

Abstract
Recent studies of the molecular mechanisms of long-term depression (LTD) suggest a crucial role for the signalling pathways of apoptosis (programmed cell death) in the weakening and elimination of synapses and dendritic spines. With this backdrop, we suggest that LTD can be considered as the electrophysiological aspect of a larger cell biological programme of synapse involution, which uses localized apoptotic mechanisms to sculpt synapses and circuits without causing cell death.
Keywords: long-term potentiation, dendritic spine, local apoptosis, caspase, long-term depression, spine elimination
1. Introduction
Long-term potentiation (LTP) and long-term depression (LTD) are much studied for their own sake as fascinating neurophysiological phenomena as well as for their crucial relevance for brain development, cognitive function and human nervous system diseases. Numerous excellent and worthy reviews have been written about the mechanisms of LTP and/or LTD, but mostly from a physiological–pharmacological and/or molecular perspective [1–6]. The majority of these reviews tend to portray synapses as largely conceptual compartments in which signalling pathways (kinases, phosphatases, etc.) are activated or inactivated, resulting in changes in the function and abundance of postsynaptic glutamate receptors. The dynamic aspects of synaptic plasticity mechanisms—such as the synaptic delivery of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in LTP and the removal/endocytosis of AMPA receptors in LTD, which are now widely accepted in the field [1,7–9]—are often conceptually divorced from the structural and cell biological changes taking place in the neuron and synapse as a whole. In this review, we offer a view of LTD from a cell biologist's perspective, emphasizing the idea that weakening of synaptic transmission and endocytosis of AMPA receptors occur not in molecular isolation, but rather in the context of an ‘involution’ of synapses, whereby synapses wither and die and their contents are mobilized and removed.
2. Caspases and apoptotic mechanisms in long-term depression
Programmed cell death (also called apoptosis) [10] removes excess cells that form during the development of an organism, sculpting a wide range of tissues and organs, not least the nervous system, into their adult form [11,12]. The process of apoptosis—which involves the coordinated regulation and action of multiple genes and proteins—results in the timely demise and tidy removal of unwanted cells with minimal inflammatory or other untoward effects on surrounding tissues. In recent years, evidence is accumulating for an involvement of the molecular mechanisms of apoptosis in processes that do not result in cell death (i.e. non-apoptotic pathways).
Examples of non-cell death roles of apoptotic mechanisms in the nervous system include the dendritic pruning of Drosophila neurons during development [13,14], the structural remodelling of hippocampal neuron synapses and Xenopus retinal growth cones [15,16], and bird song learning [17]. This topic has been reviewed recently by Hyman & Yuan [18].
The cysteine-aspartate protease caspase-3 is well established as a key ‘executioner’ enzyme in the programmed cell death of neurons and other cell types [11]. Caspase-3 (and its close relative caspase-7) lies downstream of the two major apoptotic pathways—intrinsic and extrinsic—within cells. In the intrinsic (or mitochondrial) pathway, apoptotic stimuli lead to release of proapoptotic molecules, such as cytochrome c and second mitochondria-derived activator of caspases (SMAC, also known as DIABLO), from mitochondria; these molecules lead to activation of caspase-9, which then cleaves and activates caspase-3 and caspase-7, leading to apoptosis [19]. The release of cytochrome c from mitochondria is inhibited by antiapoptotic members of the Bcl-2 family of proteins (such as Bcl-xL) and promoted by proapoptotic members (such as BAD, BAX and BAK) [20]. In the cytoplasm, activation of the caspase-9–caspase-3 cascade is restrained by inhibitor of apoptosis proteins (IAPs) which bind to and inhibit these caspases [21,22].
In addition to their crucial role in programmed cell death, recent studies show that apoptotic mechanisms, in particular caspase-3, participate in LTD and perhaps synapse elimination in the absence of cell death. Pharmacologic inhibition of caspase-3/7 or genetic disruption of caspase-3 in mice blocks induction of LTD in CA1 region of hippocampus [23]. N-methyl-d-aspartate (NMDA) receptor-dependent AMPA receptor internalization—which is an essential mechanism underlying LTD induction—is also blocked by the same manipulations in cultured neurons [23]. Thus, caspase-3 activity is required for NMDA receptor-dependent LTD. Interestingly, caspase-3 does not appear to be necessary for mGluR-dependent LTD (Kei Cho 2013, personal communication, [24]). In addition, knockout mice lacking BAD or BAX (proapoptotic Bcl-2 family proteins) are defective in AMPA receptor internalization and NMDA receptor-dependent LTD, but not in mGluR-dependent LTD [24]. In hippocampal slice cultures, postsynaptic overexpression of Bcl-xL (an antiapoptotic Bcl-2 family member) suppresses the induction of LTD [23]. Together, these data strongly support the idea that activation of the mitochondrial (intrinsic) pathway of apoptosis—which seems to occur at least in part within the postsynaptic compartment—is required for LTD. LTD in hippocampal slice cultures is also inhibited by postsynaptic overexpression of different fragments of XIAP (an IAP protein that inhibits caspase-9 and caspase-3), confirming the importance of caspase-9–caspase-3 activity in LTD [23].
3. Localized ‘synaptic apoptosis’ as a conceptual framework for long-term depression
Caspase-3 is necessary for LTD, but is it activated during LTD? This has been more difficult to ascertain, because the degree and extent of caspase-3 activation would be predicted to be less than that occurring during programmed cell death. Indeed, by staining and immunoblotting approaches, it has been shown that caspase-3 is activated, but only to a modest extent, following bath NMDA stimulation (‘chemical LTD’) [23,24]. Active caspase-3 was detectable in dendrites as well as cell body under such conditions [23].
The activation of caspase-3 (and presumably the mitochondrial apoptosis pathway leading to caspase-3 activation) may be restricted to peripheral compartments of the neuron so that death of the entire cell does not ensue (figure 1). This has given rise to the idea of localized ‘synaptic apoptosis’—a term first coined by Mattson et al. [25]—as the cell biological basis of LTD. A critical question that remains is whether local activation of the mitochondrial apoptosis pathway and of caspase-3 is sufficient—as opposed to just being necessary—for LTD. Addressing this question will require development of experimental tools to activate apoptosis mechanisms within neurons at a specific location and time. An important related unknown is the molecular mechanism(s) by which active caspase-3 contributes to LTD.
Figure 1.

A putative illustration of apoptotic pathway involvement in spine pruning. LTD induces Ca2+ influx via NMDA receptors and an increase in Ca2+ levels in mitochondria, which leads to cytochrome c release. Activation of the mitochondrial apoptosis pathway culminates in activation of caspase-3, which exerts local effects on synaptic strength (LTD) and spine size by proteolysis of proteins in the postsynaptic compartment, e.g. Akt. The ubiquitin-proteasome system (UPS) and IAPs inhibit activated caspases and limit the extent of local synaptic apoptosis. For simplicity, NMDA receptors are shown only in one spine at bottom of the dendrite, and the LTD effects of AMPA receptor depletion and spine shrinkage are shown only on the upper side of the dendrite (GSK3β, glycogen synthase kinase-3β).
Once apoptotic mechanisms including caspase-3 are locally activated by LTD-inducing stimuli, what determines the outcome between local synapse weakening versus cell-wide death? It appears that the mitochondrial apoptotic pathway is only transiently and modestly activated by LTD-inducing stimulation, whereas neuronal cell death is associated with higher levels of activation of apoptotic mechanisms and caspases [23,24,26]. We hypothesize that a greater amplitude, broader extent and/or longer duration of activation of the mitochondria-caspase pathway in neurons would be required to mediate cell death. It is possible that the dendrites of a neuron might be able to integrate depressive signals, such that only when these signals accumulate beyond a certain threshold, do they lead to apoptotic death of the whole neuron. When the apoptotic signals are below the threshold for cell death, only local synaptic apoptosis can occur. The elaborate, extended morphology of neurons is well suited for localized apoptosis to occur in restricted compartments far from the cell body. The mechanisms that ‘insulate’ the effects of apoptosis to localized peripheral regions of the cell are presently unclear but would be important to study. It would not be surprising if there were active cell biological mechanisms to restrict the spread of local synaptic apoptosis in neurons, just as there are active mechanisms to inhibit the progression of apoptotic cell death [20].
How big is the region of localized synaptic apoptosis? Most dendritic mitochondria are quite large relative to individual spines (typically 2–20 μm in length [27]). Mitochondria reside within the dendrite shaft and they span lengths that encompass multiple spines/synapses (figure 2). If the mitochondrial release of proapoptotic factors, and consequent activation of the caspase-9–caspase-3 cascade, mediates the induction of LTD, one would expect that LTD would spread over considerably more than 10 μm. Previous conclusions of the homosynaptic nature of LTD are largely based on experiments showing pathway or input specificity, in which the stimulated pathways/inputs could be hundreds of micrometres apart. In such circumstances, short-distance heterosynaptic LTD could be missed. Indeed, LTD has been reported to ‘spread’ more than LTP [28], and heterosynaptic LTD (LTD occurring at unstimulated synapses) has been described in several studies [29,30].
Figure 2.
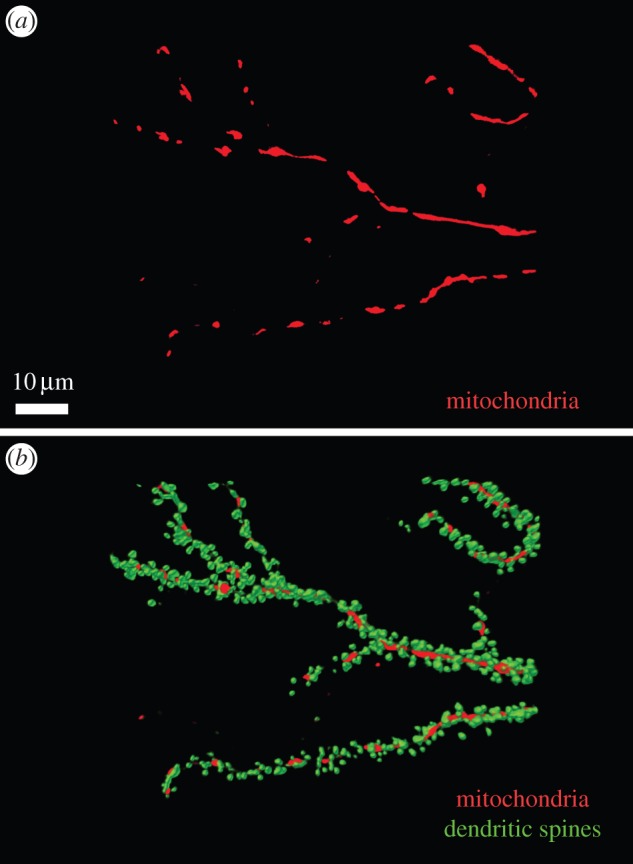
Relative size and distribution of mitochondria and spines in neuronal dendrites. Dendrites of cultured hippocampal neurons days in vitro (DIV) are shown with mitochondria labelled with mitochondria-targeted red fluorescence protein (RFP; red) (a), and in merge with dendritic spines labelled with yellow fluorescence protein (YFP)-actin (green) (b). Dendritic mitochondria range in length from 1–2 µm to 20–30 µm. In most cases, a single mitochondrion spans a distance that encompasses multiple dendritic spines.
4. Localized apoptosis and ‘synapse involution’
Interestingly, LTD is most robustly inducible during the period of two to four weeks of age in a rodent's life [31,32], which corresponds to the most active period of synapse elimination during brain development. LTD is also directly associated with shrinkage and loss of dendritic spines in live-imaging experiments [33,34], leading many to think about NMDA receptor-dependent LTD as an electrophysiological correlate (or harbinger) of excitatory synapse elimination. Given that apoptosis mechanisms seem to underlie LTD (as discussed above), it is appealing to consider LTD and the associated spine shrinkage and elimination as different facets of an overall process of synapse involution.
What are the molecular mechanisms that underlie spine shrinkage and synapse loss associated with LTD? In programmed cell death, caspase-3 and other effector caspases orchestrate dismantling of the cell structure through cleavage of specific substrates [35]. It seems reasonable to suppose that localized synaptic apoptosis, such as in LTD, might also involve local cleavage of key substrates by caspase-3, leading to local pruning of neuronal structures. Actin rearrangements underlie the structural remodelling of dendritic spines, while microtubules are the core support of dendritic shafts [36]. Various actin regulatory proteins, including ROCK1, gelsolin, spectrin and fodrin are present in dendrites [37–39] and are known caspase-3 targets [40–42]. Microtubules play an essential role in shaping the dendritic tree. Tau, a microtubule-associated protein, is also a caspase-3 target [43]. It is worth pointing out here that caspase-3 activation has also been observed not just downstream of NMDA receptor activation, but dependent on NMDA-induced AMPA receptor internalization [44].
Other prominent signalling molecules that are involved in LTD and shown to be present in dendrites are the calcium-sensitive phosphatase calcineurin and the protein kinase Akt. Calcineurin regulates ion channels and cytoskeletal proteins [45,46], inhibits synaptic function [47] and is proteolytically activated by caspase-3 [48]. Akt is an antiapoptotic and progrowth kinase that phosphorylates and inhibits glycogen synthase kinase (GSK) 3β activity, which is required for induction of LTD [49,50]. Akt is also a substrate of caspase-3 [51], and prevention of cleavage of Akt blocks LTD [23] (figure 1). Hence, caspase-3 involvement in LTD induction could involve Akt proteolysis, which would in turn increase GSK3ß activity. It is interesting that Akt is a proto-oncogene that promotes dendrite growth [52], raising the possibility that the phosphatidylinositol 3 (PI3) kinase-Akt signalling pathway is antagonistic to the mitochondrial apoptosis-caspase-3 pathway in the local regulation of synapse strength and synapse morphology.
The postsynaptic density (PSD) is a sine qua non feature of excitatory synapses; it is tightly aligned with the presynaptic active zone and typically found at the tip of dendritic spines. A complex, proteinaceous, membrane-associated organelle, the PSD is the structure in which postsynaptic glutamate receptors are concentrated and where they interact physically and functionally with scaffold proteins and associated signalling molecules (kinases, phosphatases, guanine nucleotide exchange factors, GTPase activating proteins, etc.) [53]. The size of the PSD (and of the dendritic spine) correlates closely with the strength of the synapse and the abundance of postsynaptic AMPA receptors [53]. Therefore, during LTD, the removal of postsynaptic AMPA receptors and associated shrinkage of synapses and spines must involve major molecular rearrangements, and ultimately down-sizing, of the PSD. Indeed, a recent study by Granger et al. supports the notion that structural reorganization of the synapse is a fundamental mechanism underlying synaptic plasticity [54,55].
The PSD is organized around a lattice of key scaffold proteins, of which PSD-95 is one of the most abundant and best understood [56,57]. PSD-95 overexpression boosts excitatory synaptic transmission, a function that depends on the accumulation of PSD-95 in synapses, which in turn is regulated by phosphorylation of PSD-95 on ser-295 by protein kinase JNK [58]. Ser-295 dephosphorylation of PSD-95 occurs rapidly following LTD-like stimulation and is required for AMPA receptor internalization and LTD [58]. The dephosphorylation appears to be mediated by PP1/PP2A phosphatases, which are activated during LTD [46,59]. Dephosphorylation of ser-295—as well as phosphorylation of threonine-17 by GSK-3—contributes to the destabilization of PSD-95 in the PSD [60], which should lead to mobilization and loss of PSD-95 from synapses and mediate morphological shrinkage of synapses/spines in addition to weakening of synaptic transmission.
It is likely that the size and structure of the PSD are also regulated by additional mechanisms during LTD, such as protein degradation by proteases and by proteasomes, which are recruited to spines in activity-dependent fashion [61,62]. Whether caspase-3 substrates exist in the PSD is presently unknown and an exciting question to pursue in future studies.
5. External forces in elimination of spines and synapses: microglia engulfment
In addition to caspase activation, removal of apoptotic cellular debris by phagocytic cells is a core event in programmed cell death [63]. As LTD and synapse involution by local apoptosis could depend on similar mechanisms of programmed cell death, one is forced to wonder whether phagocytosis might play a role in removal of ‘dying’ synapses.
Indeed, recent studies point to phagocytic processes for clearance of synapses and spines in the nervous system [64,65]. Microglia (resident macrophage-like cells of the central nervous system) have been shown in imaging studies to engulf remnants of synapses in the developing brain [66]. The phagocytosis of synapses appears to depend, at least in part, on complement proteins, which have been long studied in innate immunity and which are significantly expressed in the brain. A major function of complement is to tag microbes or unwanted cells and cellular debris for rapid clearance by macrophage engulfment or complement-mediated cell lysis [67]. Complement cascade components C1q and C3 can be found at synapses during the period of synapse elimination and are required for the developmental pruning of retinogeniculate synapses [65,68]. Disruption of microglia or complement function caused defects in the development of neural circuits [65–69].
These studies suggest that synapse elimination in development and in disease might involve recognition and clearance of dying synapses by professional phagocytic cells, analogous to the removal of apoptotic cell corpses in programmed cell death.
6. How is the mitochondrial apoptosis pathway activated by long-term depression stimuli?
Mitochondria are organelles well positioned to play a role in synaptic plasticity. Mitochondria are present close to and occasionally even within individual spines (figure 2), and their distribution and motility are regulated by synaptic activity [70].
Well known to act as Ca2+ buffers in cells, mitochondria take up Ca2+ after synaptic stimulation [71]. Additionally, Ca2+ uptake by mitochondria promotes their release of proapoptotic factors [72]. Thus, a dendritic mitochondrion can act as a sensor of prolonged, moderate elevation of postsynaptic Ca2+, a condition that seems to be critical for induction of LTD.
Mitochondria also often have close contacts with the endoplasmic reticulum (ER) [73], which is extensively present in postsynaptic compartments (figure 1). Activation of mitochondrial Ca2+ uptake by Ca2+ release from ER leads to Ca2+ accumulation in mitochondria [74] and cytochrome c release [75]. Ca2+ release from ER stores via ryanodine receptors and IP3 receptors may also play a role in LTD in hippocampal neurons and Purkinje cells [28,76,77], and appears to be required for caspase-9/3 activation by NMDA stimulation in neurons [23]. It is tempting to speculate that the functional interaction of ER and mitochondria—which has been recently shown to also be involved in autophagosome formation [78]—might be important for LTD induction.
7. Long-term depression in neurodegeneration
Synapse loss and reduced dendritic spine density are characteristic features of various neurodegenerative diseases [68,79,80]. NMDA receptor activity has been implicated in the neurotoxicity of epilepsy, Huntington's disease, Alzheimer's disease, stroke and traumatic brain injury [81–85]. Excessive NMDA receptor activation can induce loss of dendritic spines and neurite beading [86], and subsequent neuronal death [87]. Hence, it is possible that physiological LTD and synapse loss in neurodegeneration share common mechanisms. In line with this hypothesis, Aβ oligomers (which are probable pathogenic agents in Alzheimer's disease) can enhance LTD. Aβ oligomers can also induce or promote spine loss, AMPA receptor internalization, stimulation of calcineurin and GSK3ß and activation of caspase-3 [88–95], which are common to the features of NMDA receptor-dependent LTD. Notably, D'Amelio et al. [47] observed that active caspase-3 is elevated in the dendritic spines of a transgenic mouse model of Alzheimer's disease (Tg2576), in the absence of cell death, and this caspase-3 activation correlated with memory impairment and reduced spine density and size.
8. Are there similar apoptotic mechanisms occurring in axons and presynaptic sites?
Most of the studies reviewed here have focused on dendrites and spines, because the postsynaptic compartment is easier to manipulate and measure experimentally in molecular-genetic and electrophysiological experiments. Nevertheless, there have been a few studies showing the involvement of caspases in axon pruning in the absence of neuronal death [96,97]. Although further studies are needed, it appears that apoptotic mechanisms can also regulate axonal morphology. It will be interesting to investigate whether local activation of caspases can locally affect synapse structure and function from the presynaptic side [98,99].
9. Viewing long-term potentiation and long-term depression as growth versus apoptosis of synapses
At a very simple cell biological level, we propose that LTD and synapse loss are the opposites of LTP and synapse formation: LTD and synapse elimination can be regarded as manifestations of synapse ‘involution’, whereas LTP and new synapse formation can be considered synapse ‘growth’. Both are, of course, natural, even temporally overlapping processes in the maturation of neurons and circuits.
In the context of growth versus involution, it is notable that LTP and LTD use largely opposing signalling pathways that have been much studied in the context of oncogenesis. In contrast to the attrition of synapses in LTD, which is dependent on apoptotic signalling pathways (as discussed above), LTP relies on signalling mechanisms implicated in cell growth and proliferation. For example, activation of the proto-oncogene Ras, and its oncogenic downstream kinases PI3 kinase and ERK, favours LTP rather than LTD [100,101]. The proto-oncogene Akt, which is downstream of PI3 kinase, promotes the growth of dendrites and LTP but antagonizes LTD [49]. Phosphatase and tensin homolog (PTEN), a protein phosphatase and tumour suppressor that opposes the action of the Ras-PI3 kinase pathway, drives synaptic depression and is required for LTD [102]. The protein kinase GSK3ß is critical for NMDA receptor-dependent LTD [49] and is also known to promote cell death, at least in part via the intrinsic apoptosis pathway [103].
In conclusion, recent data from a variety of experimental systems point to an important role for apoptotic mechanisms in LTD and synapse elimination. By analogy to cancer signalling, we suggest that common molecular mechanisms that drive cell growth and proliferation in mitotic cells are used in postmitotic neurons for expansion and strengthening of specific synapses (LTP). Conversely, postmitotic neurons have adapted the molecular mechanisms of programmed cell death to weaken and eliminate unwanted synaptic connections in localized regions of the cell.
References
- 1.Malenka RC, Bear MF. 2004. LTP and LTD: an embarrassment of riches. Neuron 44, 5–21 (doi:10.1016/j.neuron.2004.09.012) [DOI] [PubMed] [Google Scholar]
- 2.Collingridge GL, Peineau S, Howland JG, Wang YT. 2010. Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473 (doi:10.1038/nrn2867) [DOI] [PubMed] [Google Scholar]
- 3.Bliss TV, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39 (doi:10.1038/361031a0) [DOI] [PubMed] [Google Scholar]
- 4.Kerchner GA, Nicoll RA. 2008. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 9, 813–825 (doi:10.1038/nrn2501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bear MF. 2003. Bidirectional synaptic plasticity: from theory to reality. Phil. Trans. R. Soc. Lond. B 358, 649–655 (doi:10.1098/rstb.2002.1255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas GM, Huganir RL. 2004. MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183 (doi:10.1038/nrn1346) [DOI] [PubMed] [Google Scholar]
- 7.Shepherd JD, Huganir RL. 2007. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 23, 613–643 (doi:10.1146/annurev.cellbio.23.090506.123516) [DOI] [PubMed] [Google Scholar]
- 8.Collingridge GL, Isaac JT, Wang YT. 2004. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962 (doi:10.1038/nrn1556) [DOI] [PubMed] [Google Scholar]
- 9.Malinow R, Malenka RC. 2002. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126 (doi:10.1146/annurev.neuro.25.112701.142758) [DOI] [PubMed] [Google Scholar]
- 10.Kerr JF, Wyllie AH, Currie AR. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 (doi:10.1038/bjc.1972.33) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuchs Y, Steller H. 2011. Programmed cell death in animal development and disease. Cell 147, 742–758 (doi:10.1016/j.cell.2011.10.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim WR, Sun W. 2011. Programmed cell death during postnatal development of the rodent nervous system. Dev. Growth Differ. 53, 225–235 (doi:10.1111/j.1440-169X.2010.01226.x) [DOI] [PubMed] [Google Scholar]
- 13.Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. 2006. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 51, 283–290 (doi:10.1016/j.neuron.2006.07.014) [DOI] [PubMed] [Google Scholar]
- 14.Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW. 2006. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236 (doi:10.1038/nn1774) [DOI] [PubMed] [Google Scholar]
- 15.Gilman CP, Mattson MP. 2002. Do apoptotic mechanisms regulate synaptic plasticity and growth-cone motility? Neuromolecular Med. 2, 197–214 (doi:10.1385/NMM:2:2:197) [DOI] [PubMed] [Google Scholar]
- 16.Campbell DS, Holt CE. 2003. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37, 939–952 (doi:10.1016/S0896-6273(03)00158-2) [DOI] [PubMed] [Google Scholar]
- 17.Huesmann GR, Clayton DF. 2006. Dynamic role of postsynaptic caspase-3 and BIRC4 in zebra finch song-response habituation. Neuron 52, 1061–1072 (doi:10.1016/j.neuron.2006.10.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyman BT, Yuan J. 2012. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 13, 395–406 (doi:10.1038/nrn3228) [DOI] [PubMed] [Google Scholar]
- 19.Tait SW, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632 (doi:10.1038/nrm2952) [DOI] [PubMed] [Google Scholar]
- 20.Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 (doi:10.1038/nrm2308) [DOI] [PubMed] [Google Scholar]
- 21.Srinivasula SM, Ashwell JD. 2008. IAPs: what's in a name? Mol. Cell 30, 123–135 (doi:10.1016/j.molcel.2008.03.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riedl SJ, Shi Y. 2004. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 5, 897–907 (doi:10.1038/nrm1496) [DOI] [PubMed] [Google Scholar]
- 23.Li Z, et al. 2010. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871 (doi:10.1016/j.cell.2010.03.053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiao S, Li Z. 2011. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 70, 758–772 (doi:10.1016/j.neuron.2011.04.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattson MP, Keller JN, Begley JG. 1998. Evidence for synaptic apoptosis. Exp. Neurol. 153, 35–48 (doi:10.1006/exnr.1998.6863) [DOI] [PubMed] [Google Scholar]
- 26.Lu C, Fu W, Salvesen GS, Mattson MP. 2002. Direct cleavage of AMPA receptor subunit GluR1 and suppression of AMPA currents by caspase-3: implications for synaptic plasticity and excitotoxic neuronal death. Neuromolecular Med. 1, 69–79 (doi:10.1385/NMM:1:1:69) [DOI] [PubMed] [Google Scholar]
- 27.Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ. 2003. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J. Neurosci. 23, 7881–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. 2000. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584–588 (doi:10.1038/35046067) [DOI] [PubMed] [Google Scholar]
- 29.Scanziani M, Nicoll RA, Malenka RC. 1996. Heterosynaptic long-term depression in the hippocampus. J. Physiol. Paris 90, 165–166 (doi:10.1016/S0928-4257(97)81416-7) [DOI] [PubMed] [Google Scholar]
- 30.Christie BR, Stellwagen D, Abraham WC. 1995. Evidence for common expression mechanisms underlying heterosynaptic and associative long-term depression in the dentate gyrus. J. Neurophysiol. 74, 1244–1247 [DOI] [PubMed] [Google Scholar]
- 31.Kemp N, McQueen J, Faulkes S, Bashir ZI. 2000. Different forms of LTD in the CA1 region of the hippocampus: role of age and stimulus protocol. Eur. J. Neurosci. 12, 360–366 (doi:10.1046/j.1460-9568.2000.00903.x) [DOI] [PubMed] [Google Scholar]
- 32.Wagner JJ, Alger BE. 1995. GABAergic and developmental influences on homosynaptic LTD and depotentiation in rat hippocampus. J. Neurosci. 15, 1577–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh WC, Hill TC, Zito K. 2013. Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc. Natl Acad. Sci. USA 110, E305–E312 (doi:10.1073/pnas.1214705110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Q, Homma KJ, Poo MM. 2004. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757 (doi:10.1016/j.neuron.2004.11.011) [DOI] [PubMed] [Google Scholar]
- 35.Crawford ED, Wells JA. 2011. Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 80, 1055–1087 (doi:10.1146/annurev-biochem-061809-121639) [DOI] [PubMed] [Google Scholar]
- 36.Hotulainen P, Hoogenraad CC. 2010. Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629 (doi:10.1083/jcb.201003008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salminen A, Suuronen T, Kaarniranta K. 2008. ROCK, PAK, and Toll of synapses in Alzheimer's disease. Biochem. Biophys. Res. Commun. 371, 587–590 (doi:10.1016/j.bbrc.2008.04.148) [DOI] [PubMed] [Google Scholar]
- 38.Furukawa K, Fu W, Li Y, Witke W, Kwiatkowski DJ, Mattson MP. 1997. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J. Neurosci. 17, 8178–8186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ursitti JA, et al. 2001. Spectrins in developing rat hippocampal cells. Brain Res. Dev. Brain Res. 129, 81–93 (doi:10.1016/S0165-3806(01)00160-2) [DOI] [PubMed] [Google Scholar]
- 40.Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. 2001. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3, 346–352 (doi:10.1038/35070019) [DOI] [PubMed] [Google Scholar]
- 41.Kothakota S, et al. 1997. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278, 294–298 (doi:10.1126/science.278.5336.294) [DOI] [PubMed] [Google Scholar]
- 42.Wang KK, Posmantur R, Nath R, McGinnis K, Whitton M, Talanian RV, Glantz SB, Morrow JS. 1998. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J. Biol. Chem. 273, 22 490–22 497 (doi:10.1074/jbc.273.35.22490) [DOI] [PubMed] [Google Scholar]
- 43.Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A. 2000. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 75, 624–633 (doi:10.1046/j.1471-4159.2000.0750624.x) [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Ju W, Liu L, Fam S, D'Souza S, Taghibiglou C, Salter M, Wang YT. 2004. α-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptor (AMPAR) endocytosis is essential for N-methyl-D-aspartate-induced neuronal apoptosis. J. Biol. Chem. 279, 41 267–41 270 (doi:10.1074/jbc.C400199200) [DOI] [PubMed] [Google Scholar]
- 45.Halpain S, Hipolito A, Saffer L. 1998. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. 18, 9835–9844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winder DG, Sweatt JD. 2001. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat. Rev. Neurosci. 2, 461–474 (doi:10.1038/35081514) [DOI] [PubMed] [Google Scholar]
- 47.D'Amelio M, et al. 2011. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat. Neurosci. 14, 69–76 (doi:10.1038/nn.2709) [DOI] [PubMed] [Google Scholar]
- 48.Mukerjee N, McGinnis KM, Park YH, Gnegy ME, Wang KK. 2000. Caspase-mediated proteolytic activation of calcineurin in thapsigargin-mediated apoptosis in SH-SY5Y neuroblastoma cells. Arch. Biochem. Biophys. 379, 337–343 (doi:10.1006/abbi.2000.1889) [DOI] [PubMed] [Google Scholar]
- 49.Peineau S, et al. 2007. LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron 53, 703–717 (doi:10.1016/j.neuron.2007.01.029) [DOI] [PubMed] [Google Scholar]
- 50.Peineau S, Bradley C, Taghibiglou C, Doherty A, Bortolotto ZA, Wang YT, Collingride GL. 2008. The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl. 1), S428–S437 (doi:10.1038/bjp.2008.2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Asselin E, Mills GB, Tsang BK. 2001. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 61, 1862–1868 [PubMed] [Google Scholar]
- 52.Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. 2005. Control of dendritic arborization by the phosphoinositide-3’-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 25, 11 300–11 312 (doi:10.1523/JNEUROSCI.2270-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheng M, Kim E. 2011. The postsynaptic organization of synapses. Cold Spring Harbor Perspect. Biol. 3, a005678 (doi:10.1101/cshperspect.a005678) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. 2013. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature 493, 495–500 (doi:10.1038/nature11775) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheng M, Malinow R, Huganir R. 2013. Neuroscience: strength in numbers. Nature 493, 482–483 (doi:10.1038/493482a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim E, Sheng M. 2004. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5, 771–781 (doi:10.1038/nrn1517) [DOI] [PubMed] [Google Scholar]
- 57.Sheng M, Hoogenraad CC. 2007. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu. Rev. Biochem. 76, 823–847 (doi:10.1146/annurev.biochem.76.060805.160029) [DOI] [PubMed] [Google Scholar]
- 58.Kim MJ, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. 2007. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 56, 488–502 (doi:10.1016/j.neuron.2007.09.007) [DOI] [PubMed] [Google Scholar]
- 59.Mulkey RM, Herron CE, Malenka RC. 1993. An essential role for protein phosphatases in hippocampal long-term depression. Science 261, 1051–1055 (doi:10.1126/science.8394601) [DOI] [PubMed] [Google Scholar]
- 60.Nelson C, et al. 2013. Phosphorylation of threonine-19 of PSD-95 by GSK-3β is required for PSD-95 mobilization and long-term depression. J. Neurosci. 33, 12 122–12 135 (doi:10.1523/JNEUROSCI.0131-13.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bingol B, Sheng M. 2011. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 69, 22–32 (doi:10.1016/j.neuron.2010.11.006) [DOI] [PubMed] [Google Scholar]
- 62.Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. 2010. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140, 567–578 (doi:10.1016/j.cell.2010.01.024) [DOI] [PubMed] [Google Scholar]
- 63.Fadok VA, Chimini G. 2001. The phagocytosis of apoptotic cells. Semin. Immunol. 13, 365–372 (doi:10.1006/smim.2001.0333) [DOI] [PubMed] [Google Scholar]
- 64.Aguzzi A, Barres BA, Bennett ML. 2013. Microglia: scapegoat, saboteur, or something else? Science 339, 156–161 (doi:10.1126/science.1227901) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stephan AH, Barres BA, Stevens B. 2012. The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–339 (doi:10.1146/annurev-neuro-061010-113810) [DOI] [PubMed] [Google Scholar]
- 66.Schafer DP, et al. 2012. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (doi:10.1016/j.neuron.2012.03.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ricklin D, Hajishengallis G, Yang K, Lambris JD. 2010. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797 (doi:10.1038/ni.1923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stevens B, et al. 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (doi:10.1016/j.cell.2007.10.036) [DOI] [PubMed] [Google Scholar]
- 69.Paolicelli RC, et al. 2011. Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458 (doi:10.1126/science.1202529) [DOI] [PubMed] [Google Scholar]
- 70.Sheng ZH, Cai Q. 2012. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93 (doi:10.1038/nrg3141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pivovarova NB, Pozzo-Miller LD, Hongpaisan J, Andrews SB. 2002. Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J. Neurosci. 22, 10 653–10 661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pacher P, Hajnoczky G. 2001. Propagation of the apoptotic signal by mitochondrial waves. Embo J. 20, 4107–4121 (doi:10.1093/emboj/20.15.4107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 (doi:10.1126/science.280.5370.1763) [DOI] [PubMed] [Google Scholar]
- 74.Csordas G, Thomas AP, Hajnoczky G. 1999. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. Embo J. 18, 96–108 (doi:10.1093/emboj/18.1.96) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nutt LK, Pataer A, Pahler J, Fang B, Roth J, McConkey DJ, Swisher SG. 2002. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 277, 9219–9225 (doi:10.1074/jbc.M106817200) [DOI] [PubMed] [Google Scholar]
- 76.Kohda K, Inoue T, Mikoshiba K. 1995. Ca2+ release from Ca2+ stores, particularly from ryanodine-sensitive Ca2+ stores, is required for the induction of LTD in cultured cerebellar Purkinje cells. J. Neurophysiol. 74, 2184–2188 [DOI] [PubMed] [Google Scholar]
- 77.Reyes M, Stanton PK. 1996. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J. Neurosci. 16, 5951–5960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hamasaki M, et al. 2013. Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 (doi:10.1038/nature11910) [DOI] [PubMed] [Google Scholar]
- 79.Selkoe DJ. 2002. Alzheimer's disease is a synaptic failure. Science 298, 789–791 (doi:10.1126/science.1074069) [DOI] [PubMed] [Google Scholar]
- 80.Luo L, O'Leary DD. 2005. Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 28, 127–156 (doi:10.1146/annurev.neuro.28.061604.135632) [DOI] [PubMed] [Google Scholar]
- 81.Urban L, Aitken PG, Friedman A, Somjen GG. 1990. An NMDA-mediated component of excitatory synaptic input to dentate granule cells in ‘epileptic’ human hippocampus studied in vitro. Brain Res. 515, 319–322 (doi:10.1016/0006-8993(90)90615-I) [DOI] [PubMed] [Google Scholar]
- 82.Milnerwood AJ, et al. 2010. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron 65, 178–190 (doi:10.1016/j.neuron.2010.01.008) [DOI] [PubMed] [Google Scholar]
- 83.Sonkusare SK, Kaul CL, Ramarao P. 2005. Dementia of Alzheimer's disease and other neurodegenerative disorders–memantine, a new hope. Pharmacol. Res. 51, 1–17 (doi:10.1016/j.phrs.2004.05.005) [DOI] [PubMed] [Google Scholar]
- 84.Faden AI, Demediuk P, Panter SS, Vink R. 1989. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800 (doi:10.1126/science.2567056) [DOI] [PubMed] [Google Scholar]
- 85.Choi DW, Rothman SM. 1990. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 13, 171–182 (doi:10.1146/annurev.ne.13.030190.001131) [DOI] [PubMed] [Google Scholar]
- 86.Isokawa M, Levesque MF. 1991. Increased NMDA responses and dendritic degeneration in human epileptic hippocampal neurons in slices. Neurosci. Lett. 132, 212–216 (doi:10.1016/0304-3940(91)90304-C) [DOI] [PubMed] [Google Scholar]
- 87.Hu NW, Ondrejcak T, Rowan MJ. 2012. Glutamate receptors in preclinical research on Alzheimer's disease: update on recent advances. Pharmacol. Biochem. Behav. 100, 855–862 (doi:10.1016/j.pbb.2011.04.013) [DOI] [PubMed] [Google Scholar]
- 88.Harada J, Sugimoto M. 1999. Activation of caspase-3 in beta-amyloid-induced apoptosis of cultured rat cortical neurons. Brain Res. 842, 311–323 (doi:10.1016/S0006-8993(99)01808-9) [DOI] [PubMed] [Google Scholar]
- 89.Jo J, et al. 2011. Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 14, 545–547 (doi:10.1038/nn.2785) [DOI] [PubMed] [Google Scholar]
- 90.Shankar GM, et al. 2008. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842 (doi:10.1038/nm1782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Knobloch M, Mansuy IM. 2008. Dendritic spine loss and synaptic alterations in Alzheimer's disease. Mol. Neurobiol. 37, 73–82 (doi:10.1007/s12035-008-8018-z) [DOI] [PubMed] [Google Scholar]
- 92.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. 2006. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843 (doi:10.1016/j.neuron.2006.10.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. 2007. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875 (doi:10.1523/JNEUROSCI.4970-06.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Snyder EM, et al. 2005. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 8, 1051–1058 (doi:10.1038/nn1503) [DOI] [PubMed] [Google Scholar]
- 95.Olsen KM, Sheng M. 2012. NMDA receptors and BAX are essential for Abeta impairment of LTP. Sci. Rep. 2, 225 (doi:10.1038/srep00225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. 2009. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 (doi:10.1038/nature07767) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 97.Simon DJ, et al. 2012. A caspase cascade regulating developmental axon degeneration. J. Neurosci. 32, 17 540–17 553 (doi:10.1523/JNEUROSCI.3012-12.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA. 2008. Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J. Neurophysiol. 99, 1515–1522 (doi:10.1152/jn.00598.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li H, et al. 2008. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl Acad. Sci. USA 105, 2169–2174 (doi:10.1073/pnas.0711647105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. 2002. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110, 443–455 (doi:10.1016/S0092-8674(02)00897-8) [DOI] [PubMed] [Google Scholar]
- 101.Qin Y, et al. 2005. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 19, 2000–2015 (doi:10.1101/gad.342205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jurado S, Benoist M, Lario A, Knafo S, Petrok CN, Esteban JA. 2010. PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J. 29, 2827–2840 (doi:10.1038/emboj.2010.160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beurel E, Jope RS. 2006. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Progr. Neurobiol. 79, 173–189 (doi:10.1016/j.pneurobio.2006.07.006) [DOI] [PMC free article] [PubMed] [Google Scholar]