

Abstract
Background
Colorectal carcinoma (CRC) is one of the leading causes of cancer-related mortality worldwide. MicroRNAs (miRNAs, miRs) play important roles in carcinogenesis. MiR-126 has been shown to be down-regulated in CRC. In this study, we identified the potential effects of miR-126 on some important biological properties of CRC cells and clarified the regulation of insulin receptor substrate 1 (IRS-1) and its possible signaling pathway by miR-126.
Methods
The effect of miR-126 on IRS-1, AKT, and ERK1/2 expression was assessed in the CRC cell lines HT-29 and HCT-116 with a miR-126 mimic or inhibitor to increase or decrease miR-126 expression. Furthermore, the roles of miR-126 in regulation of the biological properties of CRC cells were analyzed with miR-126 mimic or inhibitor-transfected cells. The 3′-untranslated region (3′-UTR) of IRS-1 regulated by miR-126 was analyzed by using a dual-luciferase reporter assay.
Results
We found that IRS-1 is the functional downstream target of miR-126 by directly targeting the 3′-UTR of IRS-1. Endogenous miR-126 and exogenous miR-126 mimic inhibited IRS-1 expression. Furthermore, gain-of-function or loss-of-function studies showed that over-expression of miR-126 down-regulated IRS-1, suppressed AKT and ERK1/2 activation, CRC cells proliferation, migration, invasion, and caused cell cycle arrest, but had no effect on cell apoptosis. Knockdown of miR-126 promoted these processes in HCT-116 cells and promoted AKT and ERK1/2 activation by up-regulating the expression of the IRS-1 protein.
Conclusions
MiR-126 may play roles in regulation of the biological behavior of CRC cells, at least in part, by targeting IRS-1 via AKT and ERK1/2 signaling pathways.
Introduction
Colorectal cancer (CRC) is one of the most common human gastrointestinal malignancies in the world with a yearly increasing incidence and mortality rate [1], [2]. It is the fourth leading cause of cancer-related death in both men and women in China [3]. The pathogenesis of CRC is not yet fully understood. It is currently proposed that colorectal carcinogenesis involves multi-step molecular processes with activation of oncogenes, mutation of mismatch repair genes or inactivation of tumor suppressor genes, which affect the proliferation, migration, invasion, apoptosis, or other aspects of cancer cells. In addition to gene activation and inactivation, increasing evidences suggest that microRNAs (miRNAs, miRs) may play roles in the development of CRC [4].
Mature miRNAs are a class of small, non-coding RNA molecules with a length of 20–25 nucleotides. They usually interact with the miRNA-recognition elements in the 3′-untranslated region (3′-UTR) of target mRNAs, regulate mRNA degradation, or repress their translation as important post-transcriptional regulators. MiRNAs have been proven to play critical roles in many biological processes such as cell differentiation, proliferation, apoptosis, inflammatory and immune responses [5], [6]. Increasing evidence has shown that miRNAs are critically involved in tumorigenesis. Depending on the cellular context and target genes that they regulate, miRNAs may function as tumor suppressors or oncogenes [7], [8]. MiR-200 and miR-155 could be involved in cancer cell migration and invasion by regulating the epithelial-to-mesenchymal transition or cellular adhesion [9], [10]. Zhang et al. reported an inverse correlation between metastasis-associated in colon cancer-1(MACC1) and miR-143 expression in colon cancer cell lines and demonstrated that the direct inhibition of metastasis-associated in colon cancer-1 mRNA translation was mediated by miR-143 [11]. Over-expression of miR-211 in HCT-116 cells altered p53 pathway-associated regulatory proteins, e.g., MDM2, Bcl-2, Bcl-xL and Bax [12]. Numerous studies found that miR-126 is significantly decreased in multiple cancer types and, thus, may play a role as tumor suppressor. For instance, low miR-126 expression was observed in non-small cell lung cancer and identified as unfavorable prognostic factor in non-small cell lung cancer patients [13]; miR-126 expression was also decreased in human breast cancer, and may play roles in tumorigenesis and growth by regulating the vascular endothelial growth factor/phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway [14]. The expression of miR-126 in CRC tissues was significantly lower than that in non-tumor tissues, and miR-126 over-expression inhibited the growth of CRC cells [15]. Guo C et al. noted loss of miR-126 expression in colon cancer cell lines when compared to normal human colon epithelia and revealed that miR-126 regulates PI3K signaling partly by targeting p85β [16]. However, the function of miR-126 and its possible signaling pathway in CRC has not been fully elucidated.
Insulin receptor substrate-1 (IRS-1) is a family member of insulin receptor substrates, which were firstly characterized as typical cytosolic adaptor proteins both in insulin receptor (IR) and insulin-like growth factor I receptor (IGF1R) signaling. Recent studies established that IRS-1 also plays roles in promoting mitosis and apoptosis resistance, malignant transformation and proliferation [17]. Chang et al. [18] found that IRS-1 was over-expressed in various types of solid tumors, including breast cancers, leiomyomas, Wilms' tumors, rhabdomyosarcomas, liposarcomas, leiomyosarcomas and adrenal cortical carcinomas. Moreover, IRS-1 is associated with CRC [19] and up-regulated in cancer cell lines [20]. Bioinformatics has shown that the 3′-UTR of IRS-1 contains a putative binding site for miR-126. However, the regulation of miR-126 in CRC and its association with IRS-1 has not been reported yet.
In this study, we aimed to characterize the roles of miR-126 and its possible signaling pathway in the pathogenesis of CRC cells. In gain-of-function studies, we found that over-expression of miR-126 down-regulated IRS-1 expression, suppressed AKT and ERK1/2 activation, CRC cells proliferation, migration, invasion, and resulted in cell cycle arrest, but had no effect on cell apoptosis. Knockdown of miR-126 promoted these processes in CRC cells and up-regulated the expression of IRS-1 protein. By using luciferase-reporter gene constructs, we identified IRS-1 as functional downstream target of miR-126.
Materials and Methods
Cell culture
The CRC cell lines HT-29, HCT-116, SW480 and SW620 were purchased from the cell bank of the tumor hospital of the Chinese Academy of Medical Sciences/Biological Detection Center (Beijing, China). All cells were maintained in RPMI-1640 (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco), 100 IU/mL penicillin, and 100 µg/mL streptomycin at 37°C in a 5% CO2 incubator.
miRNA mimic or inhibitor transfection
All cells were seeded into 6-well, 12-well, 24-well, or 96-well plates at 90% confluence and kept in an incubator at 37°C and 5% CO2 overnight. MiR-126 mimic, miR-126 negative control mimic (NC mimic), miR-126 inhibitor and miR-126 negative control inhibitor (NC inhibitor) were purchased from Ribobio (RiboBio, Guangzhou, China).Varying amounts of miR-126 mimic or NC mimic (RiBoBio) were transfected into HT-29 cells, whereas miR-126 inhibitor or NC inhibitor (RiBoBio) were transfected into HCT-116 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The transfected cells were incubated at 37°C in a 5% CO2 incubator for 24 or 48 h. Total cellular RNA and protein were harvested separately and stored at −80°C until use.
Identification of potential downstream targets of miR-126
To predict potential targets of miR-126, three in silico analysis programs were used for microRNA target prediction, i.e., MicroCosm Targets (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/), Targetscan (http://www.targetscan.org/), and PicTar (http://pictar.mdc-berlin.de/). The 3′-UTR of IRS-1 mRNA (RefSeq NM_005544) has a putative miRNA-126 binding site.
IRS-1 3′-UTR wild type and mutant constructs
In order to test the effects of miR-126 on the expression of IRS-1, we created dual-luciferase reporter constructs. The 3′-UTR of IRS-1 mRNA containing the putative miR-126 binding sequence were synthesized by Sangon Biotech (Shanghai, China). The fragments corresponding to 1-298 nucleotides of the 3′-UTR of IRS-1 mRNA (298 bp) were then subcloned into XhoI and NotI sites downstream of the Renilla luciferase in the psiCheck-2 vector (Promega). To generate a construct containing the miR-126 binding site mutant, we substituted two nucleotides corresponding to the 5′-seeding region of the miR-126 binding site on the wild type fragment.
Wild type and mutant constructs were designated as psi-IRS-1 and psi-mutIRS-1, respectively. The sequences of the constructs were verified by DNA sequencing. The constructs were transformed into DH5α cells and the plasmid DNA was purified using TIANpure MidiPrep kit (Tiangen Biotech, Beijing, China).
Construct transfection and luciferase reporter assay
To determine the specific effect of miR-126 on the 3′-UTR of IRS-1, the miR-126 mimic and the psi-IRS-1 luciferase construct (500 ng/well) were co-transfected into HT-29 cells using Lipofectamine 2000. Renilla and Firefly luciferase levels were measured at 48 h post-transfection using the Dual Luciferase Reporter Assay System (Promega Corporation, Madison, WI). Experiments were performed in quadruplicate wells and data represented the mean of three independent experiments.
Isolation of Total RNA
Cultured cells were rapidly transferred into 1.0 mL of TRIzol reagent (Invitrogen) and total RNA was extracted according to the manufacturer's instructions and stored at −80°C until use.
Real-time quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
The One Step PrimeScript® miRNA cDNA Synthesis Kit and DRR036A (Takara Bio, Inc., Tokyo, Japan) were used to reverse-transcribe the miRNA and mRNA, respectively. For qRT-PCR on transfected cells, 500 ng of total RNA was converted to cDNA. Then, 2 µL of cDNA from each sample was amplified by real-time fluorescent qPCR, using SYBR® Premix Ex Taq™ II (Perfect Real Time) (Takara Bio, Inc.). The expression of miR-126 in each group was calculated relative to that of U6B, a ubiquitously expressed small nuclear RNA used as internal control. Expression of beta-actin was used as normalization control in mRNA qPCR. For miRNA qPCR, the reverse primer was the universal qPCR primer for miRNA (Takara Bio, Inc.). Forward miRNA and mRNA primers were synthesized by Sangon Biotech, and the respective sequences were listed in Table 1. A comparative threshold cycle method was used to compare each condition with the respective controls. The reactions were performed on a LightCycler® (Roche Diagnostics, Basel, Switzerland). The PCR conditions were 30 s at 95°C, followed by 40 cycles of 5 s at 95°C and 20 s at 60°C. The 2−△Ct (2−[(Ct of gene) − (Ct of U6)]) method was used for analysis.
Table 1. Primers used for quantitative reverse transcription-polymerase chain reaction in this study.
| Name | Direction | Primer (5′-3′) |
| For microRNA qPCR | ||
| Universal qPCR primer | Reverse | The One Step PrimeScript® miRNA cDNA Synthesis Kit (Takara) |
| miR-126 | Forward | TCGTACCGTGAGTAATAATGCG |
| U6 snRNA | Forward | CTCGCTTCGGCAGCACA |
| For mRNA qPCR | ||
| IRS-1 | Forward | AGTCCTAACCGCAACCAGAGT |
| Reverse | CCTCAGCCACACATTCTCAA | |
| Beta-actin | Forward | GGCGGCAACACCATGTACCCT |
| Reverse | AGGGGCCGGACTCGTCATACT |
Western blot
Western blot was performed according to the standard protocol. Briefly, 20 µg protein per sample were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes. The membranes were probed with rabbit anti-human IRS-1 primary antibody (1∶1000 dilution; CST), rabbit anti-human p-AKT primary antibody (1∶500 dilution; CST), rabbit anti-human total-AKT primary antibody (1∶500 dilution; CST), rabbit anti-human p-ERK1/2 primary antibody (1∶500 dilution; CST), or rabbit anti-human total-ERK1/2 primary antibody (1∶500 dilution; CST) overnight at 4°C. The membranes were then incubated with horseradish peroxidase-labeled goat anti-rabbit IgG (1∶1000; Beyotime, Jiangsu, China) at room temperature (RT) for 1 h. Glyceraldehyde-3-phosphate dehydrogenase was used as loading control and a mouse anti-human glyceraldehyde-3-phosphate dehydrogenase antibody was used for its detection. The integral of the optical density of the protein bands was measured using Quantity One image analysis software (Bio-Rad, Hercules, CA).
Immunofluorescence
Immunofluorescence staining was performed according to a standard protocol (Santa Cruz Biotechnology). Briefly, HCT-116 cells on coverslips were fixed in acetone at 4°C for 10 min and blocked with 10% goat serum albumin at RT for 1 h. The cells were incubated with rabbit anti-human IRS-1 primary antibody (1∶200) (Santa Cruz Biotechnology) overnight at 4°C. After rinsing in phosphate-buffered saline (PBS) for 3 times, the cells were incubated with secondary polyclonal Cy3-labeled goat anti-rabbit-594 IgG (1∶400) (Zhongshan Goldenbridge Biotechnology, Beijing, China) for 1 h at RT in the dark and then washed with PBS. The anti-IRS-1 antibody was replaced with PBS in the negative control sample. 4′,6-diamidino-2-phenylindole was used to counter-stain the nuclei (5 min at RT). The staining was evaluated and the fluorescence intensity was measured on a Leica converted fluorescence microscope. We quantified the total Cy3 fluorescence intensity (as shown in red) as representation of the protein expression level of IRS-1. The intensity of 4′,6-diamidino-2-phenylindole staining (blue) was used as internal normalization control for adjusting fluorescence signals among different slides.
Cell cycle analysis
Cell cycle analysis was performed by using Cell Cycle and Apoptosis Analysis Kits (Beyotime). HT-29 cells were seeded at a density of approximately 5×105 cells/well into 12-well plates, cultured for 48 h after transfection as mentioned above, harvested, washed with PBS, and then fixed with 70% ethanol overnight at 4°C. Cells were treated with 20 µg/mL RNase, stained with 20 µg/mL propidium iodide for 30 min at 37°C in the dark, and then analyzed by flow cytometry (FACScan; BD Biosciences, San Diego, CA, USA).
Apoptosis assay
The measurement of apoptosis was conducted according to the manufacturer's instructions for the annexin V-fluorescein isothiocyanate apoptosis detection kit (Beyotime). At 48 h after transfection, harvested cells were suspended in 500 µL annexin V binding buffer. Then, 5 µL annexin V-fluorescein isothiocyanate and 10 µL propidium iodide were added to the wells and cells were incubated for 5 min in the dark. The samples were analyzed within 1 h after staining by flow cytometry (FACScan; BD Biosciences). Cells were discriminated as viable cells, necrotic cells, and apoptotic cells by using the Cell Quest software (BD Biosciences); then, the percentages of apoptotic cells from each group were compared. Assays were performed in triplicate.
Cell growth assay
Cell viability was examined using the Cell Counting Kit-8 assay (Dojindo Laboratories, Kumamoto, Japan). HT-29 cells were seeded at a density of 5×103 per well in a 96-well plate and cultured as described above for 48 h after transfection. On completion of incubation, cell viability was determined using Cell Counting Kit-8 according to the manufacturer's instructions. The medium was removed and replaced with 100 µL fresh medium supplemented with the Cell Counting Kit-8 reagent (10 µL) in each well and incubated for 1 h at 37°C. The absorbance (A) of each well was read at 450 nm using an enzyme-linked immunosorbent assay reader (Thermo, USA). The cell viability (% of control) was calculated using the following equation: Survival rate (%) = (Asample − Ablank)/(Acontrol − Ablank), where Asample was the optical density of miR-126-transfected cells, Acontrol was the optical density of NC-transfected cells, and Ablank was the optical density of the wells without cells. All experiments were performed in triplicate.
In vitro transwell migration and invasion assay
Transwell membranes (polycarbonic membrane, diameter 6.5 mm, pore size 8 µm) (Corning Costar, NewYork, USA) coated without or with Matrigel (BD Biosciences) were used to assay migration or invasion of cells in vitro, respectively. At 48 h after transfection, HT-29 or HCT-116 cells were detached by treatment with 0.25% trypsin-EDTA (Gibco), centrifuged, and resuspended in serum-free medium. Transfected cells (10×104 in 200 µL serum-free medium) were reseeded into the upper chamber. Then, 600 µL medium supplemented with 20% fetal bovine serum was added to the lower chamber as chemoattractant. After 48 h of incubation, non-migrating or non-invading cells on the upper surface of the membrane were removed with a cotton swab. The migrated or invaded cells, which penetrated to the lower surface of the membrane, were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet (Beyotime). The number of cells migrating or invading the membrane was counted from 5 randomly selected visual fields, with an inverted microscope at 200× magnification. Results were obtained from three independent experiments.
Statistical analysis
Experimental results are expressed as mean values ± standard error. Statistical analyses were performed with Student's t-test for 2 groups using SPSS software, v13.0 (International Business Machines Corporation). P<0.05 was considered significant.
Results
Expression of miR-126 in CRC cell lines
We analyzed the expression level of miR-126 in a panel of CRC cell lines with different degrees of differentiation and metastatic ability, including HT-29, HCT-116, SW480 and SW620 cells. We observed that the miR-126 expression was relatively higher in HCT-116 cells than in the other three cell lines. The results shown in Figure 1 suggest that miR-126 expression may be associated with the degree of CRC cells differentiation and metastatic ability. Based on this expression pattern, we chose HT-29 and HCT-116 cells for the following gain-of-function and loss-of-function studies, respectively.
Figure 1. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) of miR-126 expression in different colorectal cancer (CRC) cell lines.
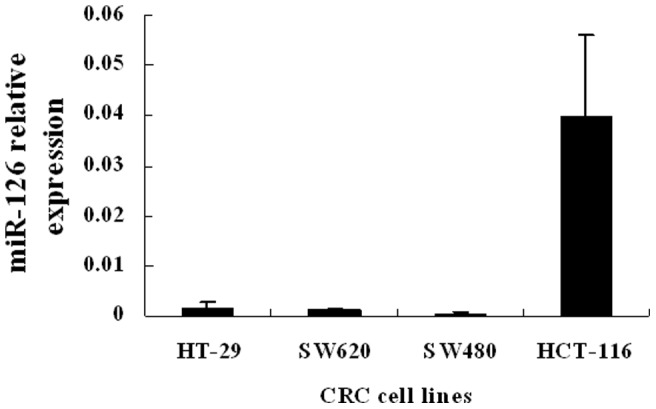
The expression of miR-126 was relatively higher in HCT-116 cells and lower in HT-29, SW620 and SW480 cells among the 4 different CRC cell lines. Data are presented as mean ± SEM of three independent experiments.
Selection of potential downstream targets of miR-126
MiR-126 was found to be down-regulated in CRC [15]. In order to identify downstream targets of miR-126, we used three miRNA target prediction programs, i.e., MicroCosm Targets, Targetscan, and PicTar, to identify potential targets. Interestingly, IRS-1, which is highly expressed in CRC cells [21], is one of the predicted targets of miR-126. A putative miR-126 binding site that encompasses 6 perfectly matched nucleotides was defined at the 3′-UTR of IRS-1 (Figure 2A).
Figure 2. MicroRNA-126 (miR-126) targeting the 3′-untranslated region (UTR) of the insulin receptor substrate 1 (IRS-1).

(A) Position of the miR-126 target site in the 3′-UTR of the IRS-1 mRNA predicted by TargetScan. HT-29 cells were transfected with either the original dual luciferase reporter vector (psi-vector) or an IRS-1 3′-UTR construct with the wild type miR-126 binding site (psi-IRS-1) or the mutated miR-126 binding site (psi-IRS-1-mut). Relative Renilla luciferase activity (normalized to Firefly luciferase activity) was measured 24 h post-transfection. (B) Relative Renilla luciferase activity was reduced in cells transfected with the wild type IRS-1 3′-UTR construct (psi-IRS-1) compared to those transfected with the psi-vector (500 ng/well of a 24-well-plate). Relative Renilla luciferase activity was restored in cells transfected with the mutant construct (psi-IRS-1mut). (*P<0.05, compared to that of psi-vector). (C) Co-transfection of HT-29 cells with 50 nM miR-126 mimic and various luciferase reporter constructs (500 ng/well) reduced the relative Renilla luciferase activity. (*P<0.05, compared to that of psi-vector). Data are presented as mean ± SEM of three independent experiments.
In an effort to delineate potential downstream targets and further understand the mechanisms of miR-126 down-regulation in the pathogenesis of CRC, we transfected HT-29 cells with an IRS-1 3′-UTR luciferase reporter construct containing a wild type miR-126 putative binding sites (psi-IRS-1) or a mutant construct bearing mutations in miR-126 binding sites (psi-mutIRS-1). The relative luciferase activity of the wild type psi-IRS-1 construct showed 45% reduction when compared to the psi-mutIRS-1 construct (P<0.05) (Figure 2B). These results indicate that endogenous miR-126 can regulate IRS-1 expression by directly targeting its 3′-UTR.
To further confirm these findings, the miR-126 mimic was co-transfected with the above luciferase reporter constructs into HT-29 cells. The miR-126 mimic dramatically reduced (>60%) the luciferase activity of the wild type IRS-1 3′-UTR reporter construct psi-IRS-1, whereas the NC mimic had no effect on the luciferase activity in any group (Figure 2C).
However, the miR-126 mimic did not reduce the luciferase activity of the mutant construct psi-mutIRS-1 (Figure 2C), indicating its specific recognition effect. These results further indicate that miR-126 can regulate IRS-1 expression by directly targeting its 3′-UTR.
Alteration of miR-126 expression changed the IRS-1 protein expression level but not the IRS-1 mRNA level
To test whether miR-126 regulates endogenous IRS-1 expression, the miR-126 mimic and inhibitor were transiently transfected into HT-29 and HCT-116 cells, respectively. MiR-126 and IRS-1 mRNA expression levels were assessed. Compared to the NC mimic, transfection with 50 nM of the miR-126 mimic in HT-29 cells led to an approximately 48-fold increase in the miR-126 expression level, as detected by qRT-PCR (Figure 3A). Although there was a decreasing trend in the IRS-1 mRNA expression level in cells transfected with miR-126 mimic, it did not reach statistical significance between the two groups tested (P>0.05) (Figure 3B). At the same time, we found that transfection with 100 nM of the miR-126 inhibitor in HCT-116 cells could decrease the mature miR-126 level noticeably (Figure 3C), while the IRS-1 mRNA level remained unchanged (Figure 3D).
Figure 3. Effects of microRNA 126 (miR-126) mimic or inhibitor on the expression of the insulin receptor substrate 1 (IRS-1) mRNA in human colorectal cancer cell lines.

(A, B) HT-29 cells were transfected with 50 nM of miR-126 mimic or negative control mimic for 48 h. Expression of miR-126 and IRS-1 mRNA in the total RNA of these cells was detected by quantitative reverse transcription-polymerase chain reaction analysis. (C, D) HCT-116 cells were transfected with 100 nM of miR-126 inhibitor or negative control inhibitor for 48 h. Expression of miR-126 and IRS-1 mRNA in the total RNA of these cells was detected by qRT-PCR analysis. Data are presented as mean ± SEM of three independent experiments (*P<0.05, compared to that of negative control mimic treatment).
Next, we determined whether the expression of IRS-1 protein was altered in HT-29 cells transfected with miR-126 mimic or NC mimic and HCT-116 cells transfected with miR-126 inhibitor or NC inhibitor. The increase in miR-126 levels also significantly decreased the IRS-1 protein expression levels as determined by western blot (P<0.05) (Figures 4A, B), whereas the mRNA levels remained unchanged (P>0.05) (Figure 3B). In contrast, to conduct loss-of-function experiments, 100 nM miR-126 inhibitor was transfected into HCT-116 cells and compared to the NC group. The results showed a decrease in miR-126 expression (Figure 3C) and an increase in IRS-1 protein expression (P<0.05) (Figures 4C, D).
Figure 4. Alteration of microRNA 126 (miR-126) influences AKT and ERK1/2 activation by targeting insulin receptor substrate 1 (IRS-1).

(A) HT-29 cells were transfected with miR-126 mimic or negative control (NC) mimic, and total proteins from the cells were used to detect IRS-1, p-AKT, total-AKT, p-ERK1/2, total-ERK1/2, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression by western blotting. (B) Relative protein levels were normalized to those of GAPDH and represented as mean ± SD from three experiments. *, indicates that the expression levels of IRS-1, p-AKT, and p-ERK1/2 were significantly lower in miR-126 mimic transfected cells than that of the negative control (NC) mimic group (P<0.05). (C) HCT-116 cells were transfected with miR-126 inhibitor or negative control (NC) inhibitor, and the above proteins were detected by western blotting. (D) Relative protein levels were normalized to those of GAPDH and represented as mean ± SD from three experiments. *, indicates that the expression levels of IRS-1, p-AKT, and p-ERK1/2 were significantly increased in the miR-126 inhibitor group when compared to the NC inhibitor group (P<0.05). (E) HCT-116 cells stained for IRS-1 by immunofluorescence. IRS-1 was expressed in the cytoplasm and the levels were significantly increased in miR-126 inhibitor-transfected cells. Red, IRS-1; blue, 4′,6-diamidino-2-phenylindole nuclear staining. Pictures were imaged at 630× magnification on a Leica converted fluorescence microscope. (F) Fluorescence intensity of IRS-1 in each group was then calculated. Data are presented as mean ± SEM of three independent experiments (*P<0.05 compared to that of NC inhibitor).
Alteration of miR-126 expression influenced AKT and ERK1/2 activation
To further understand the molecular mechanism of miR-126 in inhibiting tumorigenesis, we found that IRS-1 is a potential novel direct target of miR-126 with a binding site in its 3′-UTR region. IRS-1 can be recruited and phosphorylated by insulin-like growth factor I upon binding to its receptor, insulin-like growth factor I receptor, thus activating downstream signaling pathways such as PI3K/AKT [22]. In this study, we found that the IRS-1 protein expression in HT-29 cells transfected with 50 nM of miR-126 mimic was significantly inhibited (by 47%) as detected by western blot analysis (Figures 4A, B). Consistent with the decrease in the IRS-1 level, over-expression of miR-126 in HT-29 cells also inhibited p-AKT and p-ERK1/2 expression levels (Figures 4A, B). Moreover, transfection of HCT-116 cells with 100 nM of miR-126 inhibitor could up-regulate IRS-1, p-AKT, and p-ERK1/2 protein expression levels, but had no effect on total AKT and ERK1/2 expression levels (Figures 4C, D). These results suggest that miR-126 regulates downstream molecules via targeting IRS-1. In addition, we further performed immunofluorescent staining on HCT-116 cells transfected with miR-126 inhibitor. The staining results showed that the IRS-1 protein was clearly expressed in the cytoplasm of HCT-116 cells (Figure 4E). Its level was markedly increased in the miR-126 inhibitor group when compared to the NC inhibitor group(P<0.05) (Figure 4F), which is in agreement with the results obtained by western blotting.
MiR-126 induced G0/G1 phase arrest in CRC cells
We investigated whether the anti-proliferative activity of miR-126 in HT-29 cells correlated with cell cycle arrest. As demonstrated in Figure 5A, cell cycle analysis revealed that transfection with the miR-126 mimic increased the number of CRC cells in the G0/G1 phase, compared to the NC mimic (P<0.05).
Figure 5. MicroRNA 126 (miR-126) mimic induces G0/G1 phase arrest, but had no effect on cell apoptosis.

(A) MiR-126 mimic and NC mimic transfected cells were stained with propidium iodide (PI) and the DNA content was analyzed by flow cytometry. The number of cells in each phase was calculated using ModFit software. The results shown in the bottom graph were representative of three independent experiments (*P<0.05). (B) HT-29 cells were transfected with 50 nM miR-126 mimic or negative control mimic for 48 h. The percentage of apoptotic cells (Annexin V positive) relative to the total measured cell population is shown in the bottom right quadrant of the upper panel. The results were expressed as fold change relative to the corresponding NC mimic (bottom graph). (P>0.05, compared to that of NC mimic). Data are presented as mean ± SEM of three independent experiments.
MiR-126 had no effect on apoptosis in CRC cells
To measure the effect of miR-126 on CRC cells apoptosis, apoptosis was measured at 48 h after miR-126 mimic transfection by using flow cytometry. There was no significant difference in the number of annexin V-fluorescein isothiocyanate (+) apoptotic cells in the miR-126 mimic-transfected group compared to the NC mimic-transfected group (Figure 5B, P>0.05). These findings indicate that miR-126 might not play an anti-apoptotic role in CRC cells.
MiR-126 inhibited CRC cells proliferation
MiR-126 has been reported to be down-regulated in CRC [23], implicating its potential role in the biological properties of CRC cells. To further characterize the functional importance of miR-126 in CRC tumorigenesis, we examined the effect of miR-126 on the proliferation of HT-29 cells using the Cell Counting Kit-8 assay. We observed that over-expression of miR-126 significantly suppressed the proliferation of HT-29 cells at 48 h after transfection (P<0.05) (Figure 6A).
Figure 6. MicroRNA 126 (miR-126) inhibits cell proliferation, migration and invasion of colorectal cancer cells.

(A) HT-29 cells were transfected with various amounts of miR-126 mimic or negative control (NC) mimic and cell proliferation was analyzed using Cell Counting Kit-8. The miR-126 mimic showed dose-dependent effects on inhibiting cell proliferation after transfection for 48 h. (*P<0.05, compared to that of NC mimic). Data are presented as mean ± SEM of three independent experiments. HT-29 cells were transfected with 50 nM of miR-126 mimic or NC mimic (B) and HCT-116 cells were transfected with 100 nM of miR-126 inhibitor or NC inhibitor (C), migrated cells stained by 0.1% crystal violet were counted 48 h after transfection (×200). The number of migrated cells per field was obtained from at least three replicate wells and presented as mean ± SD (P<0.05) (bottom graph). (D, E) Invaded cells stained by 0.1% crystal violet were counted 48 h after transfection (×200). The number of invaded cells per field was obtained from at least three replicate wells and presented as mean ± SD (P<0.05) (bottom graph). *, indicates significant differences between HT-29-miR-126 mimic or HCT-116-miR-126 inhibitor group when compared to each NC group separately (P<0.05).
MiR-126 inhibited cell migration and invasion
To test the function of miR-126 in CRC cells, stable cell lines expressing miR-126 (HT-29-miR-126) and negative control (HT-29-NC) were established by Liposome 2000 transduction. Over-expression of miR-126 in HT-29 cells significantly suppressed cell migration (P<0.05) (Figure 6B) and cell invasion (P<0.05) (Figure 6D), whereas loss of its expression promoted HCT-116 cells migration (P<0.05) (Figure 6C) and cells invasion (P<0.05) (Figure 6E). These observations suggest that miR-126 plays an important role in inhibiting migration and invasion of CRC cells.
Discussion
Recent studies have demonstrated that miRNAs play important roles in tumorigenesis by a variety of mechanisms. Firstly, they have been shown to alter tumorous epithelial-mesenchymal transitions and adhesion. For example, over-expression of miR-200 could down-regulate epithelial-to-mesenchymal transition and inhibit tumor migration and invasion [5], [6], [10], [24]. Secondly, they regulate the expression of tumor suppressor genes or oncogenes. MiR-21 is highly expressed in CRC and promotes cancer cell growth by targeting programmed cell death 4 [25]. A similar observation has been made for miR-17-92, which may function as oncogene. On the contrary, under-expressed miRNAs, e.g., let-7, could control cell proliferation and apoptosis as tumor suppressors by regulating oncogenes [26]. Thirdly, miRNAs regulate the expression of metastasis-associated gene in cancers [11]. In addition, miRNAs are involved in DNA methylation and regulate vessel formation, thereby influencing tumor development [27], [28].
Numerous studies have demonstrated that miR-126 is associated with inflammation as well as cancers. Over-expression of miR-126 was found to inhibit placenta-specific 1(Plac1) and further increase the viability of gastric cancer by targeting sex-determining region Y-box 2 (SOX2) as oncogene [29]. On the other hand, miR-126 was shown to function as a tumor suppressor in breast cancer [30], lung cancer [31], [32], and bladder cancer [33], cervical cancer [34] by targeting different genes. MiR-126 was down-regulated in CRC tissues [15]. Increased miR-126 expression could suppress CRC cells proliferation [15]. These findings suggest that miR-126 regulates different molecular targets in different cancer processes. However, the function of miR-126 in CRC carcinogenesis remains unknown.
In this study, we found that the expression of miR-126 was relatively low in HT-29, SW480, SW620 cells and high in HCT-116 cells, indicating that miR-126 may be associated with the biological behavior of CRC cells. Therefore, we chose HT-29 cells for subsequent gain-of-function and HCT-116 cells for loss-of-function studies. In this study, we studied the function and potential mechanisms of miR-126 in regulating the biological behavior of CRC cells and its possible signaling pathway. Our results showed that miR-126 inhibited proliferation, migration, invasion, and resulted in cell cycle arrest of CRC cells, but had no effect on cell apoptosis in vitro. To characterize the potential mechanisms of miR-126-mediated changes in the biological properties of CRC cells, we identified molecular targets of miR-126 through target scan analysis. We found that the 3′-UTR of IRS-1 mRNA possesses a miR-126-targeted sequence.
IRS-1, a docking protein, is highly expressed in many kinds of cancers where it acts as an oncogene, e.g., pancreatic cancer [35] and breast cancer [18]; we found that IRS-1 is mainly expressed in the cytoplasm of the CRC cell line HCT-116. Transgenic mice over-expressing IRS-1 or IRS-2 in the mammary gland showed progressive mammary hyperplasia, tumorigenesis, and metastasis [36]. In addition, over-expression of IRS-1 can target upstream binding factor-1 (UBF-1) and, therefore, affect cell size [37]. These findings indicate that IRS-1 may play major roles on in cell growth, proliferation, and differentiation. We also found that insertion of the wild type of miR-126 binding site in the IRS-1 3′-UTR in a dual-luciferase reporter construct led to a reduction of the reporter gene expression and the mutant construct showed the restoration of the reporter gene expression. Moreover, over-expression of miR-126 induced the inhibition of IRS-1, specifically.
It has been reported that miRNA may play roles in CRC tumorigenesis via different signaling pathways. For example, miR-145 and miR-101 have been shown to be under-expressed and promote Wnt/β-catenin signaling in human colon cancer cells [38], [39]. In addition, miR-145 also inhibited p-ERK expression by targeting PAK4, leading to inhibition of tumor growth [40]. Another study showed that miR-31 could activate the RAS signaling pathway through inhibiting RASA1 translation [41]. Down-regulation of miR-144 leads to poor prognosis of CRC patients via activation of the mTOR signaling pathway [42]. All of these findings indicate that CRC processes are regulated by various miRNAs and signaling pathways. Guo et al. found that miR-126 may act as a tumor suppressor in CRC by down-regulating the p58β subunit through the PI3K signaling pathway [16]. Another study indicated that miR-126 suppresses colon cancer cell proliferation and invasion via inhibition of the RhoA/ROCK signaling pathway [23]. These observations raise the question: Could miR-126 be involved in CRC tumorigenesis through a third pathway?
AKT- and ERK1/2-mediated signaling pathways are complex biological processes. It has been confirmed that IRS-1 transduces extracellular signals into cells through AKT and ERK1/2 signaling pathways via miR-145 in CRC cells [21]. Here, we demonstrated that endogenous miR-126 and exogenous miR-126 mimic can inhibit IRS-1 expression. We also found that down-regulation of miR-126 could promote AKT and ERK1/2 activation by targeting IRS-1.Taken together, our results imply that up-regulation of miR-126 could down-regulate IRS-1, inhibit CRC cells proliferation, migration, invasion, and induce cell cycle arrest, but not in apoptosis of CRC cells, partly through AKT and ERK1/2 signaling pathways. Moreover, the results of our study and previous studies further confirme that miRNA and its target genes are not in a one-to-one correspondence relationship, but instead, one miRNA can regulate several pathways by targeting different mRNAs, and one target mRNA can be regulated by several miRNAs and pathways in the same human tumor.
Conclusion
In conclusion, our study demonstrated previously unknown biological functions of miR-126 in CRC cells. In addition, IRS-1 was down-regulated at the post-transcriptional level via a binding site of miR-126 in the 3′-UTR of IRS-1 mRNA. These findings suggest that miR-126 is possibly involved in the tumorigenesis of CRC, at least to some extent, by suppression of IRS-1 through AKT and ERK1/2 signaling pathways, which would have important implications for further understanding the signaling mechanisms involved in modulating tumorigenesis.
Funding Statement
This study was supported by Foundation for Distinguished Young Talents in Higher Education of Guangdong, China (grant number LYM11103). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108. [DOI] [PubMed] [Google Scholar]
- 2. Walsh JM, Terdiman JP (2003) Colorectal cancer screening: scientific review. JAMA 289: 1288–1296. [DOI] [PubMed] [Google Scholar]
- 3. Yang L, Parkin DM, Li L, Chen Y (2003) Time trends in cancer mortality in China: 1987–1999. Int J Cancer 106: 771–783. [DOI] [PubMed] [Google Scholar]
- 4. Faltejskova P, Svoboda M, Srutova K, Mlcochova J, Besse A, et al. (2012) Identification and functional screening of microRNAs highly deregulated in colorectal cancer. J Cell Mol Med 16: 2655–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carthew RW (2006) Gene regulation by microRNAs. Curr Opin Genet Dev 16: 203–208. [DOI] [PubMed] [Google Scholar]
- 6. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- 7. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, et al. (2005) MicroRNA expression profiles classify human cancers. Nature 435: 834–838. [DOI] [PubMed] [Google Scholar]
- 8. Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6: 857–866. [DOI] [PubMed] [Google Scholar]
- 9. Kong W, Yang H, He L, Zhao JJ, Coppola D, et al. (2008) MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 28: 6773–6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22: 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Wang Z, Chen M, Peng L, Wang X, et al. (2012) MicroRNA-143 targets MACC1 to inhibit cell invasion and migration in colorectal cancer. Mol Cancer 11: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cai C, Ashktorab H, Pang X, Zhao Y, Sha W, et al. (2012) MicroRNA-211 Expression Promotes Colorectal Cancer Cell Growth In Vitro and In Vivo by Targeting Tumor Suppressor CHD5. PLoS One 7: e29750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang J, Lan H, Huang X, Liu B, Tong Y (2012) MicroRNA-126 Inhibits Tumor Cell Growth and Its Expression Level Correlates with Poor Survival in Non-Small Cell Lung Cancer Patients. PLoS One 7: e42978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu N, Zhang D, Xie H, Zhou Z, Chen H, et al. (2011) Endothelial-specific intron-derived miR-126 is down-regulated in human breast cancer and targets both VEGFA and PIK3R2. Mol Cell Biochem 351: 157–164. [DOI] [PubMed] [Google Scholar]
- 15. Li XM, Wang AM, Zhang J, Yi H (2011) Down-regulation of miR-126 expression in colorectal cancer and its clinical significance. Med Oncol 28: 1054–1057. [DOI] [PubMed] [Google Scholar]
- 16. Guo C, Sah JF, Beard L, Willson JK, Markowitz SD, et al. (2008) The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer 47: 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reiss K, Del Valle L, Lassak A, Trojanek J (2012) Nuclear IRS-1 and cancer. J Cell Physiol 227: 2992–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang Q, Li Y, White MF, Fletcher JA, Xiao S (2002) Constitutive activation of insulin receptor substrate 1 is a frequent event in human tumors: therapeutic implications. Cancer Res 62: 6035–6038. [PubMed] [Google Scholar]
- 19. Slattery ML, Samowitz W, Curtin K, Ma KN, Hoffman M, et al. (2004) Associations among IRS1, IRS2, IGF1, and IGFBP3 genetic polymorphisms and colorectal cancer. Cancer Epidemiol Biomarkers Prev 13: 1206–1214. [PubMed] [Google Scholar]
- 20. Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, deAngelis T, et al. (2007) Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J Biol Chem 282: 32582–32590. [DOI] [PubMed] [Google Scholar]
- 21. Yin Y, Yan ZP, Lu NN, Xu Q, He J, et al. (2013) Downregulation of miR-145 associated with cancer progression and VEGF transcriptional activation by targeting N-RAS and IRS1. Biochim Biophys Acta 1829: 239–247. [DOI] [PubMed] [Google Scholar]
- 22. Kim EJ, Kang IJ, Cho HJ, Kim WK, Ha YL, et al. (2003) Conjugated linoleic acid downregulates insulin-like growth factor-I receptor levels in HT-29 human colon cancer cells. J Nutr 133: 2675–2681. [DOI] [PubMed] [Google Scholar]
- 23. Li N, Tang A, Huang S, Li Z, Li X, et al. (2013) MiR-126 suppresses colon cancer cell proliferation and invasion via inhibiting RhoA/ROCK signaling pathway. Mol Cell Biochem 380: 107–119. [DOI] [PubMed] [Google Scholar]
- 24. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, et al. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10: 593–601. [DOI] [PubMed] [Google Scholar]
- 25. Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, et al. (2008) MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27: 2128–2136. [DOI] [PubMed] [Google Scholar]
- 26. Zhang B, Pan X, Cobb GP, Anderson TA (2007) microRNAs as oncogenes and tumor suppressors. Dev Biol 302: 1–12. [DOI] [PubMed] [Google Scholar]
- 27. Wang H, Wu J, Meng X, Ying X, Zuo Y, et al. (2011) MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis 32: 1033–1042. [DOI] [PubMed] [Google Scholar]
- 28. Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, et al. (2012) MiR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res 40: 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otsubo T, Akiyama Y, Hashimoto Y, Shimada S, Goto K, et al. (2011) MicroRNA-126 inhibits SOX2 expression and contributes to gastric carcinogenesis. PLoS One 6: e16617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J, Du YY, Lin YF, Chen YT, Yang L, et al. (2008) The cell growth suppressor, mir-126, targets IRS-1. Biochem Biophys Res Commun 377: 136–140. [DOI] [PubMed] [Google Scholar]
- 31. Crawford M, Brawner E, Batte K, Yu L, Hunter MG, et al. (2008) MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines.Biochem Biophys Res Commun. 373: 607–612. [DOI] [PubMed] [Google Scholar]
- 32. Miko E, Czimmerer Z, Csánky E, Boros G, Buslig J, et al. (2009) Differentially expressed microRNAs in small cell lung cancer. Exp Lung Res 35: 646–664. [DOI] [PubMed] [Google Scholar]
- 33. Saito Y, Friedman JM, Chihara Y, Egger G, Chuang JC, et al. (2009) Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem Biophys Res Commun 379: 726–731. [DOI] [PubMed] [Google Scholar]
- 34. Wang X, Tang S, Le SY, Lu R, Rader JS, et al. (2008) Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth. PLoS One 3: e2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bergmann U, Funatomi H, Kornmann M, Beger HG, Korc M (1996) Increased expression of insulin receptor substrate-1 in human pancreatic cancer. Biochem Biophys Res Commun 220: 886–890. [DOI] [PubMed] [Google Scholar]
- 36. Dearth RK, Cui X, Kim HJ, Kuiatse I, Lawrence NA, et al. (2006) Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol Cell Biol 26: 9302–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun H, Tu X, Baserga R (2006) A mechanism for cell size regulation by the insulin and insulin-like growth factor-I receptors. Cancer Res 66: 11106–11109. [DOI] [PubMed] [Google Scholar]
- 38. Yamada N, Noguchi S, Mori T, Naoe T, Maruo K, et al. (2013) Tumor-suppressive microRNA-145 targets catenin δ-1 to regulate Wnt/β-catenin signaling in human colon cancer cells. Cancer Lett 335: 332–342. [DOI] [PubMed] [Google Scholar]
- 39. Strillacci A, Valerii MC, Sansone P, Caggiano C, Sgromo A, et al. (2013) Loss of miR-101 expression promotes Wnt/β-catenin signalling pathway activation and malignancy in colon cancer cells. J Pathol 229: 379–389. [DOI] [PubMed] [Google Scholar]
- 40. Wang Z, Zhang X, Yang Z, Du H, Wu Z, et al. (2012) MiR-145 regulates PAK4 via the MAPK pathway and exhibits an antitumor effect in human colon cells. Biochem Biophys Res Commun 427: 444–449. [DOI] [PubMed] [Google Scholar]
- 41. Sun D, Yu F, Ma Y, Zhao R, Chen X, et al. (2013) MicroRNA-31 activates the RAS pathway and functions as an oncogenic MicroRNA in human colorectal cancer by repressing RAS p21 GTPase activating protein 1 (RASA1). J Biol Chem 288: 9508–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Iwaya T, Yokobori T, Nishida N, Kogo R, Sudo T, et al. (2012) Down-regulation of miR-144 is associated with colorectal cancer progression via activation of mTOR signaling pathway. Carcinogenesis 33: 2391–2397. [DOI] [PubMed] [Google Scholar]