

Abstract
The development of nanotechnology has been largely inspired by the biological world. The complex, but well-organized, living system hosts an array of molecular-sized machines responsible for information processing, structure building and, sometimes, movement. We present here a novel light-powered DNA mechanical device, which is reminiscent of cellular protein motors in nature, especially those of green plants. This walking device, which is based on pyrene- assisted photolysis of disulfide bonds, is capable of autonomous locomotion, with light control of initiation, termination and velocity. Based on DNA sequence design and such physical conditions as temperature and ionic strength, this photon-fueled DNA walker exhibits the type of operational freedom and mechanical speed that may rival protein motors in the future.
Keywords: autonomous movement, DNA, light control, molecular devices
The development of nanotechnology has been largely inspired by the biological world. The complex, but well-organized, living system hosts an array of molecular-sized machines responsible for information processing, structure building and, sometimes, movement. Molecular motors are tiny protein machines that power motion in the cellular world, e.g., myosins moving on actin filaments and dyneins or kinesins walking along microtubule tracks.[1]
Artificial DNA-based motors have recently emerged to mimic molecular motors[2] and to perform tasks in cargo transport[2e] and biosynthesis[2f]. The programmable assembly and simplicity of polynucleotide interactions have made DNAs suitable for the control of progressive and directional movement at the molecular level. However, energy supply is a major concern for any motor.[3] Biological molecular motors employ free energy based on the binding and hydrolysis of ATP, while artificial DNA walkers have previously explored energy supplied by DNA hybridization[4] or hydrolysis of either the DNA/RNA backbone[5] or ATP molecules[6]. These artificial walking devices are still in their infancy, and they are not as powerful as their protein counterparts from nature. Still, identification of new types of energy supplies could play a major role in the development of the next-generation mechanical robots.
Biological motors normally operate in a constant environment without external intervention, and they remain in operation as long as a source of energy is available. Thus, these biological motors are considered to be autonomous. For artificial DNA walkers, however, non-autonomous locomotion by regular addition of fuel[7] is always much simpler from a design standpoint, compared to an autonomous device. On the other hand, the capability of autonomous motion sometimes results in decreased controllability[2,5,6] because the device cannot be stopped at a desired position or time, or once stopped, it cannot be easily restarted. Therefore, construction of free-running and autonomous “true” molecular motors,[2b] in comparison to non-autonomous designs, is still essential to the operation of nanomachines that mimic biological function. The availability of such an easily-controllable, free-running DNA walker would also be significant for the future design of nano-robots to perform multiple and complex functions.
We report here a new light energy-powered DNA walker capable of regulated autonomous movement along a nucleic acid track. It has been widely known that photochemical energy sources can serve as inputs for molecular-level switches in the operation of nanomachines.[8] We recently found that aromatic hydrocarbons (e.g., pyrene molecules) can efficiently facilitate the photolysis of disulfide bonds within artificial nucleic acid backbones[9] and that a catalytic cleavage function could be achieved through the design of pyrene-incorporated DNAzyme analogs (Figure 1a). This same pyrene-assisted photolysis reaction is also a major component of our DNA walker design (Figure 1b).
Figure 1.
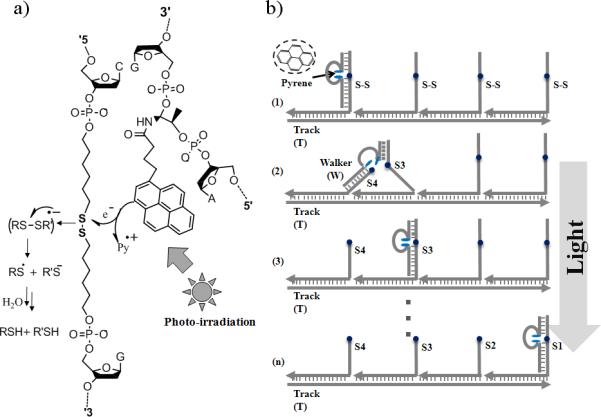
Photolysis and DNA walker system. a) Pyrene assisted photolysis of disulfide within DNA structures. b) The walking principle: walker, four anchorage sites, and track form the walking system through self-assembly; when the walker takes a step, light triggered photolysis of anchorage sites provides the energy for locomotion.
Photon radiation, which is virtually unlimited, supplies the energy (together with DNA hybridization free energy) for this type of nanomachine, and the amount of energy input to operate such machines can be readily controlled by using different intensities of excitation light. Moreover, by taking advantage of recent developments in laser and near-field techniques, high spatial and temporal resolution can be achieved from light control as well.[10] With its renewable energy supply, we envision that the light-powered DNA walker will allow us to precisely control the speed and motion of nano-robots and, hence, advance the progress of molecular biology in the future. This work represents the first light-powered DNA walking devices able to achieve controllable, autonomous and directional movement, and it expands the scope of energy supplies available to power nano-sized motion.
The walking system consists of three parts: a single-stranded DNA track (T), four anchorage sites (S1, S2, S3 and S4), and a light-sensitive walker (W) (Figure 1). Similar as previous design of processive walker by He et al,[2f,5a] the track contains four 21-nucleotide binding regions that are complementary to the recognition tag of the respective anchorage sites. Through specific base-pairing, the track organizes the four anchorage sites into one self-assembled construction, with four walker (W) binding sites extending from the track. After the assembly, the distance between two adjacent anchorage sites corresponds to two helical pitches, roughly 7nm. It is worth mentioning that, the sequence design of the walking system is not limited to this established one, and we just employ it for demonstration of the light-controlled motion.
The DNA walker contains two motion legs, a short leg (7-nt) and a long leg (16-nt), and they are linked by a pyrene moiety that works as a photo-sensitizer and is incorporated into a DNA oligomer. The two motion legs are designed to respectively bind the two extender segments, which are connected by a weak disulfide bond. The recognition event between legs and anchorage sites brings the pyrene and disulfide moieties together. Irradiation at 350nm causes pyrene-facilitated photolysis of the disulfide bond with the sufficiency required to initiate movement. Once the disulfide bond is cleaved, the shorter leg dissociates to search its surroundings for the next accessible anchorage site, while the longer leg remains bound to the extender segment, thus preventing the walker from leaving the track. Based on the toehold-mediated strand displacement,[5] the longer leg will further move onto the anchorage site that the shorter leg binding with, finally complete one step forward. The construction of the overall walking system was verified by native polyacrylamide gel electrophoresis (N-PAGE, Figure S1, Supporting Information).
In the nano-sized world, random Brownian motion is much more important than gravity and momentum, which govern movement in the macroscopic world. To overcome random motion, a “Burnt Bridge” mechanism[11] was incorporated to make the revisiting of the same anchorage site less probable. That is, by consuming the anchorage site after stepping, forward motion results in higher thermostability and statistical probability compared with the backward motion (revisiting the cleaved site), provides the basis for the directional movement. In our design, this is achieved by the light energy-powered photolysis of the anchorage extenders.
Directionality
Light-powered autonomous and directional locomotion was demonstrated by monitoring the anchorage cleavage (separation of two extender segments on opposite sides of the disulfide linker) after the DNA walker takes a step (Figure 2). Directional walking pathways, e.g., S1→S4 or S4→S1, were investigated. For example, from S4 to S1, the walker was specifically positioned through separately prepared T(track)-S1-S2-S3 and S4-W(walker) conjugates. Mixing the conjugates immediately prior to light irradiation ensured that the S4 anchorage was the initiating site (the largest distribution binding position before irradiation) for the walker. Sequential cleavage is observed in Figure 2 by the amounts of the respective cleaved anchorage strands (shorter extender segments on one side of disulfide linker). The results are consistent with the expected direction: S1>S2>S3>S4 for S1→S4 and S4>S3>S2>S1 for S4→S1. As control, negligible bond cleavage was observed in the absence of the pyrene-incorporated walker (Figure S2a, Supporting Information). To test the integrity of this system, we synthesized another DNA track (T*), with the anchorage sites in the order of S3-S2-S1-S4, and the gel results followed the nucleotide sequence from the track (Figure S2b, Supporting Information).
Figure 2.
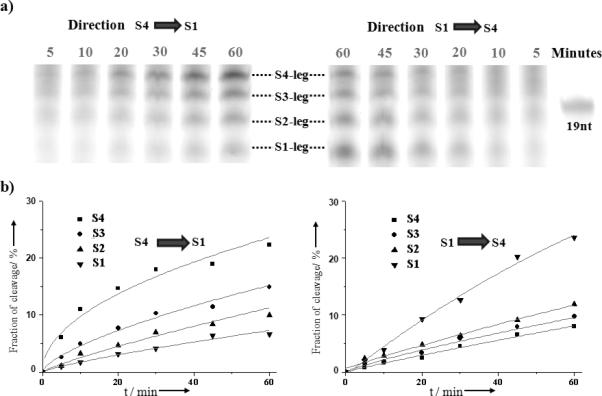
Directional and autonomous locomotion. a) Denatured PAGE analysis for walker movement in the S4→S1 and S1→S4 directions; the band shows the short cleaved substrate fragments for the respective anchorage site as a function of irradiation time. b) Quantification of the cleavage fraction using Image J software.
Progression
The progressive locomotion of the DNA walker is limited only by the spatial dimensions available to it. Specifically, after the shorter leg (7-nt) dissociates from the track, the closest anchorage site will be in the most accessible location to serve as the replacement strand. The walker will prefer the stepwise motion, instead of “hopping”, i.e., after leaving S4 site, the next step will be to the S3, not S2 site. The progressive locomotion of the DNA walker was investigated using a fluorescence resonance energy transfer (FRET) assay. As the FRET donor, 6-FAM fluorphores were labeled on the walker's longer leg, and Cy5, an acceptor for FAM fluorescence energy, was attached to anchorage site S3. Because the FRET efficiency is proportional to an inverse 6th power of the donor-acceptor distance, distance-dependent FRET is expected to occur most efficiently when the walker steps onto the S3 site, thereby bringing FAM and Cy5 together. Indeed, consistent with our expectations, the S1-S2-S3-S4 track required a longer time to reach maximal fluorescence signal than the S4-S3-S2-S1 track (longer distance and more steps for progressive motion from two-step S1→S3 than one-step S4→S3, Figure S3b, Supporting Information). Moreover, anchorage site S2 was labeled with another FRET acceptor, TAMRA, and sequential transient FRET was demonstrated following the progressive motion (Figure 3). In a control experiment, to further confirm such progressive movement, an S2* site was synthesized without a disulfide bond. Without this cleavage point to keep the walker moving, a much longer residence time on S2* was observed (Figure S3c, Supporting Information). These experiments confirmed the directionality and progressive motion of our light-powered walker.
Figure 3.
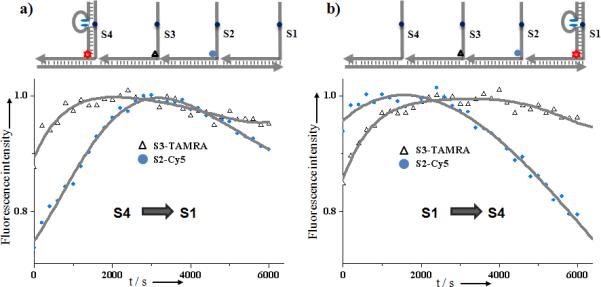
Progressive walking demonstrated by FRET assay. FAM (donor fluorophore, green star) labelled walker moves on the anchorage site labelled by FRET acceptor fluorophore TAMRA (red) and Cy5 (blue). Fluorescence intensity (662nm for Cy5, 575nm for TAMRA, λex= 488nm) was monitored during a) S4→S1 and b) S1→S4 tracks. The light source was obtained using the fluorometer, with light power of 66μW/cm2 at 350nm (spectral bandwidth=10nm, interval time=0.1s).
Controllability
The major merit of light-regulated DNA walker is its precise controllability, which is essential for nano-sized devices. In contrast to DNA walkers fueled by DNA[2a,5] or ATP hydrolysis[6], light energy can be transmitted to the system without physical contact. As a result, the initiation and termination of walking activities can be simply and immediately achieved via on/off switching of the light power source. Additionally, the motional velocity of the walker can be controlled by the intensity of the light source. As shown in Figure 4a, the motion of the DNA walker was accelerated by increasing the light intensity. Control of locomotion speed was also investigated using bulk fluorescence assay. Based on the FRET measurement after 350 nm irradiation with varied light power, more rapid movement was observed with increasing light intensity (Figure S4, Supporting Information).
Figure 4.
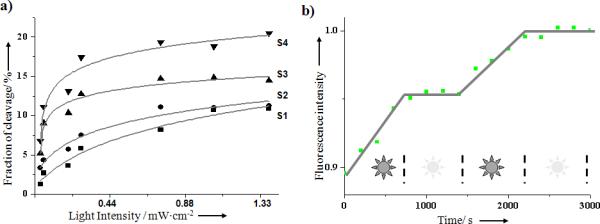
Controllable locomotion. a) Walking speed controlled by different irradiation power; the cleavage fraction of respective anchorage site (along S4→S1 direction) was analyzed after 45 min irradiation at 350 nm. b) The STOP and RESTART actions of the DNA walker are controlled by light irradiation. Monitoring the Cy5 (S2 anchorage site) fluorescence intensity (662nm, λex=488nm) while alternating the irradiation ON/OFF (using the light source in the fluorometer at 350 nm, light power 66μW/cm2). Walker moves along anchorage S1-S2’-S3-S4 on track T.
An inherent hindrance of chemically modulated nanomachines is the difficulty in controlling their STOP and RESTART actions by the addition or removal of chemicals. However, this problem is easily overcome by the light-powered walking system. Figure 4b shows that the miniature walker can be regulated by simply switching the irradiation light ON or OFF. Such controllable autonomous movement will be a significant improvement for regulating and optimizing nano-robot devices, making them potentially useful for cargo transport and other tasks.
Other properties of this light-powered nano-walker system include broad choices in design and operation. First, there is freedom in DNA sequence design. As mentioned above, the walker is comprised of three parts: two legs and one body. Irrespective of catalytic efficiency, each of these parts can be freely modified to optimize the motion. For example, the length of the body sequence, which connects the two pyrene moieties, can be readily modified to optimize the spatial dimension available to the walker and the motional style.[12] As a demonstration, two additional light-sensitive walkers, WS and WL, were synthesized with different body lengths. Both probes demonstrated programmable walking along the track, but with different motional speeds (Figure S5, Supporting Information). It should be noted that elongating the body does allow faster motion, but with increased risk of more “hopping” (more than one-sized step) than “stepping” (because of increased spatial dimensions available to it, which disturb the progressive locomotion). Therefore, a compromise between speed and “hopping” could be struck for different applications. For example, in walker-based synthesis,[2f] product purity and reaction efficiency may be more important than time requirements, thereby making the DNA walker with shorter linker length more desirable. Other operational freedoms exist with respect to physical conditions, e.g., temperature and solution components. Unlike enzyme or DNAzyme-based walkers,[5,6] which normally function at physiological temperature or certain cofactor concentration (e.g., Mg2+), the light-powered walker can theoretically be operated over a broader physical condition (as long as the DNA strands still bind together), thus enhancing the potential application area of the walking device.
In conclusion, we demonstrated in this work the feasibility of designing an autonomous, but controllable, DNA walking device by incorporating photosensitive moieties within DNAzyme analog structures. Compared with “molecular motors” in nature, such light-powered walkers are still not as fast and powerful (the fraction of walkers reach the end of track is still lower than 40%, which might due to the equlibrium between the the two disulfide molecular states, the oxidized form,S-S, and the reduced form, -SH). However, as a new type of energy supply for powering nano-sized robots, photon energy has the benefits of precise controllability and optimization freedom, while preserving autonomous, progressive motion. Considering the continuous effort to identify clean and renewable energy sources, more rationally designed light energy-powered miniature mechanical devices will arise, emulating nature's stepping rate for performing various tasks, e.g., transferring cargoes and stimulating macroscopic motion.
Experimental Section
Denatured PAGE analysis
The denatured PAGE gels were run in 15% acrylamide (containing 19/1 acrylamide/bisacrylamide and 8.3M urea), at 200V constant voltage for 1.5 hours using 1×Tris-borate-EDTA (TBE) as the separation buffer; normally 10 μL 1.0 μM DNA strands are loaded for each well. The fluorescence images of gels were recorded by FAM fluorescence, with excitation at 488nm and emission at 526nm, using a Typhoon 9410 variable mode imager (GE Healthcare, Piscataway, NJ). The band intensities were analyzed with Image J software from NIH. Origin 8.0 was used for data analysis.
Native PAGE analysis
The binding and three-dimensional structures of the DNA walking system were observed using native PAGE gel. Normally the gel was run in 8-10% acrylamide (containing 19/1 acrylamide/bisacrylamide) solution with 1×TBE/15mM Mg2+ buffer, at 100V constant voltage for 1 hour (4°C). After that, the gel was stained 30 min using Stains-All (Sigma-Aldrich) to image the position of DNA. Photographic images were obtained under visible light with a digital camera.
FRET measurements
A FluoroMax-4 Spectrofluorometer with a temperature controller (Jobin Yvon) was used for all steady-state fluorescence measurements. The locomotion of walker along the track was realized by excitation at 350nm using the spectrofluorometer source (spectral bandwidth=10nm; interval time=0.1s). After a series of irradiation time periods, fluorescence intensities at 515nm (FAM), 575nm (TAMRA) and 662nm (Cy5) were recorded using an excitation wavelength of 488nm.
Manipulation of the walking system
The oligonucleotides employed are listed in Supplementary Table S1 and Table S2. Using S4→ S1 as an example, the formation of the walking system was as follows. After separate annealing (95°C-10°C, over 20 min) in 20mM Tris buffer (pH=7.5, containing 100mM NaCl and 10mM MgCl2), T-S1-S2-S3 and S4-W conjugates were mixed with final concentration for each sequence of 1.0μM, and the sample was irradiated with a UV-B lamp (SANKO DENKI, Japan) with a 352nm photochemical optical filter (3nm half bandwidth; Oriel Instruments, Stratford, CT, Newport). The irradiation power employed was measured using a power meter (Newport Corp., Irvine, CA). After a series of irradiation times, the samples were removed and subjected to denatured PAGE for analysis and imaging. The FRET experiments were performed using a FluoroMax-4 Spectrofluorometer with a temperature controller (Jobin Yvon). 300nM final concentration of each DNA strand was mixed in the 10mM Tris buffer (pH=7.5, containing 50mM NaCl and 5mM MgCl2). Locomotion of the walker along the track was realized by irradiation at 350nm using the source in the spectrofluorometer (spectral bandwidth=10nm; interval time=0.1s; light power 66 μW/cm2). After a series of irradiation times, fluorescence intensities at 515nm (FAM), 575nm (TAMRA) and 662nm (Cy5) were recorded using excitation at 488nm (λex for FAM).
Supplementary Material
Acknowledgments
The authors would like to thank the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida for technical support. This work is supported by grants awarded by the National Institutes of Health (GM066137, GM079359 and CA133086) and by NSF.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org
Contributor Information
Mingxu You, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA).
Yan Chen, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA).
Xiaobing Zhang, State Key Laboratory for Chemo/Biosensing and Chemometrics, College of Biology and College of Chemistry and Chemical Engineering Hunan University, Changsha 410082 (P.R. China).
Haipeng Liu, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA).
Ruowen Wang, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA).
Kelong Wang, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA).
Kathryn R. Williams, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA)
Weihong Tan, Department of Chemistry and Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Shands Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida Gainesville, FL 32611-7200 (USA); State Key Laboratory for Chemo/Biosensing and Chemometrics, College of Biology and College of Chemistry and Chemical Engineering Hunan University, Changsha 410082 (P.R. China).
References
- 1.Schliwa M, Woehlke G. Nature. 2003;422:759–765. doi: 10.1038/nature01601. [DOI] [PubMed] [Google Scholar]
- 2.a Lund K, Manzo AJ, Dabby N, Michelotti N, Johnson-Buck A, Nangreave J, Taylor S, Pei R, Stojanovic MN, Walter NG, Winfree E, Yan H. Nature. 2010;465:206–210. doi: 10.1038/nature09012. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bath J, Turberfield AJ. Nat. Nanotechnol. 2007;2:275–284. doi: 10.1038/nnano.2007.104. [DOI] [PubMed] [Google Scholar]; c Von Delius M, Leigh DA. Chem. Soc. Rev. 2011;40:3656–3676. doi: 10.1039/c1cs15005g. [DOI] [PubMed] [Google Scholar]; d Krishnan Y, Simmel FC. Angew. Chem. 2011;123:3180–3215. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:3124–3156. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]; e Gu H, Chao J, Xiao S, Seeman NC. Nature. 2010;465:202–205. doi: 10.1038/nature09026. [DOI] [PMC free article] [PubMed] [Google Scholar]; f He Y, Liu DR. Nat. Nanotechnol. 2010;5:778–782. doi: 10.1038/nnano.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Barrell MJ, Campana AG, von Delius M, Geertsema EM, Leigh DA. Angew. Chem. 2011;123:299–304. doi: 10.1002/anie.201004779. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:285–290. [Google Scholar]; b Von Delius M, Geertsema EM, Leigh DA, Tang DD. J. Am. Chem. Soc. 2010;132:16134–16145. doi: 10.1021/ja106486b. [DOI] [PubMed] [Google Scholar]
- 4.a Yin P, Choi HMT, Calvert CR, Pierce NA. Nature. 2008;451:318–322. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]; b Omabegho T, Sha R, Seeman NC. Science. 2009;324:67–71. doi: 10.1126/science.1170336. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Green SJ, Bath J, Tuberfield AJ. Phy. Rev. Lett. 2008;101:238101. doi: 10.1103/PhysRevLett.101.238101. [DOI] [PubMed] [Google Scholar]
- 5.a Tian Y, He Y, Chen Y, Yin P, Mao C. Angew. Chem. 2005;117:4429–4432. doi: 10.1002/anie.200500703. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:4355–4358. doi: 10.1002/anie.200500703. [DOI] [PubMed] [Google Scholar]; b Bath J, Green SJ, Tuberfield AJ. Angew. Chem. 2005;117:4432–4435. [Google Scholar]; Angew. Chem. Int. Ed. 2005;44:4358–4361. doi: 10.1002/anie.200501262. [DOI] [PubMed] [Google Scholar]; c Wickham SFJ, Endo M, Katsuda Y, Hidaka K, Bath J, Sugiyama H, Turberfield AJ. Nat. Nanotechnol. 2011;6:166–169. doi: 10.1038/nnano.2010.284. [DOI] [PubMed] [Google Scholar]; d Pei R, Taylor SK, Stefanovic D, Rudchenko S, Mitchell TE, Stojanovic MN. J. Am. Chem. Soc. 2006;128:12693–12699. doi: 10.1021/ja058394n. [DOI] [PubMed] [Google Scholar]
- 6.Yin P, Yan H, Daniell XG, Turberfield AJ, Reif JH. Angew. Chem. 2004;116:5014–5019. doi: 10.1002/anie.200460522. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;43:4906–4911. doi: 10.1002/anie.200460522. [DOI] [PubMed] [Google Scholar]
- 7.a Shin J, Pierce NA. J. Am. Chem. Soc. 2004;126:10834–10835. doi: 10.1021/ja047543j. [DOI] [PubMed] [Google Scholar]; b Sherman WB, Seeman NC. Nano. Lett. 2004;4:1203–1207. [Google Scholar]; c Wang Z, Elbaz J, Willner I. Nano. Lett. 2011;11:304–309. doi: 10.1021/nl104088s. [DOI] [PubMed] [Google Scholar]
- 8.a Gorostiza P, Isacoff EY. Science. 2008;322:395–399. doi: 10.1126/science.1166022. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhou M, Liang X, Mochizuki T, Asanuma H. Angew. Chem. 2010;122:2213–2216. doi: 10.1002/anie.200907082. [DOI] [PubMed] [Google Scholar]; Angew.Chem., Int. Ed. 2010;49:2167–2170. doi: 10.1002/anie.200907082. [DOI] [PubMed] [Google Scholar]; c Kang H, Liu H, Phillips JA, Cao Z, Kim Y, Chen Y, Yang Z, Li J, Tan W. Nano. Lett. 2009;9:2690–2696. doi: 10.1021/nl9011694. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kim Y, Cao Z, Tan W. Proc. Natl. Acad. Sci. USA. 2008;105:5664–5669. doi: 10.1073/pnas.0711803105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.You M, Zhu Z, Liu H, Gulbakan B, Han D, Wang R, Williams KR, Tan W. ACS Appl. Mater. Interfaces. 2010;2:3601–3605. doi: 10.1021/am1007886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a Kopelman R, Tan W. Science. 1993;262:1382–1384. doi: 10.1126/science.262.5138.1382. [DOI] [PubMed] [Google Scholar]; b Tan W, Shi Z, Smith S, Birnbaum D, Kopelman R. Science. 1992;258:778–781. doi: 10.1126/science.1439785. [DOI] [PubMed] [Google Scholar]
- 11.a Mai J, Sokolov IM, Blumen A. Phys. Rev. E. 2001;64:011102. doi: 10.1103/PhysRevE.64.011102. [DOI] [PubMed] [Google Scholar]; b Saffarian S, Collier IE, Marmer BL, Elson EL, Goldberg G. Science. 2004;306:108–111. doi: 10.1126/science.1099179. [DOI] [PubMed] [Google Scholar]
- 12.Wang Z. Proc. Natl. Acad. Sci. USA. 2007;104:17921–17926. doi: 10.1073/pnas.0703639104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.