation. The nanotube heterodimer mixture are PEG-precipitated and concentrated for NMR sample preparation.
Box 1. Hydrating and pooling oligonucleotide “staple strands”.
The DNA nanotubes are designed as heterodimers of two independent six-helix bundle monomers joined together in a head to tail fashion (Fig. 1b,c). Each monomer is folded with the p7308 scaffold and unique pools of oligonucleotide staple strands. To generate these pools, we purchase desalted and lyophilized DNA oligonucleotides in 96-well plates on the 200 nmol scale from Invitrogen. Oligonucleotide staple strands are listed in Supplementary Tables 2–7. For cost-saving reasons, we generally request the entire synthesis of each staple strand; the amounts we receive vary within a two-fold range comparing any pair of strands. Once hydrated, equal volumes of each oligonucleotide are pooled in two groups corresponding to the necessary staple strands for each monomer (Fig. 3). For the front monomer, the pool includes core staples, “caps” for the head of the monomer to prevent non-specific oligomerization, and connector staples for programmed dimerization at the tail of the monomer (Fig. 1b, Supplementary Tables 2–4). For the rear monomer, the pool includes core staples, “caps” for the tail of the monomer to prevent non-specific dimerization, and connector staples for programmed heterodimerization at the head of the monomer (Fig. 1b, Supplementary Tables 2–7). Pools are hydrated to achieve an average concentration of ∼5 μM per staple strand; individual strand concentrations therefore vary within a range of ∼3.5–6.5 μM. Because we are adding a large excess of staple strands compared to scaffold strand, therefore achieving an exact excess of each strand is not needed.
Additional Materials
Procedure
To achieve near-complete resuspension of each staple strand, first apply 200 μL of dH2O to each lyophilized oligonucleotide well on 96-well plates purchased from Invitrogen.
Seal each 96-well plate and allow to sit for at least 30 minutes at room temperature. Vortex at 700 rpm for 2 minutes to actively re-suspend every 10 minutes.
Spin down the plates to collect all liquid at 500 g for 2 minutes.
Recover the material from each well with a multi-channel pipette into a common reservoir (multichannel pipetter basin), either for the front monomer or for the rear monomer (Supplementary Tables 2–7). The front monomer pool will contain staple strands for front head cap, front core, and front tail connector, while the rear monomer pool will contain staple strands for rear head connector, rear core, and rear tail cap).
Apply 50 μL dH2O to the empty wells from the 96-well plates in order to collect additional material.
Seal each plate and allow to sit for at least 30 minutes at room temperature. Vortex at 700 rpm for 2 minutes to actively re-suspend every 10 minutes.
Spin down the plates to collect additional material, pool the wells appropriately as before and mix the solution with the original solution from step 4.
Estimate the concentrations of the pooled staple stocks using a UV spectrophotometer (A260 = 1 for 30 μg/mL oligonucleotides in 1 cm path length). If necessary, add water to achieve a desired target concentration. If the estimated concentration falls below the desired ∼5 μM average, a greater volume of each pool can be used for folding.
-
Label the appropriate pools “rear monomer staples stock” and “front monomer staples stock.”
<PAUSE POINT> The DNA oligonucleotide pools are very stable and can be stored at -20 °C for at least twelve months.
Box 2. Nanomole-Scale Production of M13 Bacteriophage single-stranded DNA “scaffold”.
We use a modified 7,308-base bacteriophage M13 genome [Please supply a hyperlink to the sequence (annotated sequence of this genome)?] as described previously for DNA origami13. Hyperlink: www.pnas.org/content/suppl/2007/04/02/0700930104.DC1/00930SuppAppendix2.pdf, To achieve sufficient quantities of this single-stranded DNA scaffold, production of the bacteriophage that bears the modified 7,308-base genome is progressively scaled-up in a series of steps that yield “pre-inoculation” phage, then “inoculation” phage, and finally nanomole-scale phage. Inoculation phage are produced in two steps (“pre-inoculation” and “inoculation”) to ensure sufficient quality and quantity.
Additional Materials
Luria Broth (Research Products International, cat. no. L24041-500.0)
Bacto agar (General Stores, cat. no. 4236)
Petri dishes, 100 × 15 mm CRITICAL All the equipment used for growing cells should be sterilized.
JM109 bacteria (New England Biolabs, cat. no. E4107S)
M13mp18 Single-stranded DNA New (England Biolabs, cat. no. N4040S)
Procedure
Transform the recombinant M13 bacteriophage RF dsDNA into JM109 Escherichia coli cells.
Pre-warm an LB agar plate at 37 °C for ∼30 minutes.
Streak the pre-warmed LB agar plate for single colonies and incubate the LB agar plate overnight at 37 °C.
After overnight incubation, pick a single colony from the LB agar plate and use to inoculate a 50 mL LB culture. Incubate for 8 hours at 37 °C and ∼250 rpm.
Collect bacteria by centrifugation at 6,000 g for 20 minutes at 4°C.
Recover the supernatant (bacterial pellet can be discarded) and precipitate the bacteriophage that bears the 7,308-base scaffold by adding polyethylene glycol (average MW=8,000) and NaCl to final concentrations of 4% and 0.5 M, respectively.
Incubate on ice for 30 minutes, then collect the precipitated bacteriophage by centrifugation at 6,000 g for 20 minutes.
-
Re-suspend pelleted bacteriophage in 100 mL of 10 mM Tris (pH∼8.5), 1 mM EDTA. This is “pre-inoculation” bacteriophage that will be used in following steps to scale up production of the 7,308-base scaffold.
<PAUSE POINT> The pre-inoculation bacteriophage can be stored at -20 °C for at least 12 months.
To generate the “inoculation” phage, pre-warm an LB agar plate at 37 °C for ∼30 minutes.
Streak JM109 Escherichia coli cells on the pre-warmed LB agar plate to generate single colonies.
Incubate the LB agar plate overnight at 37 °C.
After overnight incubation, pick a single colony from the LB agar plate and use to inoculate a 3 mL 2xYT culture. Incubate overnight at 37 °C and ∼250 rpm.
Use all 3 mL of the overnight culture from the previous step to inoculate a 2 L Erlenmeyer flask containing 300 mL of 2xYT medium supplemented with 5 mM MgCl2.
Shake at 280 rpm and 37 °C until OD650nm = 0.5.
Add 50 mL of the pre-inoculation phage stock. Continue shaking at 37 °C for 4 hours at 280 rpm.
Recover the bacteriophage as described in steps 6–9 above. Resuspend the pelleted bacteriophage in 3 mL of 10 mM Tris (pH ∼8.5), 1 mM EDTA and store as 50 μL aliquots at −20 °C. This is the inoculation phage.
For nanomole-scale production of phage, obtain a single JM109 colony as described in steps 11–13, and use the colony to inoculate 50 mL 2xYT. Incubate overnight at 37 °C and shake at 250 rpm.
Using 3 mL of the starter culture from the previous step, inoculate each of twelve 300 mL 2xYT cultures supplemented with 5 mM MgCl2 in 2 L Erlenmeyer flasks.
Shake at 280 rpm and 37 °C until OD650nm = 0.4.
To one 50 μL aliquot of inoculation phage from step 18, add 600 μL 10 mM Tris (pH ∼8.5), 1 mM EDTA and add 50 μL of the resulting solution to each of the twelve cultures for a multiplicity of infection (MOI) of 1.
Continue shaking for 4 hours at 37 °C and 280 rpm.
After the 4 hours incubation, harvest the bacteriophage by centrifuging the cultures in 4 × 1 L bottles (∼900 mL per bottle) at 6,000 g and 4 °C for 15 minutes.
Recover supernatant (bacterial cells can be discarded) to fresh centrifuge bottles. Again, there should be ∼900 mL supernatant in each bottle. Directly to these centrifuge bottles, add dry NaCl to 30 g/L and dry PEG8000 to 40 g/L.
Mix with magnetic stir bar until all PEG8000 has dissolved. CAUTION At this point, the supernatant should be a cloudy suspension. If it is still clear, it is likely that there are little or no phage present.
After mixing, incubate supernatant on ice for 30 minutes.
Collect the precipitated phage by centrifugation at 4,500 g and 4 °C for 15 minutes. CAUTION The pelleted phage is the fraction of interest, but it is best to save the supernatant in case some of the pellet is dislodged into this fraction.
Allow bottles to sit at an angle for a few minutes, and then remove any additional supernatant with a pipette.
Actively re-suspend pelleted phage into 1/100 the original culture volume (9 mL) with 10 mM Tris (pH∼8.5), 1 mM EDTA. To maximize bacteriophage yield, first resuspend the pellet in each bottle with 5 mL Tris-EDTA buffer, and then rinse with an additional 4 mL buffer.
Transfer resuspended phage to 2 × 40 mL centrifuge tubes (there will be roughly 18 mL per tube) and spin for 15 minutes at 6,000 g and 4 °C to remove residual bacterial cells.
Recover supernatant to a 50 mL conical tube. PAUSE POINT The sample can be stored at -20 °C overnight.
Thaw the harvested phage. When thawed, split into 2 × 250 mL centrifuge bottles.
Add 2 volumes PPB2 per bottle to strip phage protein. PPB2: 0.2 M NaOH, 1% SDS.
Mix gently by inverting tube three successive times.
Add 1.5 volumes PPB3 to each bottle to neutralize the NaOH. PPB3: 3 M KOAc pH 5.5.
Again, mix gently by inverting bottles three successive times.
Incubate centrifuge bottles on ice for 15 minutes.
Spin down bottles at 16,000 g and 4 °C for 10 minutes to remove precipitated SDS and proteinaceous bacteriophage components.
Transfer supernatant to two fresh 250 mL centrifuge bottles.
Precipitate the 7,308-base scaffold by adding 1 volume of 200-proof ethanol. Mix by swirling.
Incubate on ice for 30 minutes, then pellet the DNA scaffold by spinning at 16,000 g and 4 °C for 30 minutes.
Remove supernatant with pipette to minimize loss.
Wash each DNA pellet with 20 mL of 75% ethanol.
Pellet DNA once more at 16,000 g and 4 °C for 10 minutes.
To each pellet, apply 10 mL of 10 mM Tris (pH∼8.5), 1 mM EDTA.
Allow pellet to sit in buffer for 20–30 minutes, and then actively resuspend any remaining pellet by pipetting.
Wash each bottle with an additional 5 mL of 10 mM Tris (pH∼8.5), 1 mM EDTA to collect residual DNA. Note: residual DNA can cling to the side wall of the bottle facing away from the center of the rotor.
Estimate the concentrations of the resuspended scaffold DNA using a UV spectrophotometer (A260 = 1 for 37.5 μg/mL p7308 in 1 cm path length).
The resuspended scaffold DNA (p7308) can be stored at -20 °C. Scaffold DNA are very stable and can be stored at −20 °C for at least twelve months.
Limits of applicability and practical considerations
There are two main considerations in implementing the presented protocol.
First, we must consider the reproducibility and cost-effectiveness of nanomole-scale nanotube production. Any researcher with experience in protein purification should find this protocol quite easy to follow and should be able to obtain reliable results on the first try; all steps besides bacterial growth at 37 °C can be carried out at room temperature, and no knowledge of DNA nanotechnology is required. With this version of the protocol, enough M13-based ssDNA scaffold can be produced for a dozen NMR samples by expression in E. coli by a researcher working for about two days full-time. This scaffold can be stockpiled and frozen away for later use. Assembly and purification of DNA nanotubes sufficient for about five NMR samples requires another five days by a researcher working half-time. Thus the total hands-on labor time required per NMR sample, including scaffold preparation and DNA-nanotube assembly/purification, averages in the long-run to about two-thirds of a day. The most expensive material component is the set of oligodeoxyribonucleotide staple strands at a cost of ∼$300 per NMR sample (according to list price for 200 nmol scale synthesis from Invitrogen).
Second, as with other negatively charged alignment systems such as Pf1 filamentous phage, high pI proteins could bind nonspecifically to DNA nanotubes. The negatively charged DNA nanotubes may slow down the tumbling rate of positively charged protein-micelle complexes. Six-helix DNA nanotubes have ∼24 surface-exposed phosphates per nanometer of their length. With a 7 nm diameter, the surface-charge density then is ∼1.76.10−19 C per nm2. This compares with a surface-charge density of 8.01.10−20 C per nm2 for Pf1 phage37. Slowdown of tumbling due to charge:charge attraction can reduce the accuracy of RDC measurement. Clearly, multiple strategies and new sample conditions to generalize further the method and to allow compatibility with highly positively-charged protein-micelle complexes would be advantageous. Toward this goal, we took advantage of a large library of highly specific DNA binding molecules previously developed for medicinal purposes, which can be used to shield the negative charge of the DNA nanotubes. Reduction in surface negative charge can increase compatibility with protein-micelle complexes that carry significant positive charge and also lead to induction of alignment tensors that are linearly independent to that induced by the original medium39, as has been shown with Pf1 phage alignment of protein G at low versus high ionic strengths40. We have found two approaches that in some cases can help in this pair of concerns:
Measure RDCs at higher-salt concentrations to shield charge-charge interactions
Even with a highly positively charged protein such as ubiquitin or UCP2 membrane protein, it is possible to obtain a reasonable spectrum in the presence of a liquid crystal of DNA nanotubes when salt concentrations are raised above 100 mM. However, some amount of slowdown of tumbling of the protein may occur, thus somewhat compromising the resolution of the acquired RDCs. Furthermore, high ionic strength has an adverse effect on the sensitivity gains of NMR experiments in general. To maximize the advantage of cryogenic probes, the salt concentration should be less than 50 mM41. The nature of this effect is attributed to the increased ionic and dielectric conductivity of the sample, which leads to dissipation of the RF power and appearance of ring currents in the sample tube42-44. We observed that the negative charge of the DNA can be screened as effectively by 20 mM divalent cations (e.g. Mg2+) as by 200 mM monovalent cations (Na+), in terms of eliminating nonspecific protein binding to DNA nanotubes. The advantage is that 20 mM MgCl2 leads to much lower dissipation of RF power and therefore to higher signal-to-noise. Herein, we demonstrate an application to the protein ubiquitin which carries significant positive charge, and thus is an especially good test case for assessing compatibility between DNA nanotubes and positively charged proteins (ubiquitin possesses a positive net charge at pH 7, q = 0.53.10−19 C). It has been shown that ubiquitin interacts nonspecifically with negatively charged media such as phage45. In our experience, interactions between ubiquitin and DNA nanotubes can be adequately minimized with either 20 mM MgCl2 or with 200 mM NaCl. However, time of acquisition of couplings with the same signal-to-noise takes twice as long in 200 mM NaCl compared to 20 mM MgCl2. In addition, the alignment tensor induced under 200 mM NaCl was slightly different than that under 20 mM MgCl2. The parameters describing the alignment tensors are reported in Supplementary Table 1. The normalized scalar product was calculated for the tensor with 20 mM MgCl2 relative to the tensor with 200 mM NaCl. The normalized scalar product is 0.97 between the different alignment media conditions, indicating a co-linearity between the alignment tensors. In this case, the difference was not large enough to enable independent information to be extracted.
Weak alignment in the presence of small DNA-binding molecules
Some specific detergents can be incompatible with the presence of magnesium. In order to generalize the approach, we screened different small positively charged molecules to replace the magnesium ion. Binding modes fall into two categories: intercalation and specific hydrogen-bonding interactions in the DNA grooves46. We screened different positively charged groove binders and intercalators that can potentially shield the negative charge of the nanotubes47. Of the four molecules tested (ethidium, 9-aminoacridine, DAPI, and Hoechst 33258), we found the best performance with Hoechst 33258, a DNA groove binder48,49. As with Mg2+ screening, Hoechst-33258 screening substantially reduces line broadening within NMR spectra of ubiquitin, but more significantly alters the alignment tensors compared to the 200 mM NaCl screened sample Supplementary Table 1. The normalized scalar product for the two alignment tensors measured in presence of 200 mM NaCl and Hoechst 33258 is 0.83, indicating a lower co-linearity compared to the 200 mM NaCl screened sample, although still not enough to enable independent structural information to be obtained. However, perhaps for some proteins with the appropriate surface charge distribution, this difference may be large enough so that additional information can be obtained by repeating under the two conditions.
Thus modification of charged surface DNA can be an effective approach to change molecular alignment with the same alignment media. This is significant because NMR structure determination is more accurate when multiple alignment media are used. For many difficult membrane protein targets, multiple alignment media will be needed for reliable structure determination. The detailed protocol here describes a method where charge compatibility and variations in molecular alignment can be introduced merely by changing the charge distribution of DNA nanotubes via counter ions and small molecules.
Future challenges
While the DNA-nanotube technology is expected to be used for a wide variety of membrane proteins, it will be important to facilitate measurement of linearly independent restraints to achieve more structural information. Additional DNA-nanostructure-based alignment media will be needed. Currently, covalently modified surface charge of nanotubes and the construction of different shape objects are being developed in our laboratories.
Materials
Reagents
Ampicillin sodium salt (Sigma, cat. no. A0166)
Isopropyl β-D-1-thiogalactopyranoside (IPTG; Sigma, cat. no. I6758). ! CAUTION Do not breathe the dust. Avoid contact with skin and eyes.
Triton X-100 (Sigma, cat. no. T8787) ! CAUTION Harmful. Avoid contact with eyes.
EDTA (Fisher Scientific, cat. no. E478-1)
Magnesium chloride hexahydrate 99.995 % (Sigma-Aldrich, cat. no. 255777-25G)
MOPS (VWR International, cat. no. BDH4522-500)
2xYT Broth Capsules Microbial Medium (Research Products International, cat. no. X15640-500.0)
Sodium phosphate dibasic anhydrous (Fisher Scientific, cat. no. S375-500)
Sodium phosphate monobasic anhydrous (Fisher Scientific, cat. no. S397-500)
Polyethylene glycol 8,000 (PEG8000), (Sigma Aldrich, cat. no. P4463-1)
Sodium chloride (Fisher Scientific, cat. no. S271-10)
Tris base (Fisher Scientific, cat. no. BP152-10)
UltraPure Agarose (General Stores, cat. no. 7012)
Glacial acetic acid (Fisher Scientific, cat. no. A491-212) ! CAUTION It is evaporative and corrosive. Wear goggles, lab coat and face mask during experiments. Handle acetic acid inside a hood.
Isopropanol (Fisher Scientific, cat. no. A415-4) ! CAUTION Flammable. Perform all manipulations under a fume hood.
Ethanol 200 proof (General Stores, cat. no. 78880)
Hoechst 33258, 2'-(4-hydroxyphenyl)-5-[5-(4-methylpiperazine-1-yl)benzimidazo-2-yl]bezimidazole, is a synthetic bis-(benzimidazole) derivative developed by the Hoechst Pharmaceutical Co.158.
Equipment
Shaker incubator, 37 °C
BioProducts 96-Well PCR Plate (Fisher Scientific, cat. no. 21-402-441)
Aluminum sealing tape for 96-well plates (Fisher Scientific, cat. no. 11806)
Disposable Multichannel Pipetter Basins (Fisher Scientific, cat. no. 13-681-500)
Gilder fine bar grids (Ted Pella, cat. no. G400)
Qiagen-tip 10000 (Qiagen, cat. no. 10091)
Teflon tube FEP (Thomas Scientific, cat. no. 9567K10)
Shigemi NMR tube
Loading buffer “QBT”: 50 mM MOPS pH 7.0, 750 mM NaCl, 15 % (v/v) Isopropanol, 0.15 % (v/v) Triton X-100. Can be stored at room temperature for up to 6 months. CRITICAL It is highly recommended that all buffers used for chromatography applications be filtered.
Wash buffer “QC”: 50 mM MOPS pH 7.0, 1 M NaCl, 15 % (v/v) Isopropanol. Wash buffer can be stored at room temperature for up to 6 months.
Elution buffer “QF”: 50 mM Tris pH 8.5, 1.25 M NaCl, 15 % (v/v) Isopropanol. Elution buffer can be stored at room temperature for up to 6 months.
Folding Buffer 20x: 100 mM Tris pH ∼8.0, 20 mM EDTA, 200 mM MgCl2. Folding buffer can be stored at room temperature for up to 6 months.
“staple strands” (see Box 1).
M13 Bacteriophage single-stranded DNA “scaffold” (see Box 2).
Protein sample conditions: Typically protein samples in the range 0.1-1mM protein concentration are required. Detergents needed to solubilize membrane proteins have to be compatible with 2mM of MgCl2. Positively charged detergent can be incompatible with DNA nanotube. Large range of pH (stable from pH 4 to 9) and temperatures (stable until 60 °C) can be used.
List of the proteins that have already analysed successfully using this protocol: a mitochondrial uncoupling protein 2 (UCP2) protein7, a 40 kDa tetrameric BM2 channel15, a homologous transmembrane-signaling dimer DAP12, a covalently linked NKG2C-DAP12 trimeric38 and a transmembrane domain homodimer ζ-ζ of the T-cell receptor complex6.
Equipment Setup
Microscope with polarizer and rotating analyzer.
Thermal cycler (MJ Research)
NMR spectrometer equipped with a triple resonance probehead.
RDCs were recorded on protein labelled with 15N, 13C.
NMR spectra were processed and analyzed by using NMRPipe and nmrDraw50.
Fitting of the dipolar couplings to the known ubiquitin structure was done by singular-value decomposition (SVD), using the program PALES51,39. The goodness of fit was assessed by both Pearson correlation coefficient (r) and the quality factor (Q)52.
Procedure
Nanomole-Scale folding of the DNA Nanotube monomers. TIMING: ∼26 h
In a multichannel pipette basin, prepare a 37.8 mL master mix for each monomer containing the following: 120 nM scaffold p7308 (Box 2), 720 nM (average) each staple (Box 1), 20 mM MgCl2, 1 mM EDTA and 5 mM Tris (pH 8.0). Stock concentrations of scaffold and staples will vary, but an example of such a master mix is provided in Table 1. This volume is intended for 240 folding reactions at 150 μL per reaction and includes a 5% excess to account for pipetting error.
Table 1. Nanomole-scale folding of DNA Nanotube monomer.
Example of mix reagents and their concentrations for nanomole-scale folding of DNA nanotube monomers.
| Rear or Front monomer folding | 1× reaction (μL) | 252× reactions (mL) |
|---|---|---|
| Folding Buffer 20× | 7.5 | 1.890 |
| “Scaffold” p7308 (540 nM) | 33.3 | 8.392 |
| “Staples” mix oligos (3.6 μM each) | 30.0 | 7.560 |
| 1 M MgCl2 | 1.5 | 0.378 |
| dH2O | 77.7 | 19.580 |
| Total | 150 | 37.800 |
CRITICAL STEP Magnesium concentrations have been observed to have a drastic effect on the quality of nanotube folding. Optimal concentrations of MgCl2 vary with the design of the structure and with the vendor of the oligonucleotide staple strands. For the six-helix bundle nanotube described in this protocol and for staple strands provided as described by Invitrogen, 20 mM MgCl2 is optimal. Modified nanotubes or nanotubes folded with staple strands purchased from a different vendor may have slightly different optimal concentrations of MgCl2.
CRITICAL It is highly recommended to use pure magnesium chloride hexahydrate (99.995%) during the folding process. EDTA is added to 1 mM final concentration in the master mix to chelate divalent ion impurities that can compete with magnesium during the folding process.
2. After preparing the master mix, use a multichannel pipette to distribute 150 μL aliquots into 96-well plates (BioProducts 96-Well PCR Plate). CAUTION In order to prevent any evaporation during the folding step it is highly recommended to leave an empty “border” of wells on each plate. These border wells will be filled with water, leaving 60 wells per plate for nanotube folding reactions.
3. Fill 60 wells on each of 4 plates for a total of 240 reactions. CRITICAL STEP Make sure that there are no air bubbles trapped in the wells, since they could promote the formation of artifacts during folding. This can be done after aliquoting by gently pipetting the wells up and down.
4. Seal the plates with an aluminum sealing tape for 96-well plates. CAUTION Ensure the plates are very well sealed to prevent any evaporation during the thermal annealing step.
5. Load the 96-well plates into the thermal cycler and set up the thermal annealing ramp as follows: hold at 80 °C for five minutes, then decrease by one degree Celsius every five minutes to 65 °C, then decrease by one degree Celsius every 40 minutes to 20 °C. Use a heated lid to minimize evaporation.
Nanomole-Scale Purification of DNA Nanotube monomer. TIMING: ∼3-4 h
6. Pool all 240 completed reactions for each DNA nanotube monomer into a multichannel pipette basin (i.e. one basin for each of the two monomers) and transfer into delegated 50 mL conical tubes. CAUTION Be careful not to pipette wells filled with water on the border of each plate.
7. Once pooled, bring each sample volume to 100 mL with Buffer QBT (50 mM MOPS pH 7.0, 750 mM NaCl, 15% (v/v) Isopropanol, 0.15% (v/v) Triton X-100). CAUTION Be sure to mix the sample after the addition of QBT to ensure homogenous distribution of DNA.
8. Remove 50 μL of each of the two pools for an analytical agarose gel (Box 3).
9. Column equilibration: Use one Qiagen-tip 10000 ion exchange column per monomer. Label each column as “rear” and “front” monomer. Equilibrate each column with 75 mL Buffer QBT. CRITICAL Allow to flow through completely.
10. Column loading: Apply 100 mL of each monomer to the appropriate column and allow to flow through completely. CRITICAL STEP Collect all flow through to ensure recovery of material.
11. Column washing: After the nanotube pools have completely flowed through the column, wash the column 6 times with 100 mL Buffer QC (50 mM MOPS pH 7.0, 1 M NaCl, 15% (v/v) Isopropanol). CAUTION To improve the wash step, allowing each wash to flow through entirely before applying subsequent washes. Save the washes.
12. Nanotubes elution: Elute each monomer from the column with 100 mL Buffer QF (50 mM Tris-HCl pH 8.5, 1.25 M NaCl, 15% (v/v) Isopropanol). At this stage, the DNA solution should be homogenous and clear.
13. Remove 50 μL of each of the two eluted samples for an analytical agarose gel (Box 3).
14. Add MgCl2 to a final concentration of 25 mM. CRITICAL Magnesium stabilizes the DNA nanotubes after folding. Some precipitate may appear but does not interfere with subsequent steps.
Box 3. Agarose Gel Electrophoresis adapted for DNA origami nanostructures.
Agarose-gel electrophoresis currently provides the most effective method available for high-resolution analysis and separation of well-folded DNA nanostructures. The divalent cation magnesium is a co-factor in DNA-based molecular self-assembly reactions. When separating folded DNA origami nanostructures with agarose gel electrophoresis additions of 11 mM MgCl2 in both the gel and running buffer are required. This promoted a tighter folding of the nanostructures and ensured that minimal changes in the structure conformation took place during the separation.
Additional Materials
12 × 14 cm gel box, OWL Easycast B2 apparatus (Thermo Scientific)
Procedure
For a large 1.5% agarose gel with a total volume of 120 mL gel (12 × 14 cm OWL Easycast B2 apparatus), measure 1.8 g agarose in 600 mL beaker.
Add 0.5× TBE on the balance to total mass of 120 g.
Add additional dH2O to 150 g total mass to account for evaporation while agarose melts.
Microwave on high for 2 minutes, swirl briefly, then microwave an additional minute. CAUTION Wear well-insulated gloves while handling boiling solutions of agarose.
Gently swirl in an ice water bath until steam no longer rises from the beaker. CRITICAL After cooling, add 1 mL of 1.32 M MgCl2 for a final MgCl2 concentration of 11 mM.
Add 6 μL 10 mg/mL ethidium bromide (EtBr) for a final concentration of 0.5 μg/mL. CAUTION It is highly recommended to add ethidium bromide once the solution is below roughly 40 °C to minimize the toxic vapor.
Gently swirl until EtBr is no longer visible.
Pour gel into casting tray and insert comb.
Once gel is solid, fill the gel box with 0.5× TBE containing 11 mM MgCl2.
Remove comb and load the sample.
Set to 60 V, run for 2–3 hours before imaging.
PAUSE POINT The sample can be stored at 4 °C for at least two days.
?TROUBLESHOOTING
Heterodimerization of DNA nanotube monomers TIMING: ∼2 h
15. The DNA nanotubes to be used for NMR alignment experiments are heterodimers of a rear monomer and a front monomer. The DNA nanotubes need to be heterodimerized prior to further purification. Using the material that was eluted from the Qiagen-Tip 10000 ion exchange columns, mix equal volumes of the rear and front monomer elutions. CRITICAL We are assuming that the Qiagen-Tip 10000 purification yielded roughly equimolar quantities of each monomer, thus we need only consider volume. Equimolar amounts of each monomer have to be mixed to form 100% of heterodimer. If one of the monomers is formed in excess, its amount should be reduced to a stoichiometric quantity before mixing. CAUTION Be sure to mix by swirling after combining the two monomers.
16. Warm the mixture by incubation in a 37 °C water bath for 15 minutes. CRITICAL The heterodimerization is performed at 37 °C to improve the kinetics of the reaction.
17. Incubate the mixture in a 37 °C warm room for an additional 1 hour, 45 minutes for a total of 2 hours at 37 °C.
PAUSE POINT At this point, the heterodimerized mixture can be stored at 4 °C if it is necessary to return to the precipitation at a later time. After few hours at 4 °C the sample can turn turbid. This is typical behavior of DNA nanostructure stored in buffer QF at 4 °C, and does not harm the sample.
18. Remove 50 μL of the mixture for an analytical agarose gel (Box 3).
19. The purity and purification efficiency can be checked using agarose-gel electrophoresis (Box 3). 1.5% agarose gels are recommended for use with this nanostructure.
?TROUBLESHOOTING
Concentration of DNA-nanotubes and formation of DNA-nanotube liquid crystals TIMING: ∼4-5 h
20. Add 0.25 volumes of 20% PEG8000 to the heterodimerized nanotubes.
21. Mix gently, and incubate at room temperature for 15 minutes.
22. Spin down the nanotubes for 30 minutes at 15,000 g and 4 °C.
23. Carefully decant the supernatant into another bottle. CRITICAL Save the supernatant in case the nanotube pellet becomes dislodged from the bottle.
24. Spin the pellet once more for only 1 minute at 15,000 g and 4 °C to collect additional supernatant.
25. Carefully remove all remaining supernatant with a pipette.
26. To the nanotube pellet, add sufficient 0.5× Folding Buffer to achieve a concentration of 3 mg/mL assuming 80% recovery from the Qiagen Tip ion exchange columns. For the volumes described in this protocol, this will be about 6 mL. CRITICAL STEP Do not disturb the pellet initially. Simply add the buffer to the tube, and allow the buffer to diffuse into the pellet. Actively re-suspend loose portions of the pellet periodically by swirling. Care should be taken to avoid extremely vigorous mixing at this step. It is highly recommended to let the buffer slowly dissolve the pellet to prevent damage to the nanotubes.
27. Once the pellet has dissolved, mix the nanotube sample gently and transfer to a 50 mL conical tube.
28. Estimate the concentration of the nanotubes.
PAUSE POINT The nanotubes can be stored in 0.5x Folding Buffer at 4 °C for at least six days until one is ready to proceed with the concentration step.
29. Concentrate the nanotubes to ∼30 mg/mL using Centricon-100 concentrator units. Pre-rinse the Centricon-100 concentrator units by adding 2 mL water. Spin at 2,000 g for 5 minutes to achieve concentration.
30. Remove excess water by inverting tubes and spinning at 900 g for 2 minutes.
31. Weigh the Centricon-100 concentrator units then apply DNA nanotube samples and record the mass of the concentrator unit with the DNA nanotubes.
32. Spin the nanotubes in 15 minute increments at 1,500 g and 15 °C. Estimate the concentration by periodically recording the mass of the concentrators with the DNA nanotubes. The beginning concentration of the DNA (3 mg/mL) is 10× lower than the desired concentration (30 mg/ml), therefore a 10× decrease in the mass of the sample gives a good approximation of the desired concentration. CAUTION To prevent damage to the nanotube structure it is recommended that all spins be at speeds less than 2,000 g. Between 15 minute spins, mix the concentrated solution by pipetting up and down gently with a P1000 tip. This will help prevent the buildup of extremely high local concentrations of the nanotubes near the Centricon membrane.
33. When the 10× decrease in sample mass is achieved, recover DNA by inverting tubes into collection vials and spinning 3 minutes, 1,000 g, 20 °C. The final total volume will typically be between 1 mL and 1.5 mL. Concentrated to 30 mg/mL, the nanotube sample will be homogeneous, clear, and viscous. If the DNA-nanotube solution doesn't appear viscous, it is recommended to check the birefringence (step 34). If the sample is not birefringent, spin the nanotubes in 15 minute increments at 1,500 g and 15 °C until the sample appears viscous.
34. Place 1 μL drop solution of DNA-nanotube liquid crystal on a glass microscope slide. Examine the drop at room temperature using a dissecting microscope under normal and crossed polarized light. The nanotubes will appear birefringent between crossed polarizers with characteristic textures of the type shown in Figure 4a.
Figure 4.
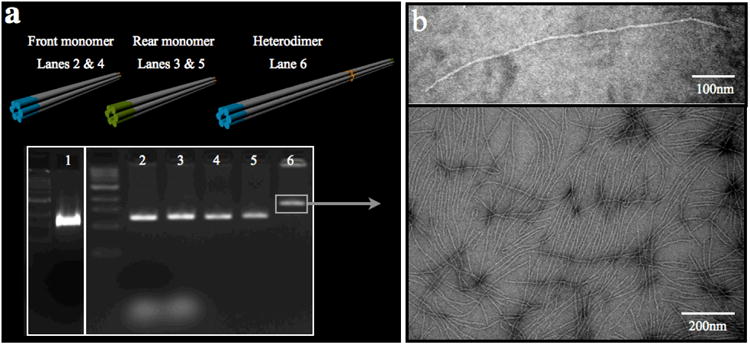
Folding, purification and characterization of DNA six-helix bundle.
a, Cylinder models of 400 nm-long six-helix bundle monomers, 800 nm-long six-helix bundle heterodimer (not-to-scale) and gel analysis of the six-helix bundles after folding and purification. 1 kb ladder. 1, p7308 “scaffold”. 2 and 3, front and rear monomers folded before purification. 4 and 5, front and rear monomers after purification. 6, heterodimer. b, Negative-stain transmission electron micrograph of a purified sample of six-helix bundle heterodimer.
PAUSE POINT DNA nanotubes are very stable and can be stored at 4 °C for at least twelve months.
Measuring residual 2H2O quadrupole coupling in the presence of DNA nanotubes TIMING: ∼1 h
35. Add D2O to 250 μL of the DNA-nanotube liquid crystal to a final concentration of 10%. Mix slowly by pipetting.
36. Use a low DNA affinity Teflon tube to transfer 250 μL of the nanotube sample with 10% D2O into a Shigemi NMR tube. CRITICAL To minimize the lose of DNA, transfer the DNA sample in several steps by pipetting only 40 μL into the NMR tube at a time.
37. Spin down the NMR sample at 500 g for 2 minutes and add the Shigemi plunger. CRITICAL At 30 mg/mL, the DNA-nanotube liquid crystal solution appears viscous. Despite the viscosity, conventional pipettes or teflon tube work well to transfer the liquid crystals to an NMR tube. A uniform and bubble-free sample is obtained by slow centrifugation (100–200 g) after transferring the sample to the tube, inserting the plunger slowly to the bottom of the tube and pulling the plunger to the desired height.
38. Record 1D NMR spectrum at 2H frequency.
39. Process 1D NMR spectra and measure D2O splittings.
Preparation of NMR protein samples with DNA nanotubes TIMING: ∼2 h
40. Pre-rinse a Centricon-100 concentrator unit as in steps 29–31.
41. Weigh the Centricon-100 concentrator unit empty and apply 250 μL + 10 % of DNA nanotube sample at a concentration of ∼25 mg/mL. Weigh the Centricon-100 concentrator unit with the DNA sample.
42. Exchange the DNA nanotubes into the desired protein buffer by diluting the nanotubes 2-fold with the protein buffer.
43. Mix the 2-fold diluted sample slowly by pipetting up and down. Spin the nanotubes in 5 minute increments at 1,500 g, 15 °C.
44. Between each spin, mix the sample slowly by pipetting up and down. Stop the concentration when the columns reach roughly the starting weight.
45. Repeat steps 42–44 three times to achieve sufficient exchange.
46. Once the DNA nanotubes are in the appropriate buffer, an appropriate amount of protein is added to the DNA-nanotube solution. The final NMR sample is then prepared by concentrating down to the appropriate sample volume using a series of 5 minute spins at 1,500 g and 15 °C. CRITICAL During the course of concentration, a local concentration of both protein and nanotubes around the Centricon membrane could appear. As a consequence, there is a much more favorable environment locally for interaction between the nanotubes and the protein. It is recommended to periodically homogenize the DNA/protein concentration between each spin by pipetting slowly up and down.
47. Recover NMR sample from the Centricon concentrator unit. CRITICAL DNA material may stick to the Centricon membrane. It is possible to recover more than 95% of the DNA sample by inverting tubes into collection vials and spinning for 3 minutes at 1,000 g and 20 °C.
<PAUSE POINT> Store at 4 °C or temperature appropriate for protein of interest.
Long-term stability of DNA nanotubes with positively charged proteins
48. Much like other negatively charged alignment systems such Pf1 filamentous phage, high pI proteins could bind nonspecifically to DNA nanotubes. However, we have developed various approaches that can increase compatibility with protein-micelle complexes that carry significant positive charge. The following approaches can be routinely used to record accurate RDC constraints for NMR applications: (A) weak alignment at higher ionic strength (Fig. 5a), (B) weak alignment with magnesium chloride (Fig. 5b), (C) weak alignment in presence of small DNA-binding molecules (Fig. 5c). In option B, positively charged magnesium ions reduce the effective charge of the nanotubes by binding with greater electrostatic affinity to the negatively charged phosphate groups of the DNA (Fig. 5b). Of the four small-molecules tested (ethidium bromide, 9-aminoacridine, DAPI, and Hoechst 33258) that specifically bind DNA and shield the charge, we found the best performance with Hoechst 33258, a DNA groove binder, and this is used in option C.
Figure 5.
Residual Dipolar Couplings measurement in presence of small molecule.
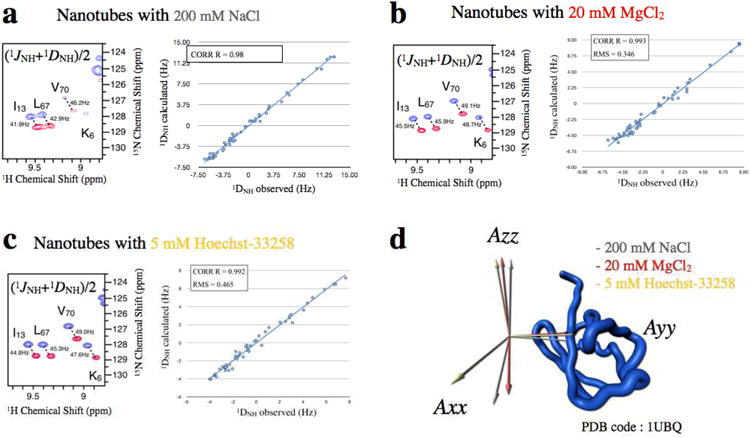
1JNH/2 splittings for the anisotropic samples. Superposition of the 1H-15N HSQC (blue peaks) and TROSY (red peaks) spectra recorded at 25 °C at 1H frequency of 600 MHz. The sample condition is 500 μM uniformly 15N labeled ubiquitin dissolved in 20 mg/mL DNA nanotubes with different buffer conditions. a, With 200 mM NaCl: 20 mM tris, 200 mM NaCl, pH 7.5. With 20 mM magnesium: 20 mM tris, 50 mM NaCl, pH 7, 20 mM MgCl2. b, With 5 mM Hoechst-33258: 20 mM tris, 50 mM NaCl, pH 7, 5 mM Hoechst-33258. The 15N–1HN splittings (in hertz) are marked together with the sequential assignment. Correlation between measured 1H-15N RDCs of ubiquitin in 20 mg/mL DNA nanotubes and couplings calculated on the basis of X-ray crystal structure of Ubiquitin pdb code: 1UBQ are shown for each buffer condition. Linear regression applied to the data gives a correlation coefficient of 0.98, 0.993 and 0.992 respectively. d, Structure of Ubiquitin (pdb code: 1UBQ) shown with the orientation of the principal components of the alignment tensors in grey 200 mM NaCl, in red 20 mM MgCl2, in yellow 5 mM Hoechst-33258.
(A) Weak alignment at higher ionic strength
Perform a small-scale screen of increasing NaCl concentrations for compatibility with the relevant protein/detergent complex. In each well of a 24-well plate, mix 1 μL each of purified protein and DNA nanotubes in a common buffer with specific NaCl concentrations (50, 100, 150, 200 and 300 mM NaCl). Incubate for 24–48 hours at 18 °C.
Using a microscope at 100× magnification, evaluate the amount of precipitate formed for each NaCl condition. The lowest NaCl concentration in which precipitates no longer form is optimal.
Exchange 180 μL of the DNA nanotube liquid crystal at 25 mg/mL into the relevant protein buffer containing the optimal concentration of NaCl (See steps 42–44 for nanotube buffer exchange).
If the purified protein is not in a buffer containing the optimal concentration of NaCl, add an appropriate amount of a concentrated NaCl stock to achieve the optimal concentration. Once the DNA nanotubes are in the appropriate buffer, the NMR samples are prepared by mixing the protein and the DNA 1:1 and then re-concentrating down to the appropriate volume and protein concentration as described previously (50–52).
-
Measure the isotropic and anisotropic splittings in the unaligned and aligned protein samples using the pulse sequences appropriate to the protein as described previously (42–43).
? TROUBLESHOOTING
(B) Weak alignment with magnesium chloride
Exchange 180 μL of the DNA nanotube liquid crystal at 25 mg/mL into the relevant protein buffer containing 20 mM of MgCl2 (See steps 42–44 for nanotube buffer exchange).
If the purified protein is not in a buffer containing 20 mM MgCl2, add an appropriate amount of a concentrated MgCl2 stock to achieve the 20 mM. Once the DNA nanotubes are in the appropriate buffer, the NMR samples are prepared by mixing the protein and the DNA 1:1 by volume and then re-concentrating to the appropriate volume and protein concentration as described above (see steps 46–47).
Measure the isotropic and anisotropic splittings in the unaligned versus aligned protein samples using the pulse sequences appropriate to the protein as described above (see steps 38–39).
? TROUBLESHOOTING
(C) Weak alignment in presence of di-cationic Hoechst-33258 molecule
Exchange 180 μL of the DNA nanotube liquid crystal at 25 mg/mL into the relevant protein buffer containing 5 mM Hoechst-33258 (See steps 42–44 for nanotube buffer exchange). After the initial dilution of the nanotubes with the protein buffer containing 5 mM Hoechst-33258, incubate at room temperature for 15 minutes.
Once the DNA nanotubes are in the appropriate buffer, the NMR samples are prepared by mixing the protein and the DNA 1:1 by volume and then reconcentrate down to the appropriate volume and protein concentration as described above (see steps 46–47).
Measure the isotropic and anisotropic splittings in the unaligned versus aligned protein samples using the pulse sequences appropriate to the protein as described above (see steps 38–39).
Timing
Nanomole-Scale folding of the DNA Nanotube monomers: ∼26h
Steps 1-4, master mix preparation: ∼1h
Step 5, thermal annealing: ∼25h
Nanomole-Scale Purification of DNA Nanotube monomer: ∼3-4h
Steps 6-14, column chromatography: ∼3-4h
Heterodimerization of DNA nanotube monomers: ∼2h
Steps 15-17, heterodimerization: ∼1h
Steps 18-19, agarose-gel electrophoresis: ∼1h
Concentration of DNA-nanotubes and formation of DNA-nanotube liquid crystals: ∼4-5h
Steps 20-28, PEG-precipitation: ∼2-3h
Steps 29-34, concentration of DNA-nanotubes: ∼2h
Measuring residual 2H2O quadrupole coupling in the presence of DNA nanotubes: ∼1h
Steps 35-39, measuring residual 2H2O quadrupole coupling: ∼1h
Preparation of NMR protein samples with DNA nanotubes: ∼2h
Steps 40-47, NMR sample preparation: ∼2h
Troubleshooting
Here is an example TROUBLESHOOTING table: http://www.nature.com/nprot/journal/v7/n9/fig_tab/nprot.2012.081_T3.html
Nanomole-Scale Purification of DNA Nanotube monomer
Steps 10-12, Problem: low purification yield. Possible reason: column was overloaded. Solution: check the sample volume and yield against the capacity of the QIAGEN-tip 10000. The maximum DNA binding capacities of the QIAGEN-tips 10000 are at least 10 mg. Inappropriate salt or pH conditions in buffers may lower this capacity dramatically. Ensure that any buffers prepared in the laboratory were prepared according to the instructions provided for each purification step. Above pH 7, the column exhibits reduced affinity for DNA nanotubes.
Heterodimerization of DNA nanotube monomers
Steps 15-17, Problem: low dimerization yield. Possible reason: ensure after the purification step that each monomer is produced in a 1:1 stoichiometric ratio. Solution: Equimolar amounts of each monomer have to be mixed to form 100% of heterodimer. If one of the monomers is formed in excess, its amount should be reduced to a stoichiometric quantity before mixing.
Weak alignment at higher ionic strength
Step 48, Problem: long-term stability of DNA nanotubes with positively charged proteins. Possible reason: highly positively charged proteins may have an inherent tendency to stick to the negatively charged DNA nanotubes. Solution: Salt concentrations above 100 mM are sufficient in many of these cases for enabling the acquisition of a reasonable spectrum. However, the tumbling of the proteins may slow down slightly, which in turn may compromise the resolution of the acquired RDCs. As an example, ubiquitin has several peaks of weak intensity while in a 20 mg/mL DNA nanotube liquid crystal (See lysine residue 6 in Figure 5a). Additionally, high ionic strength has an adverse effect on the sensitivity gains of NMR experiments in general. Using NMR buffers made of ions with low mobility provides a way to improve the sensitivity of NMR experiments at the high salt concentrations that may be necessary to prevent non-specific binding to DNA nanotubes. This can be done by using salts of alkaline earth metals such as magnesium, that have a high affinity for the DNA nanostructure, low conductivity, and low ion mobility. This will yield a significant shortening of the pulse length and improvement of the charge compatibility between the protein and DNA (Fig. 5a).
Weak alignment with magnesium chloride
Step 48, Problem: long-term stability of detergents with magnesium ions. Possible reason: some detergents can be incompatible with magnesium ions. Solution: To accommodate such detergents, charge shielding can be done via positively charged small molecules that specifically bind DNA.
Anticipated Results
Production of the DNA scaffold
Figure 4a, lane 1, shows typical results obtained from the p7308 scaffold purification. With the protocol described, usually between 20 to 25 nmol of p7308 scaffold can be obtained per 3.6 L of culture with a purity greater than 95%.
Heterodimerization of DNA nanotube monomers
Figure 4a shows typical results obtained from six-helix bundle after folding, purification and dimerization. With the protocol described, 25 to 40 mg of DNA nanotube can be obtained with a purity greater than 95 %. This purification protocol has been used consistently successful in generating nanotubes of sufficient quality and quantity for subsequent NMR experiments. The scale of the preparation can be easily scaled by modifying master mix quantities and Qiagen Tip column capacities.
Measuring residual 2H2O quadrupole coupling in the presence of DNA nanotubes
A DNA-nanotube liquid crystal with 10% D2O yields stable residual quadrupole coupling (RQC) for the 2H lock signal (Fig. 2b). Splitting of ∼6 Hz is routinely observed for large scale preparations at ∼25 mg/mL, though greater splitting can be achieved at higher nanotube concentrations (Fig. 4c).
Weak alignment at higher ionic strength
The optimal NaCl concentrations will vary from protein to protein depending on the extent of the positive charge. For ubiquitin, which is highly positively charged, the optimal NaCl concentration is 200 mM.
Weak alignment with magnesium chloride
The critical DNA concentration required to promote anisotropic protein behavior is unchanged in presence of 20 mM MgCl2, which leads to a much lower dissipation of RF power and therefore a higher signal-to-noise ratio relative to NaCl. To achieve the same signal-to-noise ratio requires an acquisition time twice as long in 200 mM NaCl compared to 20 mM MgCl2 (Fig. 5b). Additionally, the alignment tensor induced under 200 mM NaCl is slightly different than that under 20 mM MgCl2 (Fig. 5d and Supplementary table 1). Though this approach provides a general solution for the spectral optimization of many other proteins that suffer from interfacial line broadening caused by non-specific binding to the DNA nanotubes, some detergents can be incompatible with magnesium ions. To accommodate such detergents, refer to the following section in which we describe charge shielding via positively charged small molecules that specifically bind DNA.
Weak alignment in presence of di-cationic Hoechst-33258 molecule
The critical DNA concentration required to promote anisotropic protein behavior is in the same in the presence of 5 mM Hoechst-33258 and the nanotubes show similar deuterium splitting. As with Mg2+ charge shielding, Hoechst-33258 shielding substantially reduces observed line broadening in ubiquitin spectra, but more significantly alters the alignment tensors compared to the sample shielded with 200 mM NaCl (Fig. 5c,d and Supplementary Table 1). Modification of the charged surface of the DNA nanotube is not only an useful means of making positively charged proteins more compatible with the system, but also an effective approach to achieve multiple alignment tensors with the same alignment medium.
Acknowledgments
We thank Isabel Ayala and Jerôme Boisbouvier for the gift of the ubiquitin vector and Rémy Sounier for helpful discussions. The work was supported by NIH Grants 1U54GM094608 (to J.J.C.) and NIH grants 1DP2OD004641 and 1U54GM094608 to W.M.S.
Footnotes
Competing financial interests J.J.C. and W.M.S. declare competing financial interests. A patent entitled “Nucleic-acid-nanotube liquid crystals and use for NMR structure determination of detergent-solubilized membrane proteins” was filed in 2007 on behalf of the Dana–Farber Cancer Institute and Harvard Medical School by Edwards Angell Palmer &Dodge LLP, listing J.J.C. and W.M.S. as two of the co-inventors.
Contributor Information
Gaëtan Bellot, Email: Gaetan.Bellot@igf.cnrs.fr.
Mark A. McClintock, Email: Mark_McClintock@hms.harvard.edu.
James J Chou, Email: james_chou@hms.harvard.edu.
References
- 1.Seeman NC. Nucleic acid junctions and lattices. J Theor Biol. 1982;99:237–247. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 2.Seeman NC. Nanomaterials based on DNA. Annu Rev Biochem. 2010;79:65–87. doi: 10.1146/annurev-biochem-060308-102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothemund PWK. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- 4.Douglas SM, et al. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature. 2009;459:414–418. doi: 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dietz H, Douglas SM, Shih WM. Folding DNA into twisted and curved nanoscale shapes. Science. 2009;325:725–730. doi: 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Douglas SM, Chou JJ, Shih WM. DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc Natl Acad Sci U S A. 2007;104:6644–6648. doi: 10.1073/pnas.0700930104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berardi MJ, Shih WM, Harrison SC, Chou JJ. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109–113. doi: 10.1038/nature10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallin E, Heijne von G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goffeau A, et al. Life with 6000 genes. Science. 1996;274:546, 563–7. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 10.Landry Y, Gies J. Drugs and their molecular targets: an updated overview. Fundam Clin Pharmacol. 2008;22:1–18. doi: 10.1111/j.1472-8206.2007.00548.x. [DOI] [PubMed] [Google Scholar]
- 11.Myers JK, Beihoffer LA, Sanders CR. Phenotology of disease-linked proteins. Hum Mutat. 2005;25:90–97. doi: 10.1002/humu.20118. [DOI] [PubMed] [Google Scholar]
- 12.Groom CR, Hopkins AL. Protein kinase drugs--optimism doesn't wait on facts. Drug Discov Today. 2002;7:801–802. doi: 10.1016/s1359-6446(02)02359-0. [DOI] [PubMed] [Google Scholar]
- 13.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–595. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hiller S, et al. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–1210. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Pielak RM, McClintock MA, Chou JJ. Solution structure and functional analysis of the influenza B proton channel. Nat Struct Mol Biol. 2009;16:1267–1271. doi: 10.1038/nsmb.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, et al. NMR solution structure of the integral membrane enzyme DsbB: functional insights into DsbB-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Horn WD, et al. Solution nuclear magnetic resonance structure of membrane-integral diacylglycerol kinase. Science. 2009;324:1726–1729. doi: 10.1126/science.1171716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautier A, Mott HR, Bostock MJ, Kirkpatrick JP, Nietlispach D. Structure determination of the seven-helix transmembrane receptor sensory rhodopsin II by solution NMR spectroscopy. Nat Struct Mol Biol. 2010;17:768–774. doi: 10.1038/nsmb.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang C, Li Q. Solution NMR study of integral membrane proteins. Curr Opin Chem Biol. 2011;15:560–9. doi: 10.1016/j.cbpa.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Pervushin K, Riek R, Wider G, Wüthric K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prestegard JH. New techniques in structural NMR--anisotropic interactions. Nat Struct Biol. 1998;(5 Suppl):517–522. doi: 10.1038/756. [DOI] [PubMed] [Google Scholar]
- 22.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 23.Bax A, Kontaxis G, Tjandra N. Dipolar couplings in macromolecular structure determination. Meth Enzymol. 2001;339:127–174. doi: 10.1016/s0076-6879(01)39313-8. [DOI] [PubMed] [Google Scholar]
- 24.Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc Natl Acad Sci U S A. 1995;92:9279–9283. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- 26.Rückert M, Otting G. Alignment of Biological Macromolecules in Novel Nonionic Liquid Crystalline Media for NMR Experiments. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- 27.Prosser RS, Losonczi JA, Shiyanovskaya IV. Use of a Novel Aqueous Liquid Crystalline Medium for High-Resolution NMR of Macromolecules in Solution. J Am Chem Soc. 1998;120:11010–11011. [Google Scholar]
- 28.Fleming K, Gray D, Prasannan S, Matthews S. Cellulose Crystallites: A New and Robust Liquid Crystalline Medium for the Measurement of Residual Dipolar Couplings. J Am Chem Soc. 2000;122:5224–5225. [Google Scholar]
- 29.Tycko R, Blanco FJ, Ishii Y. Alignment of Biopolymers in Strained Gels: A New Way To Create Detectable Dipole–Dipole Couplings in High-Resolution Biomolecular NMR. J Am Chem Soc. 2000;122:9340–9341. [Google Scholar]
- 30.Jones DH, Opella SJ. Weak alignment of membrane proteins in stressed polyacrylamide gels. Journal of Magnetic Resonance. 2004;171:258–269. doi: 10.1016/j.jmr.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 31.Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci U S A. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou JJ, Kaufman JD, Stahl SJ, Wingfield PT, Bax A. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J Am Chem Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- 33.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 34.Park SH, Son WS, Mukhopadhyay R, Valafar H, Opella SJ. Phage-Induced Alignment of Membrane Proteins Enables the Measurement and Structural Analysis of Residual Dipolar Couplings with Dipolar Waves and λ-Maps. J Am Chem Soc. 2009;131:14140–14141. doi: 10.1021/ja905640d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma J, Goldberg GI, Tjandra N. Weak alignment of biomacromolecules in collagen gels: an alternative way to yield residual dipolar couplings for NMR measurements. J Am Chem Soc. 2008;130:16148–16149. doi: 10.1021/ja807064k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lorieau J, Yao L, Bax A. Liquid crystalline phase of G-tetrad DNA for NMR study of detergent-solubilized proteins. J Am Chem Soc. 2008;130:7536–7537. doi: 10.1021/ja801729f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marvin DA. Filamentous phage structure, infection and assembly. Curr Opin Struct Biol. 1998;8:150–158. doi: 10.1016/s0959-440x(98)80032-8. [DOI] [PubMed] [Google Scholar]
- 38.Call ME, Wucherpfennig KW, Chou JJ. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol. 2010;11:1023–1029. doi: 10.1038/ni.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zweckstetter M. NMR: prediction of molecular alignment from structure using the PALES software. Nat Protoc. 2008;3:679–690. doi: 10.1038/nprot.2008.36. [DOI] [PubMed] [Google Scholar]
- 40.Zweckstetter M, Bax A. Characterization of molecular alignment in aqueous suspensions of Pf1 bacteriophage. J Biomol NMR. 2001;20:365–377. doi: 10.1023/a:1011263920003. [DOI] [PubMed] [Google Scholar]
- 41.Flynn PF, Mattiello DL, Hill HDW, Wand AJ. Optimal Use of Cryogenic Probe Technology in NMR Studies of Proteins. J Am Chem Soc. 2000;122:4823–4824. [Google Scholar]
- 42.Kelly AE, Ou HD, Withers R, Dötsch V. Low-conductivity buffers for high-sensitivity NMR measurements. J Am Chem Soc. 2002;124:12013–12019. doi: 10.1021/ja026121b. [DOI] [PubMed] [Google Scholar]
- 43.Robosky LC, Reily MD, Avizonis D. Improving NMR sensitivity by use of salt-tolerant cryogenically cooled probes. Anal Bioanal Chem. 2007;387:529–532. doi: 10.1007/s00216-006-0982-4. [DOI] [PubMed] [Google Scholar]
- 44.Voehler MW, Collier G, Young JK, Stone MP, Germann MW. Performance of cryogenic probes as a function of ionic strength and sample tube geometry. J Magn Reson. 2006;183:102–109. doi: 10.1016/j.jmr.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zweckstetter M, Hummer G, Bax A. Prediction of charge-induced molecular alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys J. 2004;86:3444–3460. doi: 10.1529/biophysj.103.035790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neto BAD, Lapis AAM. Recent Developments in the Chemistry of Deoxyribonucleic Acid (DNA) Intercalators: Principles, Design, Synthesis, Applications and Trends. Molecules. 2009;14:1725–1746. doi: 10.3390/molecules14051725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nafisi S, Saboury AA, Keramat N, Neault J, Tajmir-Riahi H. Stability and structural features of DNA intercalation with ethidium bromide, acridine orange and methylene blue. Journal of Molecular Structure. 2007;827:35–43. [Google Scholar]
- 48.Furse KE, Corcelli SA. The Dynamics of Water at DNA Interfaces: Computational Studies of Hoechst 33258 Bound to DNA. J Am Chem Soc. 2008;130:13103–13109. doi: 10.1021/ja803728g. [DOI] [PubMed] [Google Scholar]
- 49.Banerjee D, Pal SK. Ultrafast charge transfer and solvation of DNA minor groove binder: Hoechst 33258 in restricted environments. Chemical Physics Letters. 2006;432:257–262. [Google Scholar]
- 50.Delaglio F, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 51.Zweckstetter M, Bax A. Prediction of Sterically Induced Alignment in a Dilute Liquid Crystalline Phase: Aid to Protein Structure Determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 52.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]