

Abstract
In acute hippocampal slices, we found that the presence of extracellular brain-derived neurotrophic factor (BDNF) is essential for the induction of spike-timing-dependent long-term potentiation (tLTP). To determine whether BDNF could be secreted from postsynaptic dendrites in a spike-timing-dependent manner, we used a reduced system of dissociated hippocampal neurons in culture. Repetitive pairing of iontophoretically applied glutamate pulses at the dendrite with neuronal spikes could induce persistent alterations of glutamate-induced responses at the same dendritic site in a manner that mimics spike-timing-dependent plasticity (STDP)—the glutamate-induced responses were potentiated and depressed when the glutamate pulses were applied 20 ms before and after neuronal spiking, respectively. By monitoring changes in the green fluorescent protein (GFP) fluorescence at the dendrite of hippocampal neurons expressing GFP-tagged BDNF, we found that pairing of iontophoretic glutamate pulses with neuronal spiking resulted in BDNF secretion from the dendrite at the iontophoretic site only when the glutamate pulses were applied within a time window of approximately 40 ms prior to neuronal spiking, consistent with the timing requirement of synaptic potentiation via STDP. Thus, BDNF is required for tLTP and BDNF secretion could be triggered in a spike-timing-dependent manner from the postsynaptic dendrite.
Keywords: STDP, BDNF, synaptic plasticity, tLTP
1. Introduction
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family of proteins, was initially identified as a factor critical for neuronal survival and neurite growth during early neural development [1]. Extensive studies in the past two decades have shown that neurotrophins are also involved in regulating diverse neuronal functions, including modulation of synaptic efficacy and plasticity in the mature brain [2,3]. Exogenous application of BDNF was shown to potentiate the efficacy of basal synaptic transmission by enhancing presynaptic transmitter secretion [4,5], leading to a higher capacity for some excitatory synapses to undergo activity-induced long-term potentiation (LTP) [6,7]. Depleting endogenous BDNF by genetic means [8] or acutely with extracellular-specific chelating agents, e.g. TrkB–IgG [6,9] and BDNF antibodies [10], impaired LTP induction by high-frequency stimulation (HFS), indicating that secreted BDNF is critical for the induction or stabilization of activity-dependent synaptic plasticity.
The interest in BDNF in the field of synaptic plasticity was greatly stimulated by the findings from cultured neurons that the secretion of neurotrophins is activity-dependent [11] and LTP-inducing HFS is most effective in triggering BDNF secretion [12–15]. This led to the idea that synaptic activity could trigger secretion of BDNF locally at the synapse and the subsequent actions of BDNF mediates synaptic changes underlying LTP. The evidence cumulated so far supports this idea. For example, long-term structural changes, including increased number and volume of postsynaptic spines caused by high-frequency synaptic activity, was abolished by applying chelating agent TrkB–IgG (tropomyosin receptor kinase B–immunoglobulin G) prior to the induction of spike-timing-dependent potentiation [16].
Two issues associated with synaptic secretion of BDNF remain unresolved: first, the source of BDNF—whether the BDNF is secreted from the pre- or postsynaptic neurons and what compartments are mainly responsible for storing synaptic BDNF; second, whether all forms of LTP of excitatory synapses require local secretion of BDNF. In particular, whether persistent synaptic potentiation induced by low-frequency (1 Hz) pairing of pre- and postsynaptic spiking, a form of spike-timing-dependent LTP (tLTP), also requires BDNF signalling, and whether BDNF secretion can be triggered by low-frequency synaptic activity.
In this study, we first examined whether BDNF plays a role in the induction of tLTP in hippocampal slices. We found that depletion of extracellular BDNF with TrkB–IgG, a soluble scavenger that binds to BDNF, completely abolished tLTP induction. We then used hippocampal neurons in dissociated cultures to examine whether BDNF secretion from dendrites could be triggered by low-frequency synaptic excitation in a spike-timing-dependent manner. To monitor BDNF secretion, we transfected the cultured neurons with a viral construct that expresses green fluorescent protein (GFP)-tagged BDNF, a method previously used for studying Ca2+-dependent BDNF secretion from both synaptic and extrasynaptic sites along the dendrite [17,18]. To simplify the system and interpretation of the results, we simulated the spike-induced glutamate release from the presynaptic axon terminal with iontophoretic ejection of glutamate pulse (150 mM) at the postsynaptic dendrite, as marked by the immobile BDNF fluorescent puncta in the dendrite. This allowed us to determine whether glutamate pulses are sufficient to serve as the presynaptic signal to trigger the postsynaptic BDNF secretion. Finally, we tested the dependence of BDNF secretion on the time interval between glutamate pulses and neuronal spiking, and compared the time window required for effective BDNF secretion with that of spike-timing-dependent plasticity (STDP) in this culture system [19]. The results showed that tLTP indeed requires the action of extracellular BDNF and that low-frequency pairing of glutamate release and postsynaptic spiking is sufficient to trigger the release of BDNF in a spike-timing-dependent manner.
2. Material and methods
(a). Slice preparation and electrophysiology
Acutely isolated hippocampal slices were prepared from Sprague Dawley rats (Charles River) as previously described [20]. Animal protocols were approved by the Animal Care and Use Committee of UC Berkeley. Coronal slices (250 µm thick) containing medial hippocampus were cut with a vibratome (Leica, Germany) in a solution containing (in mM) 110 choline-Cl, 25 NaHCO3, 25 d-glucose, 11.6 Na ascorbate, 7 MgSO4, 3.1 Na pyruvate, 2.5 KCl, 1.25 NaH2PO4 and 0.5 CaCl2. Before use, slices were incubated in artificial cerebrospinal fluid containing (in mM): 125 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 1.25 mM NaH2PO4, 26 NaHCO3 and 10 glucose at room temperature, with the solution bubbled with 95% O2 and 5% CO2.
Whole-cell recording was made from CA1 pyramidal neurons in the hippocampus using a patch-clamp amplifier (MultiClamp 700B, Molecular Devices, Sunnyvale, CA, USA) under infrared DIC optics at 30 (±1)°C. Data were acquired with a digitizer (DigiData 1322A, Molecular Devices) and analyzed with pClamp 10 software (Molecular Devices). Extracellular stimulation pulses (50 μs duration) were applied with a bipolar electrode (WPI Inc., Sarasota, FL, USA) placed along the Schaffer collaterals using a stimulator system (Master 8, Jerusalem, Israel). Evoked excitatory postsynaptic potentials (EPSPs) were recorded at −70 mV in current clamp in the presence of SR 95531 (10 μM). The spike-timing protocol for LTP induction consisted of 80 EPSP-spike pairs delivered at 1 s intervals (1 Hz). The postsynaptic spikes were evoked by injection of depolarizing current pulses (1–2 nA, 2–3 ms), with the onset of EPSPs preceding the peak of postsynaptic spikes by 20 ms. The intracellular solution for whole-cell recording contained (in mM): 140 K gluconate, 5 KCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 4 MgATP, 0.3 Na2GTP and 10 Na2-phosphocreatine (at pH 7.2 with KOH). Data were discarded when the series resistance changed by more than 20% during the course of the experiment.
(b). Cell culture preparation and transfection
Low-density cultures of dissociated embryonic hippocampal neurons were prepared as described previously [21]. Briefly, hippocampi were removed from embryonic rats (embryonic days 20–21) of a Sprague Dawley rat and treated with trypsin for 15 min at 37°C, followed by gentle trituration. The dissociated cells were plated at a density of 10 000–15 000 cm−2 on poly-l-lysine-coated glass coverslips. The culturing medium was Neurobasal (Invitrogen) supplemented with B-27 (Invitrogen) and glutamine (Invitrogen). We used pyramidal neurons on 14–21 days in vitro (DIV).
Cells were transfected on 5 DIV using Lipofectamine 2000 (Invitrogen) in Opti-MEM 1 medium (Invitrogen) according to the instructions of the manufacturer. We used a Gal4–UAS bipartite system to drive the expression of the BDNF probes. Approximately 3 kb genomic fragment from the zebrafish HuC putative promoter was used to drive the expression of Gal4 activator protein, which in turn binds to the upstream activation sequence (UAS) and induces the expression of mouse BDNF–EGFP (enhanced GFP) proteins. The cDNA for BDNF–EGFP was kindly provided by Dr M. Kojima (National Institute of Advanced Industrial Science and Technology, Osaka, Japan).
(c). Iontophoresis, whole-cell recording and imaging
The iontophoretic method followed that described previously [22]. Briefly, a sharp iontophoretic pipette with a resistance of 150–300 MΩ was filled with 150 mM glutamate (pH 8.0, adjusted with NaOH). The pipette tip was coated with Sylgard-184 (Dow Corning, Midland, MI, USA) to reduce the pipette capacitance. Both the holding current (1.5–2.0 nA) and iontophoretic current (10–100 nA, with a duration of 0.2–1.0 ms) were applied through the Multiclamp 700B amplifier. The pipette capacitance was compensated for a built-in function of the amplifier. The tip of the pipette was put 2–5 μm away from the targeted dendritic spine of the recorded neuron. Whole-cell recordings were made on infected cultured hippocampal neurons with fluorescence. The recording pipette was filled with the same intracellular solution as above. The bath solution contained (in mM): 145 NaCl, 3 KCl, 10 HEPES, 3 CaCl2, 8 glucose and 2 MgCl2 (at pH 7.40 with HCl). Throughout the experiment, the neurons were constantly perfused with fresh bath solution at a slow rate. All experiments were done at room temperature (22–24°C). Coverslips with transfected cells were loaded into a custom-made chamber and mounted on the stage of a Nikon E600FN microscope with 40× (NA 0.8) water-immersion objectives. Cells were perfused at 1 ml min−1 with a normal extracellular solution (in mM: 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 30 glucose and 25 HEPES). Cells were illuminated with a 100 W mercury arc lamp with neutral density filter attenuation. The filter set for EGFP comprised a 470AF40 excitation filter, a 495DRLP dichroic mirror and a 525AF50 emission filter (Chroma Technology). Time-lapse images were acquired (acquisition time, 500 ms) with a 12-bit cooled CCD camera (ProgRes) at 1 Hz. A Master-8 stimulator (AMPI, Israel) was used to control the onset timing of the iontophoretic current, postsynaptic whole-cell recording and image acquisition. The STDP protocols with different time intervals were applied randomly to the cell during the experiment. For the treatment with d(-)-amino-5-phosphonopentanoic acid (d-APV, 50 μM, Tocris), data were taken 10 min after the onset of drug perfusion.
(d). Image analysis
After acquisition, the images were processed for viewing with NIH ImageJ. We set a region of interest with a circle over the targeted BDNF–EGFP spine observed on the dendrite of the recorded neuron. To cancel out possible variation of expression levels of BDNF–EGFP and image acquisition conditions among different preparations, we presented data as normalized fluorescence changes (ΔF/F0), in which fluorescence changes (ΔF) at a given time were divided by the baseline fluorescence before stimulation (F0).
(e). Statistical analysis
For statistical comparison between two datasets, we first examined whether the data in each set were normally distributed (Jarque–Bera test). If both datasets showed normal distribution, we used the parametric test (t test); otherwise, we used the non-parametric test (Kolmogorov–Smirnov test) or one-way ANOVA. Summary data are given as means ± s.e.m.
3. Results
(a). Induction of tLTP requires extracellular BDNF
Whole-cell recording was made from CA1 pyramidal cells in acutely isolated hippocampal slices from postnatal day 18 (P18) rats, and induction of tLTP was performed by pairing extracellular stimulation of Schaffer collaterals from CA3 with the spiking of pyramidal cells induced by injection of brief (approx. 1 ms) depolarizing current pulses. Following repetitive pairing presynaptic stimulation at 20 ms before postsynaptic spiking at a frequency of 1 Hz (with the total number of 80 stimuli), we found that there was an immediate increase in the amplitude of excitatory postsynaptic potentials (EPSPs), and this potentiation persisted for as long as the recording was made (normally more than 40 min). An example of a recording is shown in figure 1a. The average results from five similar experiments showed that tLTP could be induced reliably at these CA1 synapses. These results are consistent with the finding of STDP in this slice preparation [23], although the stimulation frequency used here was lower than that used in the latter study.
Figure 1.
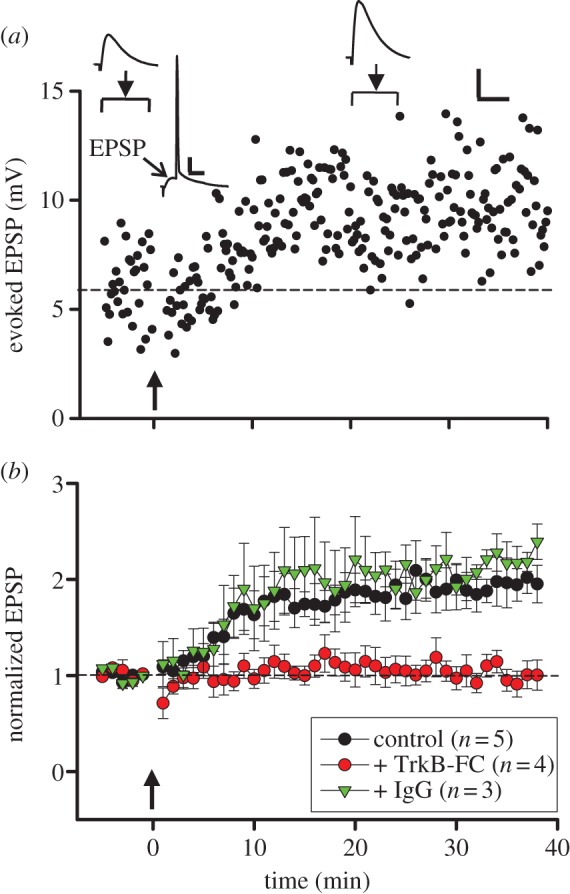
LTP induction in rat hippocampus depends on extracellular BDNF. (a) Example data on changes in the EPSP amplitude induced by paired spiking of pre- and postsynaptic neurons (pre-before-post, Δt = 20 ms, arrow) recorded in hippocampal CA1 pyramidal cells in the brain slice from a rat at P18. Sample traces above: averages of 30 EPSPs (arrowhead) and averaged trace of 80 neuronal responses triggered by paired spiking (Δt = 20 ms). Arrowhead: peak of EPSP. Scales: 4 mV, 50 ms for averaged EPSCs and 10 mV, 20 ms for averaged trace of neuronal responses. (b) Summary of all experiments similar to those in (a), showing EPSP amplitude (mean ± s.e.m., normalized by the mean baseline amplitude) before and after paired pre- and postsynaptic spiking (Δt = 20 ms, arrow) in control artificial cerebrospinal fluid (aCSF; black circle) or in the presence of TrkB–Fc (5 μg ml−1, red circle) or IgG (5 μg ml−1, green triangle).
Previous studies of HFS-induced LTP at these CA3-CA1 synapses have shown that the presence of extracellular BDNF is required [6]. In this study, the same strategy was used: The slice was perfused with solution containing TrkB–IgG (5 μg ml−1), a soluble ligand that chelates extracellular BDNF, for 10 min prior to the application of pre- and postsynaptic pairing of spikes as described above. We found that no change in the EPSP amplitude was induced. As control, incubation with non-specific IgG at the same concentration [6] had no effect on the induction of tLTP. These results indicate that extracellular presence of BDNF during the time of induction is required for induction of tLTP. As discussed below, these experiments could not distinguish whether constitutively secreted BDNF or pairing-induced secretion of BDNF is required for tLTP.
(b). Spike-timing-dependent enhancement of glutamate responses
In cultures of dissociated hippocampal neurons, potentiation and depression of excitatory synapses could be induced by pairing pre- and postsynaptic spiking, with pre-before-post spiking and post-before-pre spiking within an interval of about 20 ms resulting in LTP and LTD, respectively [19]. For the induction of LTP at hippocampal CA1 excitatory synapses via either HFS or STDP, the requirement of postsynaptic activation of N-Methyl-d-aspartate (NMDA) receptors is well documented [19,23,24]. It remains unclear whether the contribution by presynaptic neurons to LTP induction is to provide timely release of only glutamate or other substances also, e.g. BDNF. We thus decided to use a ‘reduced’ version of the hippocampal synapse, substituting the presynaptic nerve terminal with an iontophoretic pipette that delivered pulses of glutamate to simulate presynaptic glutamate release (figure 2a). Using a sharp glass microelectrode (containing 150 mM of glutamate) and brief current injections (duration 0.2–1.0 ms), iontophoretic injection of glutamate pulses near the surface of dendritic spine in cultured neurons resulted in transient inward membrane currents in the neuron (under whole-cell voltage-clamp mode) that were similar in amplitude and time course to excitatory postsynaptic currents (hereafter referred to as simulated EPSCs, or sEPSCs). The amplitude of these sEPSCs was highly sensitive to the positioning of the iontophoretic pipette. A movement of approximately 1 μm away from the dendritic site could result in a marked reduction in the sEPSC amplitude (figure 2b), consistent with the clustered distribution of glutamate receptors at synaptic sites.
Figure 2.

Glutamate iontophoresis and spiking-timing-dependent potentiation and depression of glutamate responses. (a) Bright field image of a days in vitro 14 (DIV 14) cultured hippocampal neuron. R, recording electrode; I-G, puffing microelectrode for iontophoresis of glutamate. (b) Dependence of response on the position of pipette tip relative to a spine. The response was evoked by iontophoretic application of 150 mM glutamate (pH 8), from a micropipette with a tip less than 1 μm (approx. 200 mΩ). The corresponding current traces include iontophoretic stimulus artefacts (small outward-going pulses) and inward currents mediated by glutamate receptors. Iontophoretic pulse was −150 nA, 0.6 ms. Arrows indicate the spine. Maximal response was obtained when the tip of the puffing microelectrode was pointed to the spine. The response decreased greatly when the pipette tip was moved along the dendrite to 2 μm away from the spine. (c–d) An example (top) and averaged results (bottom) of LTP-like potentiation (c) and LTD-like depression (d) of glutamate-induced responses, induced by paired iontophoretic glutamate and postsynaptic spikes at intervals of 20 ms (c) and −20 ms (d), respectively. Arrows indicate the onset of paired stimulation. (Online version in colour.)
We then repetitively paired the iontophoretic glutamate pulses with postsynaptic spikes (initiated by brief current injection) at a frequency of 1 Hz, 80 times, with each iontophoretic pulse followed by a postysynaptic spike at an interval of 20 ms. We observed an persistent increase of the amplitude of the sEPSCs immediately after the paired stimulation. An example of a recording from one experiment is shown in figure 2c, together with changes in the average sEPSC amplitude from all five experiments, normalized by the mean amplitude prior to the paired stimulation in each experiment. Furthermore, pairing of iontophoretic glutamate pulses with postsynaptic spikes but in the opposite temporal order, i.e. each postsynaptic spike preceded the glutamate pulse by 20 ms, led to a persistent reduction of the amplitude of sEPSCs (figure 2d). These results are fully consistent with STDP reported at hippocampal synapses, using paired stimulation of pre- and postsynaptic neurons via dual whole-cell recording [19], and suggest that presynaptic contribution of glutamate is sufficient for the induction of tLTP at these glutamate synapses.
(c). Spike-timing-dependent release of BDNF
Using iontophoretic application of glutamate, we then examined whether pairing of glutamate pulses with neuronal spiking at a low frequency of 1 Hz, a condition that resulted in spike-timing-dependent persistent potentiation and depression of glutamate-induced responses, also leads to dendritic secretion of BDNF. The neurons were transfected with a construct expressing BDNF–enhanced GFP (figure 3a), and the release of BDNF monitored by the reduction of EGFP fluorescence locally at the spine. As shown in the example recordings from a neuron, repetitive glutamate application (1 Hz, 80 pulses) at dendritic spines at 20 ms preceding neuronal spiking (induced by brief injection of depolarizing currents) led to a reduction of EGFP fluorescence at the spines within 2 min (figure 3b), whereas similar pairing with the order of glutamate pulse and neuronal spiking reversed resulted in no significant change in the spine EGFP fluorescence. Notably, either inducing neuronal spiking or applying glutamate pulses alone at the same frequency for the same total number of stimuli had no significant effect on the EGFP fluorescence, indicating the release of BDNF depends on the pairing of glutamate and spiking. Results from 12 experiments on the BDNF release under +20 ms and −20 ms intervals showed that spike-timing-dependent release of BDNF was highly significant (figure 3e, p < 0.01, paired t-test).
Figure 3.

Spike-timing-dependent secretion of BDNF from dendritic spines. (a) Left, an image of a cultured hippocampal neuron (DIV 16) expressing BDNF–enhanced GFP (EGFP). Right, fluorescent images of a dendrite of the neuron expressing BDNF-GFP, taken at 5 s before, 30, 60 or 140 s after the start of iontophoretic application of glutamate paired with spike triggered by an injected current (2 nA, 1 ms). The red and yellow arrows indicate a spine exposed to iontophoretic micropipette and a nearby control spine, respectively. (b–c) Fluorescence changes induced by paired iontophoretic glutamate–spike stimulation. Eighty pairs of glutamate–spike stimulation with an interval of 20 ms (glutamate before spike, n = 28) or −20 ms (glutamate after spike, n = 23) were applied at 1 Hz. Arrow: onset of stimulation at 0 s. Grey lines are traces from individual cases. Thicker red (b) or blue (c) lines are averaged traces. (d) Fluorescence changes induced by spike only (top, n = 8) or iontophoretic glutamate only (bottom, n = 10) stimulation. Eighty iontophoretic glutamate or spike stimuli were applied at 1 Hz, starting at 0 s indicated by the arrow. Coloured lines are averaged traces with grey lines which represent s.e.m. (e) Summarized results of BDNF secretion induced by paired glutamate–spike stimulation. Connected dots represent the results from the same spine (n = 12; **p < 0.005, paired t-test).
We also examined the dependence of BDNF release on the total number of glutamate–spike pairings, with the repetition rate kept a constant of 1 Hz. As shown in figure 4a–d, there was a progressive increase in the amplitude of EGFP fluorescence reduction at the spines, with a minimal number of stimuli for inducing detectable signals of between 40 and 80 stimuli. Furthermore, we examined whether BDNF release depends on the activation of NMDA receptors, similar to that required for the induction of tLTP. As shown in figure 4e–h, the reduction of EGFP fluorescence in response to 80 pairs of glutamate pulses and neuronal spikes (at the interval of +20 ms) was greatly diminished when the NMDA receptor antagonist d-(−)-2-amino-5-phosphonopentanoate (d-APV, 50 μM) was present in the recording medium. Thus, spike-timing-dependent BDNF secretion from these neurons depends on the activation of NMDA receptors.
Figure 4.

Spike-timing-dependent secretion of BDNF from dendritic spines. (a–c) Averaged fluorescence changes induced by paired iontophoretic glutamate-spike stimulation. Twenty (a, n = 10), 40 (b, n = 11) and 160 (c, n = 8) pairs of glutamate–spike stimulation with an interval of 20 ms (glutamate before spike) were applied at 1 Hz, starting at 0 s indicated by the arrow. Coloured lines are averaged traces with grey lines which represent s.e.m. (d) Summarized results of BDNF secretion during 40 s right after the stimulation induced by different pairs of iontophoretic glutamate–spike stimulation (*p < 0.05; **p < 0.01; paired t-test to corresponding averaged baseline value before the application of paired stimuli). (e,g) Averaged fluorescence changes induced by 80 (e) or 160 (g) pairs of iontophoretic glutamate-spike stimulation in the absence or presence of APV (2-amino-5-phosphonopentanoate, 50 μM). Paired glutamate-spike stimulation with an interval of 20 ms (glutamate before spike) was applied at 1 Hz, starting at 0 s indicated by the arrow. Grey lines represent s.e.m. (f,h) Cumulative percentage plot of BDNF secretion induced by 80 (f) or 160 (h) pairs of iontophoretic glutamate and postsynaptic spikes at interval of 20 ms. The magnitude of tLTP-like potentiation or tLTD-like depression is defined as the mean normalized EPSP amplitude at 20–25 min after paired stimulation (***p < 0.001; *p < 0.05; Kolmogorov–Smirnov or K–S test).
To further examine the spike-timing dependence in the dendritic secretion of BDNF, we varied the time interval between the iontophoretic application of glutamate and induction of postsynaptic spiking, with the frequency (1 Hz) and the total number of glutamate–spike pairings (80) kept constant. We found a significant reduction of EGFP fluorescence at the site of glutamate application following repetitive pairing when the interval was 0 or +20 ms, but not when the interval was +50, +100, −20 or −100 ms. These results showed a clear temporal window for BDNF secretion that coincides with the time window for tLTP induction, but not that for tLTD induction (figure 5).
Figure 5.
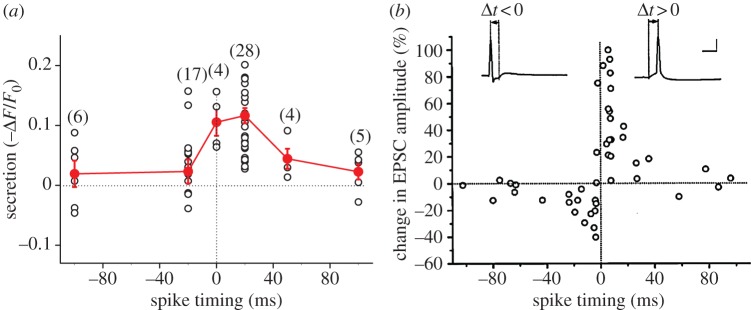
Spike-timing-dependent secretion of BDNF from dendritic spines. (a) Summary of secretion of BDNF induced by paired glutamate-spike stimulation at different intervals. Numbers in parentheses represent the number of cases under each condition. (b) Critical window for the induction of potentiation and depression of glutamate responses. The percentage change in the EPSC amplitude at 20–30 min after the repetitive correlated spiking (60 pulses at 1 Hz) was plotted against the spike timing. Spike timing was defined by the time interval (Δt) between the onset of the glutamate response and the peak of the postsynaptic spike during each cycle of repetitive stimulation, as illustrated by the traces above. Only synapses with initial amplitude of less than 500 pA were included, and all responses were subthreshold for data associated with negatively correlated spiking. Calibration: 50 mV, 10 ms. (Adapted from [19].) (Online version in colour.)
4. Discussion
Synaptic secretion of BDNF is known to modulate the efficacy and activity-dependent plasticity of many synapses [2,3]. In this study, we showed that BDNF is not only required for LTP induced by high-frequency tetanic stimulation at Schaffer collateral–CA1 synapses, as demonstrated previously [6,9], it is also required for tLTP at these synapses [23]. By using a reduced system of cultured hippocampal neurons to eliminate the possible contribution of presynaptic BDNF release for tLTP formation, we showed that tLTP- and tLTD-like changes in glutamate responses at postsynaptic sites could be induced when glutamate pulses were paired with neuronal spiking. Because the amount of BDNF secretion from single postsynaptic sites showed a similar spike-timing dependency in a manner that correlated with the induction of tLTP/tLTD (figure 5), our results suggest that presynaptic BDNF is not necessary for the induction of tLTP, and postsynaptically secreted BDNF may act in an autocrine manner for inducing tLTP. Recent study also showed that STDP formation in the prefrontal cortical synapse is impaired in the mouse with Val66Met polymorphism that reduces BDNF secretion [25], indicating a requirement of activity-dependent BDNF secretion for tLTP formation.
In the study of tLTP in hippocampal slices, TrkB–IgG was applied to the recording medium 10 min prior to the paired stimulation. This treatment presumably depletes the availability of BDNF to the synapses during the induction and maintenance of synaptic potentiation. The absence of any potentiated responses immediately following the paired stimulation suggests that extracellular BDNF is immediately required for the induction process, although previous studies have shown that BDNF is also required for the late-phase LTP (L-LTP) that involves structural changes and new protein synthesis [26]. Our results support the notion that there is a rapid protein synthesis-independent action of secreted BDNF during tLTP induction, and depleting extracellular BDNF could immediately terminate LTP induction. As neurotrophins are highly positively charged at physiological pH and readily bind to cell surfaces and the extracellular matrix upon release [27], extracellular diffusional spread of secreted BDNF is likely to be rather limited. Thus, the action of secreted BDNF may be confined to the specific synapse that receives positively timed glutamate pulses and postsynaptic spiking, without affecting neighbouring synapses. However, extracellular BDNF may be a permissive factor required at the synapse, e.g. for maintaining the presynaptic efficacy of transmitter secretion [6,7,27–29] and BDNF secretion specifically triggered by the paired spiking of pre- and postsynaptic neurons may not be necessary. To distinguish these two possibilities, it is necessary to eliminate selectively BDNF constitutively secreted at the synapse versus BDNF owing to pairing activity-induced secretion. A new experimental strategy for such selective elimination of BDNF remains to be further developed.
In our experiments on BDNF secretion, EGFP-tagged BDNF in the hippocampal neuron is presumably stored in post-Golgi dense-core vesicles, similar to those of endogenously synthesized BDNF [28]. A previous study has shown that the density of BDNF-GFP puncta was comparable with that of immunostained endogenous BDNF puncta both in dendrites and axons. However, the apparent size of BDNF-GFP-containing puncta appeared to be larger, suggesting a larger amount of BDNF-GFP in individual puncta. The same neuronal activity (or Ca2+ elevation) at the release site may evoke a higher amount of BDNF-GFP release from BDNF-GFP-expressing dendrites than of endogenous BDNF release from untransfected dendrites [17]. Our results are in general agreement with previous studies using overexpression of pH-sensitive GFP-tagged BDNF in neuronal cultures [17], indicating the importance of synaptic activation of NMDA receptors for BDNF secretion. Activation of these receptors allows substantial subsynaptic Ca2+ elevation that could promote exocytosis of BDNF-containing vesicles. However, whether activity-induced BDNF secretion in vivo indeed occurs at postsynaptic dendrites remains to be examined, because it remains unclear whether pre- or postsynaptic neurons provide the main source of BDNF in vivo. Dendritic targeting of specific forms of bdnf mRNAs has been observed in cultured neurons [29], but whether BDNF is synthesized locally and whether newly synthesized BDNF in the dendrite could undergo activity-dependent synaptic secretion in vivo remain unclear. This issue is further confounded by the recent finding that in knock-in mice expressing myc-tagged BDNF, immunogold staining of mature hippocampal tissues showed BDNF-containing organelles in the Shaffer-collaterals but not in the dendrite of CA1 pyramidal cells [30]. However, it is still possible that BDNF could be stored in CA1 dendritic compartments during development and secreted in an activity-dependent manner for LTP-like modification during neural circuit maturation and refinement. Furthermore, given the ample evidence of anterograde and retrograde transport and intercellular transfer of neurotrophins in vivo [31–34] and in vitro [35], a neuron that does not synthesize BDNF itself may acquire the protein via endocytic uptake from source cells, and acquired BDNF may undergo activity-induced secretion in a manner similar to that found for exocytosis of false transmitters [36,37].
In summary, our results support the notion that BDNF is required for tLTP and provide evidence that postsynaptic BDNF could be secreted by low-frequency synaptic activity when paired with neuronal spiking within the time window (approx. 20 ms) that allows tLTP induction. Although underlying mechanisms for spike-timing-induced BDNF secretion need to be further elucidated and the exact sites of BDNF secretion in vivo remain unclear, our study indicates that the NMDA receptor-dependent pathway in the postsynaptic cell is critical for spike-timing-dependent BDNF secretion from the dendrite during low-frequency synaptic activation.
Acknowledgement
This work was supported by grants from NIH (NS36999) and CHDI (A3794).
References
- 1.Barde YA, Edgar D, Thoenen H. 1982. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poo M-M. 2001. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2, 24–32 (doi:10.1038/35049004) [DOI] [PubMed] [Google Scholar]
- 3.Park H, Poo MM. 2013. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23 (doi:10.1038/nrn3379) [DOI] [PubMed] [Google Scholar]
- 4.Lessmann V, Gottmann K, Heumann R. 1994. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport 6, 21–25 (doi:10.1097/00001756-199412300-00007) [DOI] [PubMed] [Google Scholar]
- 5.Lohof AM, Ip NY, Poo MM. 1993. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 363, 350–353 (doi:10.1038/363350a0) [DOI] [PubMed] [Google Scholar]
- 6.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. 1996. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381, 706–709 (doi:10.1038/381706a0) [DOI] [PubMed] [Google Scholar]
- 7.Pu L, Liu QS, Poo MM. 2006. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat. Neurosci. 9, 605–607 (doi:10.1038/nn1687) [DOI] [PubMed] [Google Scholar]
- 8.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. 1995. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl Acad. Sci. USA 92, 8856–8860 (doi:10.1073/Pnas.92.19.8856) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang H, Welcher AA, Shelton D, Schuman EM. 1997. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19, 653–664 (doi:10.1016/S0896-6273(00)80378-5) [DOI] [PubMed] [Google Scholar]
- 10.Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A. 1999. Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J. Neurosci. 19, 7983–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blochl A, Thoenen H. 1995. Characterization of nerve growth-factor (NGF) release from hippocampal-neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur. J. Neurosci. 7, 1220–1228 (doi:10.1111/J.1460-9568.1995.Tb01112.X) [DOI] [PubMed] [Google Scholar]
- 12.Balkowiec A, Katz DM. 2000. Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J. Neurosci. 20, 7417–7423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gartner A, Staiger V. 2002. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc. Natl Acad. Sci. USA 99, 6386–6391 (doi:10.1073/pnas.092129699) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartmann M, Heumann R, Lessmann V. 2001. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 20, 5887–5897 (doi:10.1093/emboj/20.21.5887) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H. 2001. Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity-dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J. Neurosci. Res. 64, 1–10 (doi:10.1002/jnr.1080) [DOI] [PubMed] [Google Scholar]
- 16.Tanaka J-I, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GCR, Kasai H. 2008. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science 319, 1683–1687 (doi:10.1126/science.1152864) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM. 2009. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J. Neurosci. 29, 14 185–14 198 (doi:10.1523/JNEUROSCI.1863-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolarow R, Brigadski T, Lessmann V. 2007. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase ii signaling and proceeds via delayed fusion pore opening. J. Neurosci. 27, 10 350–10 364 (doi:10.1523/JNEUROSCI.0692-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bi GQ, Poo MM. 1998. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10 464–10 472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu H, Lim B, Poo MM. 2009. Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J. Neurosci. 29, 12 664–12 674 (doi:10.1523/JNEUROSCI.1984-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen W, Wu B, Zhang Z, Dou Y, Rao ZR, Chen YR, Duan S. 2006. Activity-induced rapid synaptic maturation mediated by presynaptic cdc42 signaling. Neuron 50, 401–414 (doi:10.1016/j.neuron.2006.03.017) [DOI] [PubMed] [Google Scholar]
- 22.Hao J, Wang XD, Dan Y, Poo MM, Zhang XH. 2009. An arithmetic rule for spatial summation of excitatory and inhibitory inputs in pyramidal neurons. Proc. Natl Acad. Sci. USA 106, 21 906–21 911 (doi:10.1073/pnas.0912022106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. 2000. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584–588 (doi:10.1038/35046067) [DOI] [PubMed] [Google Scholar]
- 24.Malenka RC, Nicoll RA. 1993. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 16, 521–527 (doi:10.1016/0166-2236(93)90197-T) [DOI] [PubMed] [Google Scholar]
- 25.Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. 2012. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J. Neurosci. 32, 2410–2421 (doi:10.1523/JNEUROSCI.5205-11.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Y, Christian K, Lu B. 2008. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol. Learn. Mem. 89, 312–323 (doi:10.1016/j.nlm.2007.08.018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blochl A, Thoenen H. 1996. Localization of cellular storage compartments and sites of constitutive and activity-dependent release of nerve growth factor (NGF) in primary cultures of hippocampal neurons. Mol. Cell. Neurosci. 7, 173–190 (doi:10.1006/Mcne.1996.0014) [DOI] [PubMed] [Google Scholar]
- 28.Haubensak W, Narz F, Heumann R, Lessmann V. 1998. BDNF-GFP containing secretory granules are localized in the vicinity of synaptic junctions of cultured cortical neurons. J. Cell Sci. 111, 1483–1493 [DOI] [PubMed] [Google Scholar]
- 29.An JJ, et al. 2008. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134, 175–187 (doi:10.1016/j.cell.2008.05.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dieni S, et al. 2012. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol. 196, 775–788 (doi:10.1083/jcb.201201038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lom B, Cogen J, Sanchez AL, Vu T, Cohen-Cory S. 2002. Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J. Neurosci. 22, 7639–7649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caleo M, Medini P, von Bartheld CS, Maffei L. 2003. Provision of brain-derived neurotrophic factor via anterograde transport from the eye preserves the physiological responses of axotomized geniculate neurons. J. Neurosci. 23, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du JL, Wei HP, Wang ZR, Wong ST, Poo MM. 2009. Long-range retrograde spread of LTP and LTD from optic tectum to retina. Proc. Natl Acad. Sci. USA 106, 18 890–18 896 (doi:10.1073/pnas.0910659106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du JL, Poo MM. 2004. Rapid BDNF-induced retrograde synaptic modification in a developing retinotectal system. Nature 429, 878–883 (doi:10.1038/nature02618) [DOI] [PubMed] [Google Scholar]
- 35.Kohara K, Kitamura A, Morishima M, Tsumoto T. 2001. Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291, 2419–2423 (doi:10.1126/science.1057415) [DOI] [PubMed] [Google Scholar]
- 36.Dan Y, Poo MM. 1992. Quantal transmitter secretion from myocytes loaded with acetylcholine. Nature 359, 733–736 (doi:10.1038/359733a0) [DOI] [PubMed] [Google Scholar]
- 37.Dan Y, Song HJ, Poo MM. 1994. Evoked neuronal secretion of false transmitters. Neuron 13, 909–917 (doi:10.1016/0896-6273(94)90256-9) [DOI] [PubMed] [Google Scholar]